Mechanism of the Dual Activities of Human CYP17A1 and Binding to Anti-Prostate Cancer Drug Abiraterone Revealed by a Novel V366M Mutation Causing 17,20 Lyase Deficiency

 ,
, 
Abstract
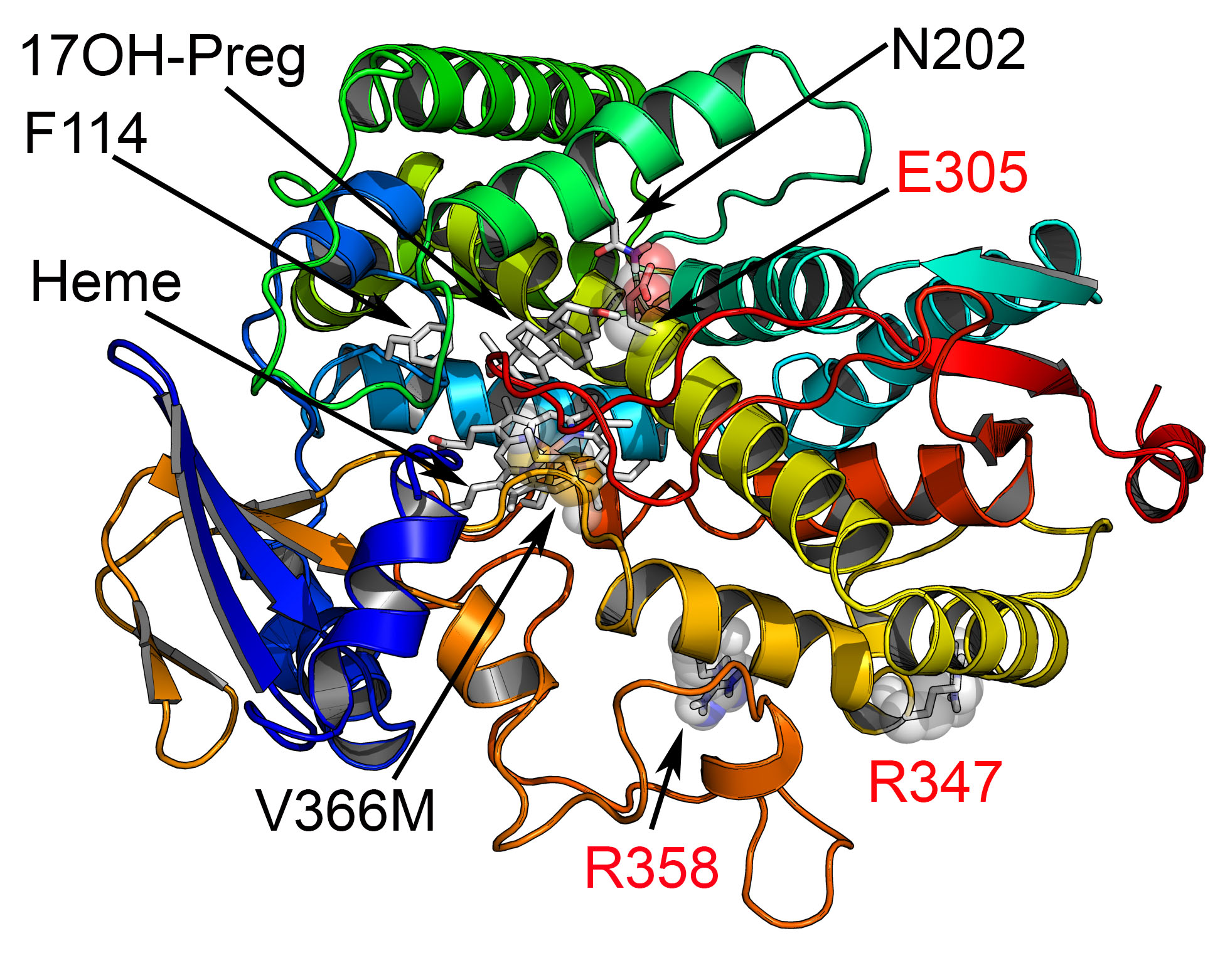
1. Introduction
2. Results
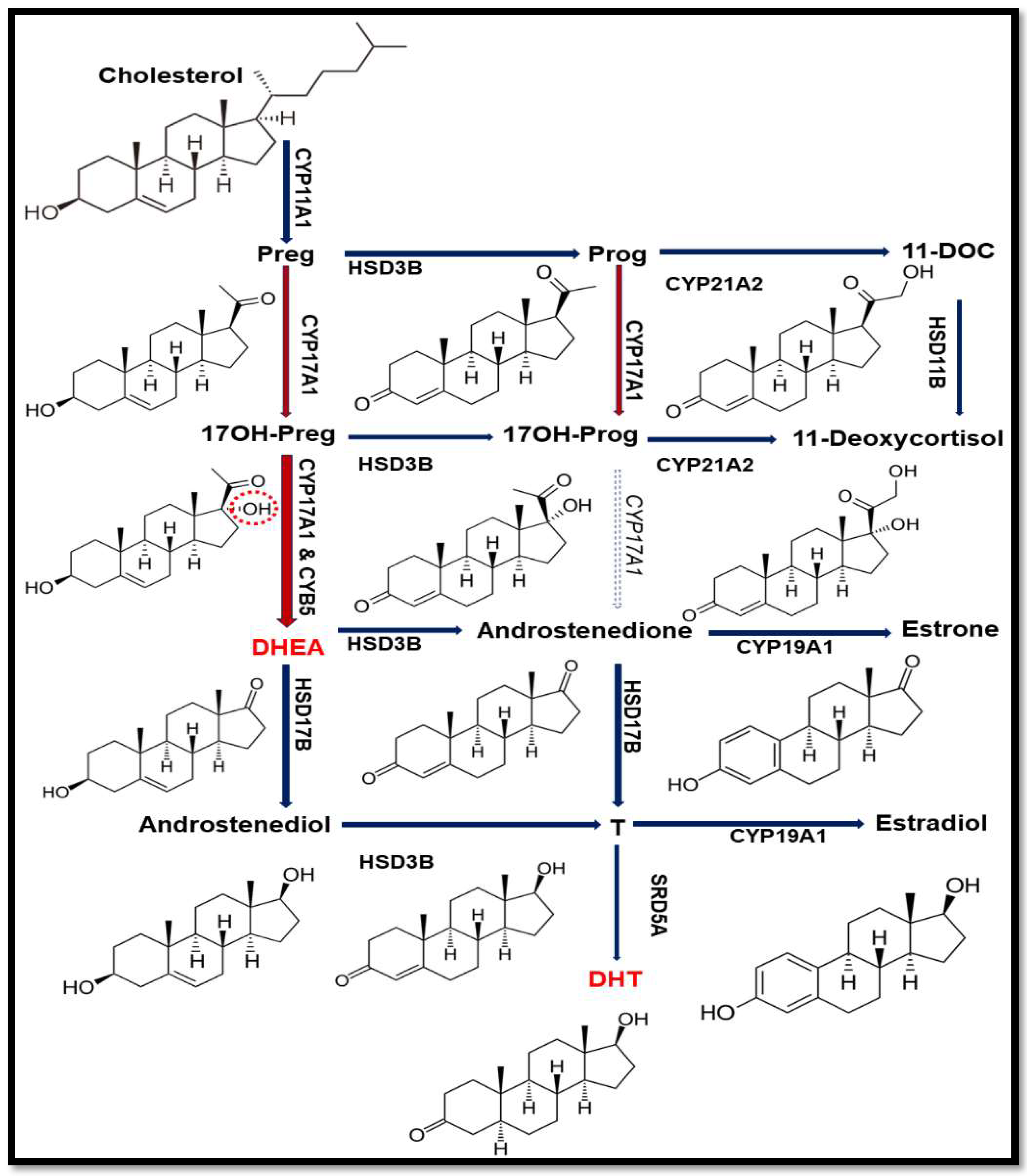
2.1. Case Report and Genetic Analysis of the Patient
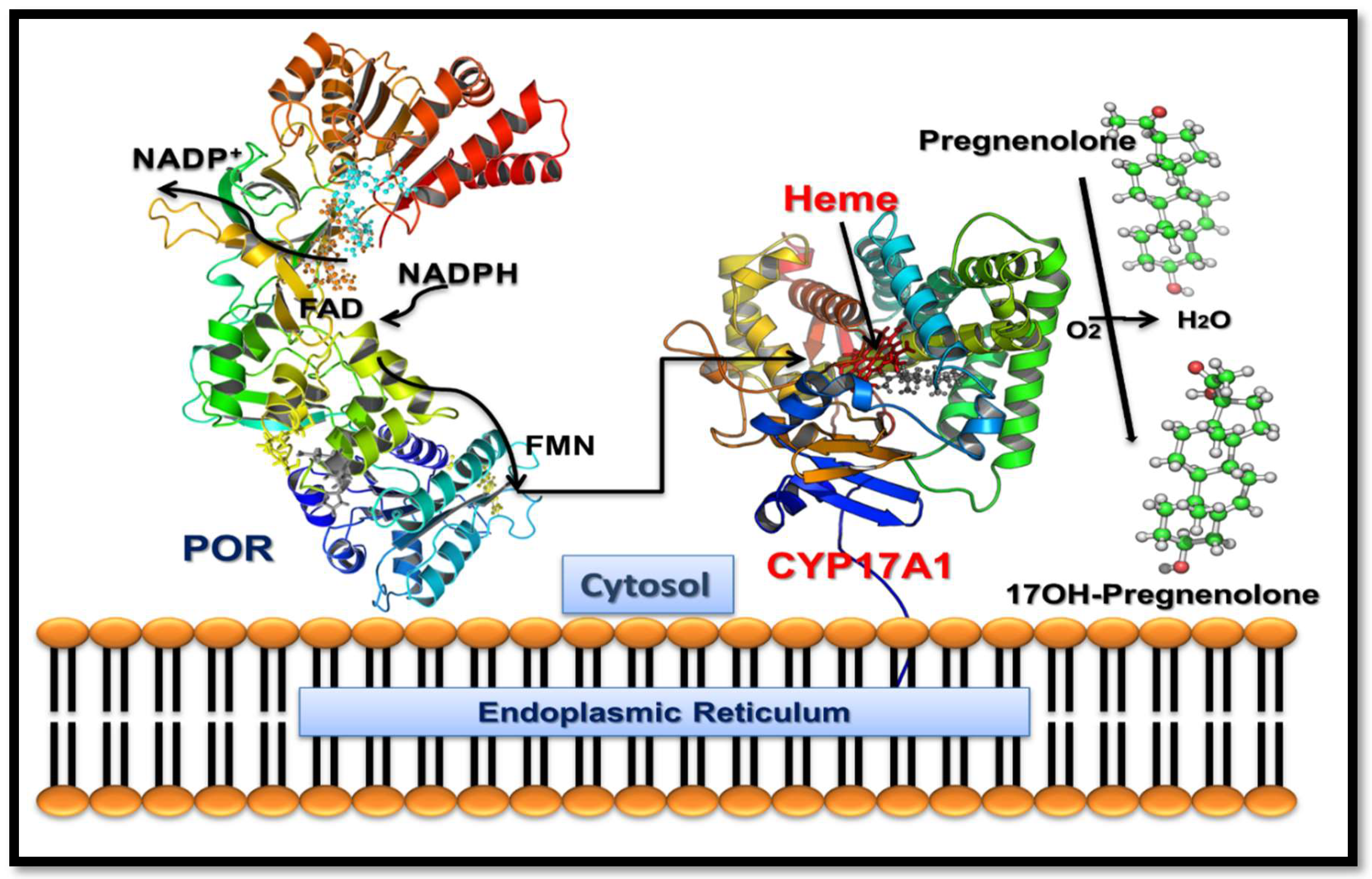
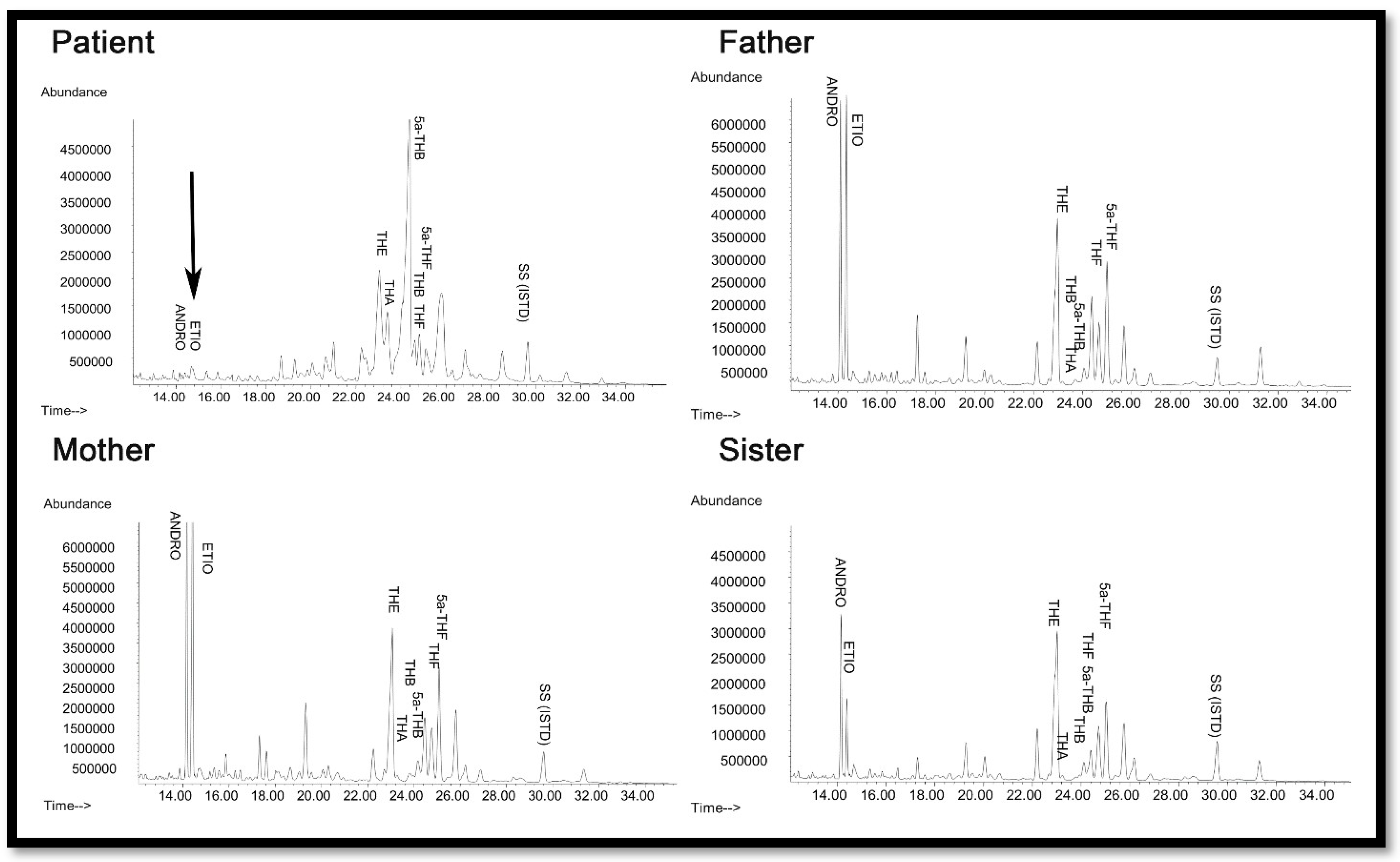
2.2. Steroid Analysis
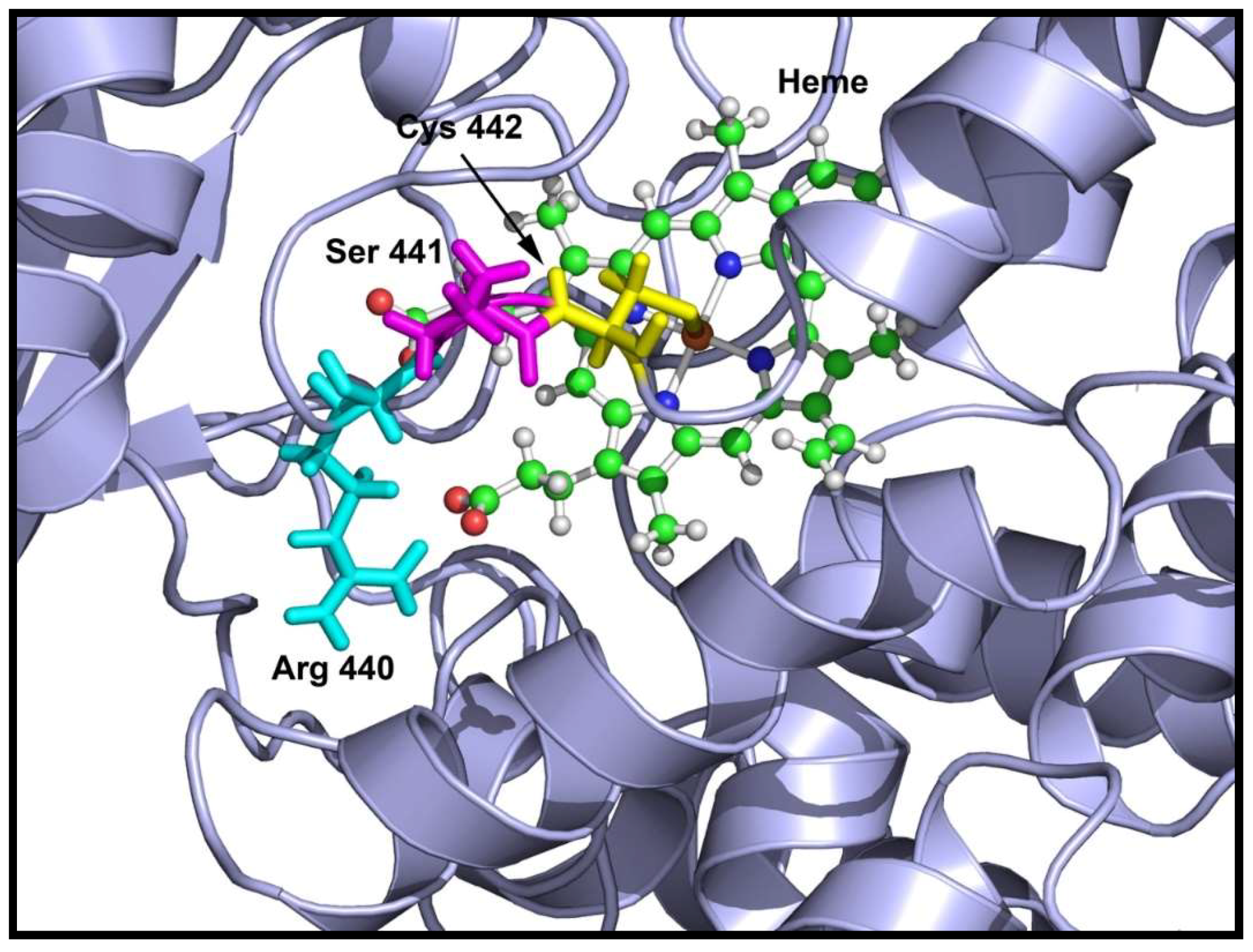
2.3. Loss of 17,20 Lyase Activity of CYP17A1 by the V366M Mutation
2.4. The 17OH-PREG is Not an Effective Inhibitor of 17α-Hydroxylase Reaction by the V366M Mutant
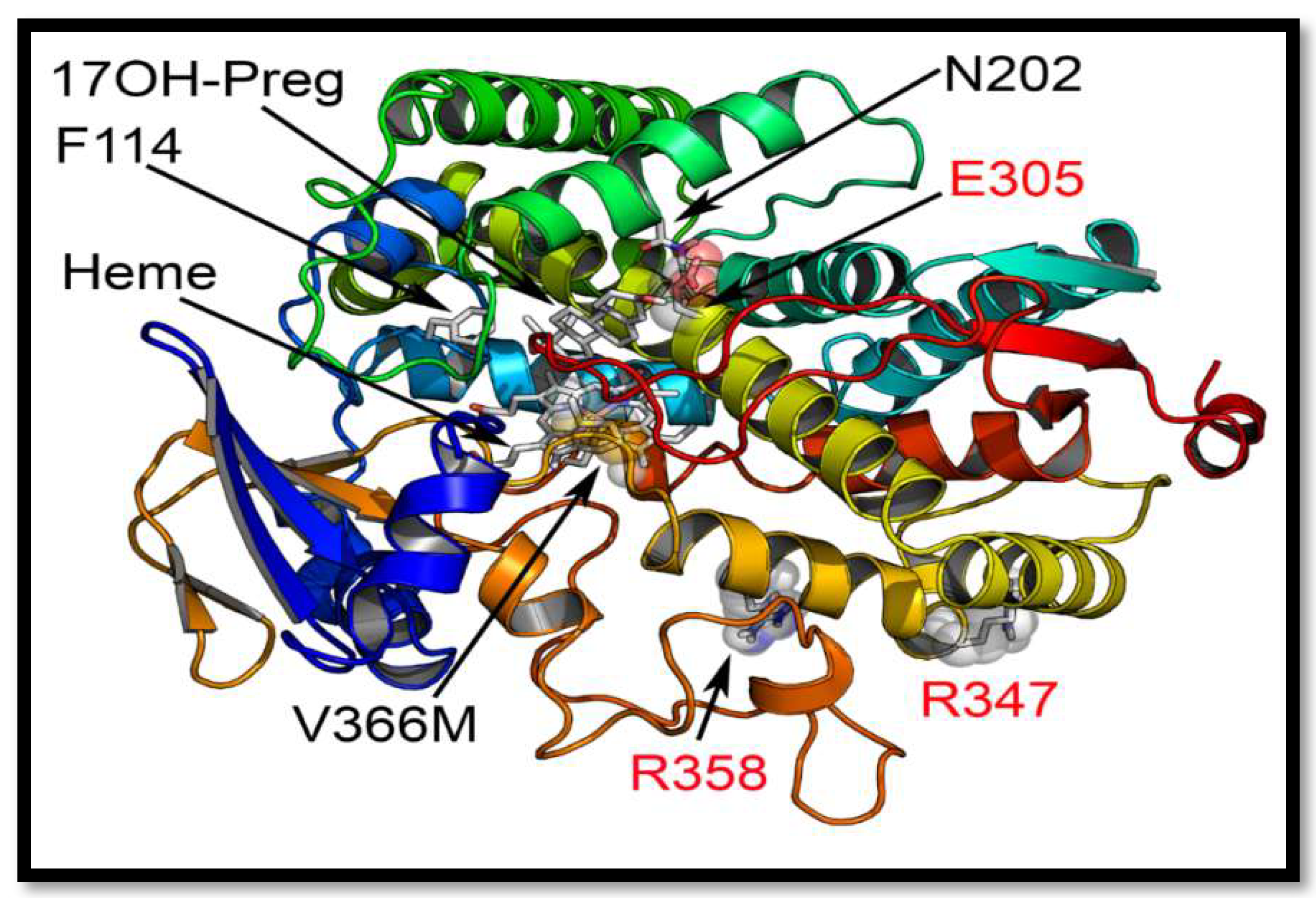
2.5. Computational Structural Analysis by Molecular Dynamics
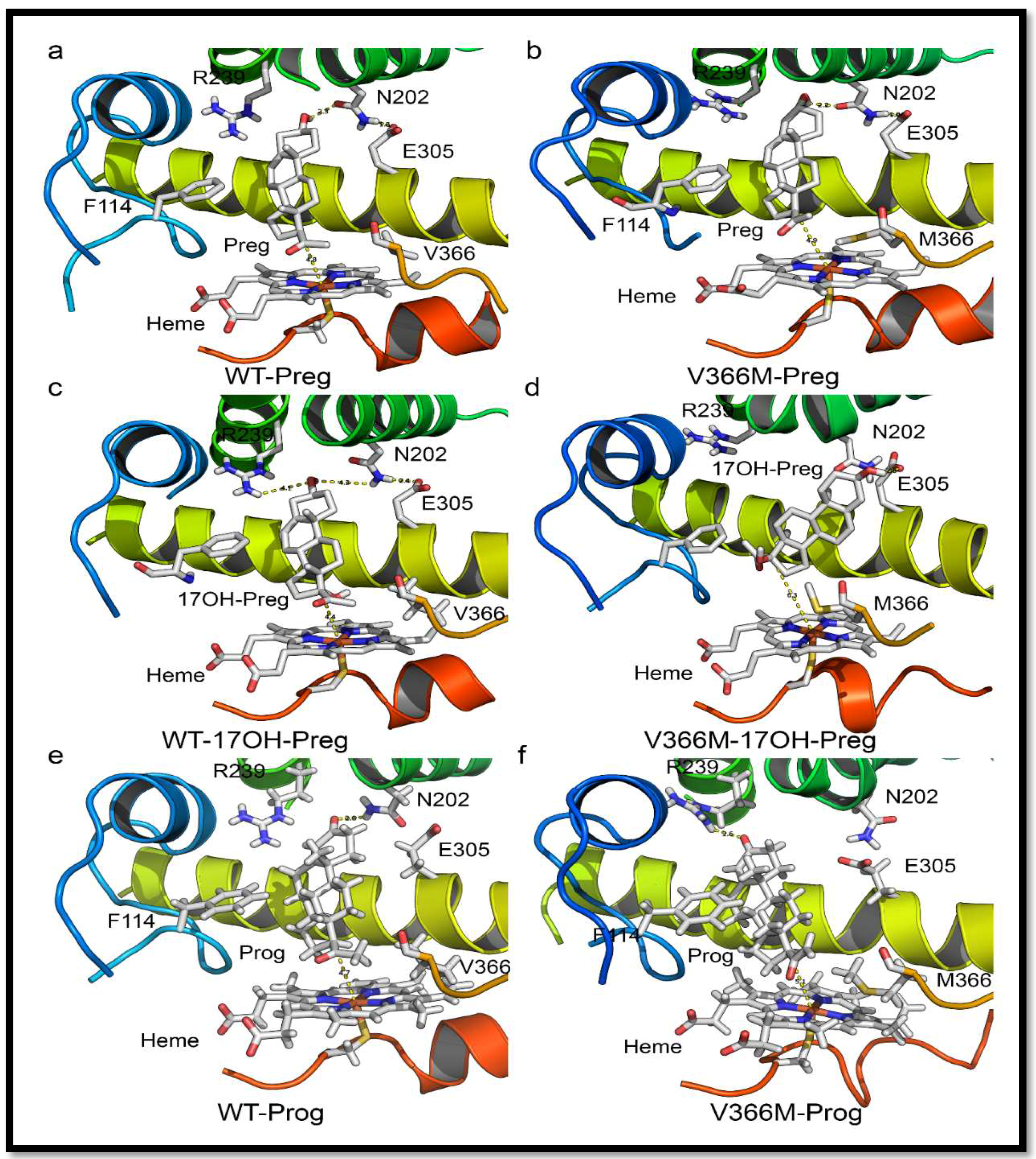
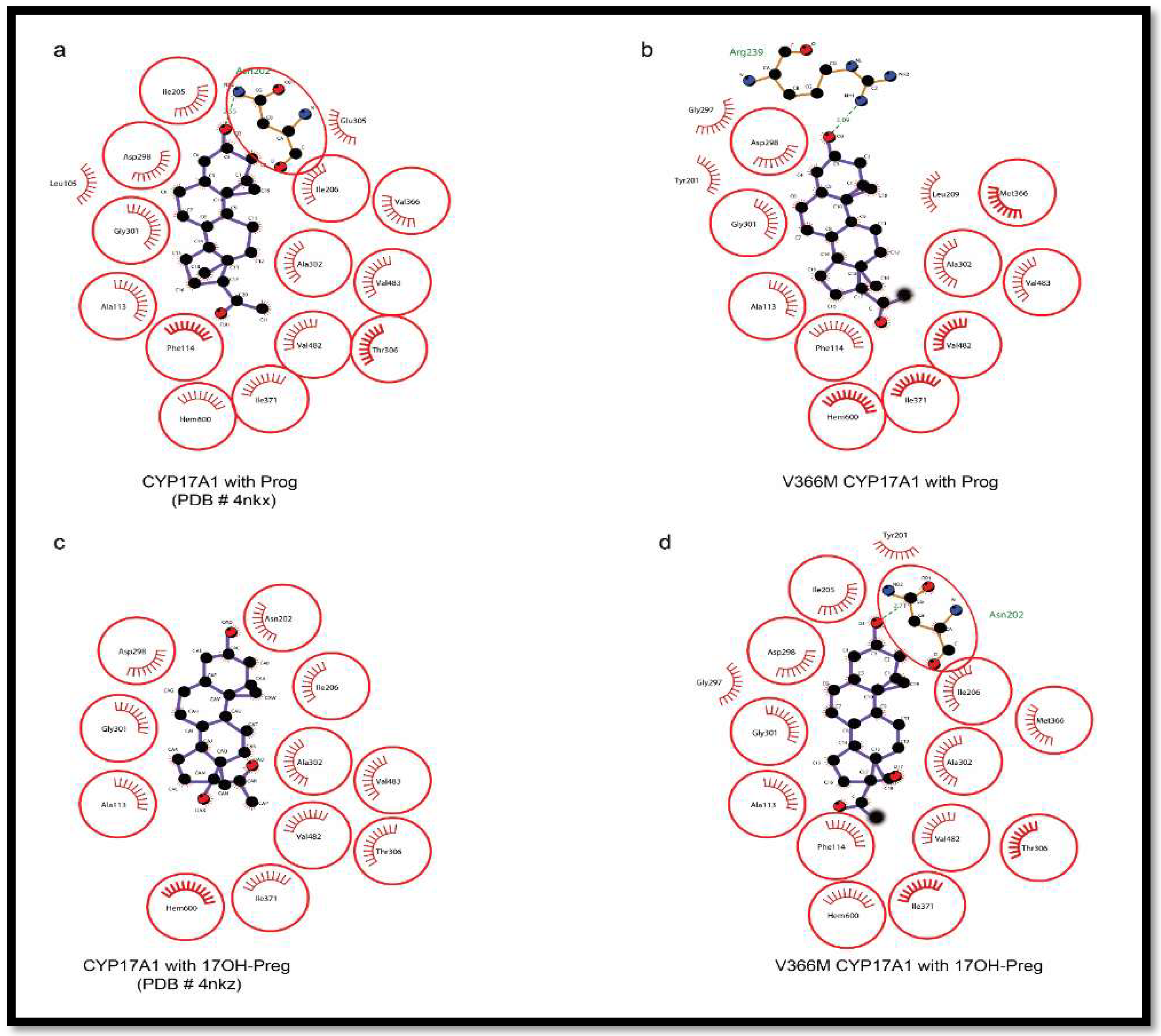
2.6. Substrate- and Inhibitor-Binding Analysis
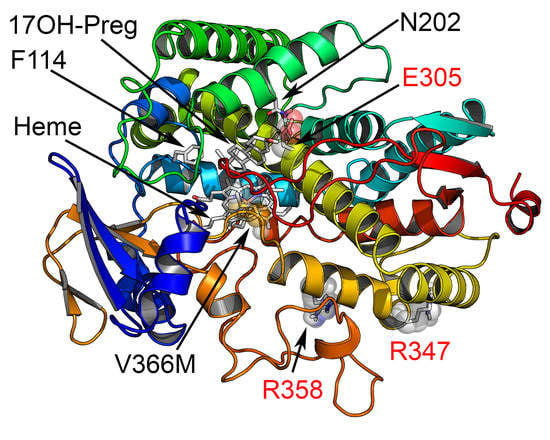
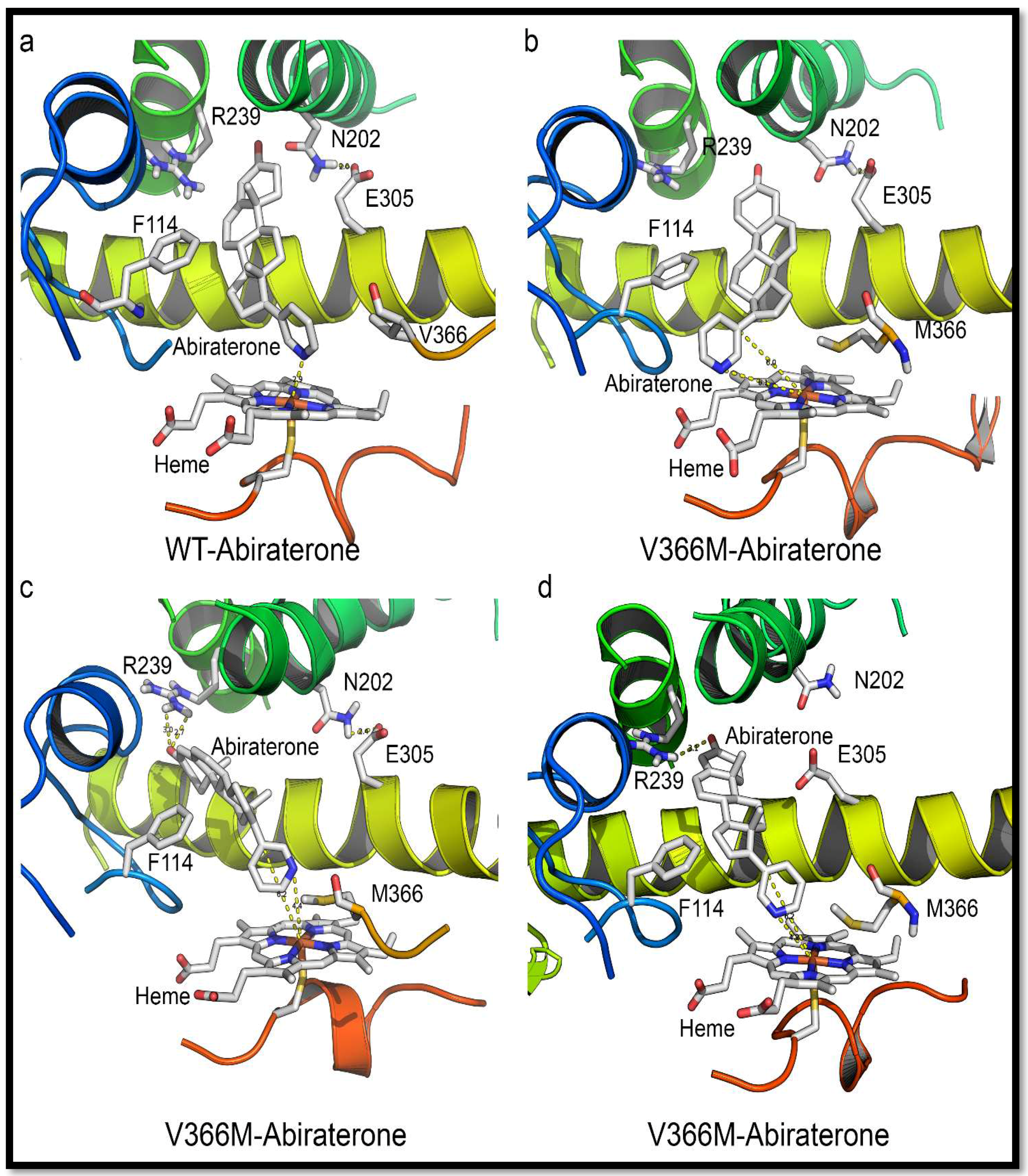
2.7. Mechanism of Steroid/Abiraterone Binding and Action in Relation to the V366M Mutation
3. Discussion
4. Materials and Methods
4.1. Human Subjects
4.2. Genetic Analysis
4.3. Steroid Profiling from 24-h Urine Samples
4.4. Recombinant Protein Expression
4.5. In Vitro Enzyme Kinetic Analysis of Identified CYP17A1 Mutations
4.6. Heme and P450 Measurements
4.7. Substrate-Binding Assay
4.8. Protein Structure Analysis of WT and Mutant CYP17A1
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Omura, T.; Sato, R. The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J. Biol. Chem. 1964, 239, 2370–2378. [Google Scholar] [PubMed]
- Pandey, A.V.; Flück, C.E. NADPH P450 oxidoreductase: Structure, function, and pathology of diseases. Pharmacol. Ther. 2013, 138, 229–254. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [PubMed]
- Zuber, M.X.; Simpson, E.R.; Waterman, M.R. Expression of bovine 17 alpha-hydroxylase cytochrome P-450 cDNA in nonsteroidogenic (COS 1) cells. Science 1986, 234, 1258–1261. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.C.; Picado-Leonard, J.; Haniu, M.; Bienkowski, M.; Hall, P.F.; Shively, J.E.; Miller, W.L. Cytochrome P450c17 (steroid 17 alpha-hydroxylase/17,20 lyase): Cloning of human adrenal and testis cDNAs indicates the same gene is expressed in both tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Nakajin, S.; Shinoda, M.; Haniu, M.; Shively, J.E.; Hall, P.F. C21 steroid side chain cleavage enzyme from porcine adrenal microsomes. Purification and characterization of the 17 alpha-hydroxylase/C17,20-lyase cytochrome P-450. J. Biol. Chem. 1984, 259, 3971–3976. [Google Scholar] [PubMed]
- Vasaitis, T.S.; Bruno, R.D.; Njar, V.C. CYP17 inhibitors for prostate cancer therapy. J. Steroid Biochem. Mol. Biol. 2011, 125, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Reid, A.H.M.; Auchus, R.; Hughes, B.A.; Cassidy, A.M.; Thompson, E.; Oommen, N.B.; Folkerd, E.; Dowsett, M.; Arlt, W.; et al. Clinical and biochemical consequences of CYP17A1 inhibition with abiraterone given with and without exogenous glucocorticoids in castrate men with advanced prostate cancer. J. Clin. Endocrinol. Metab. 2012, 97, 507–516. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; Logothetiset, C.J.; Molinaal, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B.; Saad, F.; et al. Abiraterone and Increased Survival in Metastatic Prostate Cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Reid, A.H.; Yap, T.A.; Raynaud, F.; Dowsett, M.; Settatree, S.; Barrett, M.; Parker, C.; Martins, V.; Folkerd, E.; et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J. Clin. Oncol. 2008, 26, 4563–4571. [Google Scholar] [CrossRef] [PubMed]
- Malikova, J.; Brixius-Anderko, S.; Udhane, S.S.; Parween, S.; Dick, B.; Bernhardt, R.; Pandey, A.V. CYP17A1 inhibitor abiraterone, an anti-prostate cancer drug, also inhibits the 21-hydroxylase activity of CYP21A2. J. Steroid Biochem. Mol. Biol. 2017, 174, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.Y.; Junk, K.W.; Coon, M.J. Resolution of the cytochrome P-450-containing w-hydroxylation system of liver microsomes into three components. J. Biol. Chem. 1969, 244, 3714–3721. [Google Scholar] [PubMed]
- Flück, C.E.; Tajima, T.; Pandey, A.V.; Arlt, W.; Okuhara, K.; Verge, C.F.; Jabs, E.W.; Mendonça, B.B.; Fujieda, K.; Miller, W.L. Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat. Genet. 2004, 36, 228–230. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Globerman, H.; Gertner, J.M.; Kagawa, N.; Waterman, M.R. Expression and purification of functional human 17 alpha-hydroxylase/17,20-lyase (P450c17) in Escherichia coli. Use of this system for study of a novel form of combined 17 alpha-hydroxylase/17,20-lyase deficiency. J. Biol. Chem. 1993, 268, 19681–19689. [Google Scholar] [PubMed]
- Burkhard, F.Z.; Parween, S.; Udhane, S.S.; Flück, C.E.; Pandey, A.V. P450 Oxidoreductase deficiency: Analysis of mutations and polymorphisms. J. Steroid Biochem. Mol. Biol. 2017, 165 Pt A, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.V.; Miller, W.L. Regulation of 17,20 lyase activity by cytochrome b5 and by serine phosphorylation of P450c17. J. Biol. Chem. 2005, 280, 13265–13271. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.V.; Mellon, S.H.; Miller, W.L. Protein phosphatase 2A and phosphoprotein SET regulate androgen production by P450c17. J. Biol. Chem. 2003, 278, 2837–2844. [Google Scholar] [CrossRef] [PubMed]
- Auchus, R.J.; Lee, T.C.; Miller, W.L. Cytochrome b5 augments the 17,20-lyase activity of human P450c17 without direct electron transfer. J. Biol. Chem. 1998, 273, 3158–3165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.H.; Rodriguez, H.; Ohno, S.; Miller, W.L. Serine phosphorylation of human P450c17 increases 17,20-lyase activity: Implications for adrenarche and the polycystic ovary syndrome. Proc. Natl. Acad. Sci. USA 1995, 92, 10619–10623. [Google Scholar] [CrossRef] [PubMed]
- Idkowiak, J.; Randell, T.; Dhir, V.; Patel, P.; Shackleton, C.H.; Taylor, N.F.; Krone, N.; Arlt, W. A missense mutation in the human cytochrome b5 gene causes 46,XY disorder of sex development due to true isolated 17,20 lyase deficiency. J. Clin. Endocrinol. Metab. 2012, 97, E465–E475. [Google Scholar] [CrossRef] [PubMed]
- DeVore, N.M.; Scott, E.E. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature 2012, 482, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Petrunak, E.M.; DeVore, N.M.; Porubsky, P.R.; Scott, E.E. Structures of human steroidogenic cytochrome P450 17A1 with substrates. J. Biol. Chem. 2014, 289, 32952–32964. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Petrunak, E.M.; Estrada, D.F.; Scott, E.E. Structural insights into the function of steroidogenic cytochrome P450 17A1. Mol. Cell Endocrinol. 2017, 441, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Auchus, R.J. Steroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic. J. Steroid Biochem. Mol. Biol. 2017, 165, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Mak, P.J.; Gregory, M.C.; Denisov, I.G.; Sligar, S.G.; Kincaid, J.R. Unveiling the crucial intermediates in androgen production. Proc. Natl. Acad. Sci. USA 2015, 112, 15856–15861. [Google Scholar] [CrossRef] [PubMed]
- Geller, D.H.; Auchus, R.J.; Mendonça, B.B.; Miller, W.L. The genetic and functional basis of isolated 17,20-lyase deficiency. Nat. Genet. 1997, 17, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Sherbet, D.P.; Tiosano, D.; Kwist, K.M.; Hochberg, Z.; Auchus, R.J. CYP17 mutation E305G causes isolated 17,20-lyase deficiency by selectively altering substrate binding. J. Biol. Chem. 2003, 278, 48563–48569. [Google Scholar] [CrossRef] [PubMed]
- Van Den Akker, E.L.; Koper, J.W.; Boehmer, A.L.; Themmen, A.P.; Verhoef-Post, M.; Timmerman, M.A.; Otten, B.J.; Drop, S.L.; De Jong, F.H. Differential inhibition of 17alpha-hydroxylase and 17,20-lyase activities by three novel missense CYP17 mutations identified in patients with P450c17 deficiency. J. Clin. Endocrinol. Metab. 2002, 87, 5714–5721. [Google Scholar] [CrossRef] [PubMed]
- Tiosano, D.; Knopf, C.; Koren, I.; Wudy, S.A. Metabolic evidence for impaired 17alpha-hydroxylase activity in a kindred bearing the E305G mutation for isolate 17,20-lyase activity. Eur. J. Endocrinol. 2008, 158, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Parween, S.; Roucher-Boulez, F.; Flück, C.E.; Lienhardt-Roussie, A.; Mallet, D.; Morel, Y.; Pandey, A.V. P450 Oxidoreductase Deficiency: Loss of Activity Caused by Protein Instability From a Novel L374H Mutation. J. Clin. Endocrinol. Metab. 2016, 101, 4789–4798. [Google Scholar] [CrossRef] [PubMed]
- Flück, C.E.; Pandey, A.V. Impact on CYP19A1 activity by mutations in NADPH cytochrome P450 oxidoreductase. J. Steroid Biochem. Mol. Biol. 2017, 165 Pt A, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.V.; Sproll, P. Pharmacogenomics of human P450 oxidoreductase. Front. Pharmacol. 2014, 5, 103. [Google Scholar] [CrossRef] [PubMed]
- Udhane, S.S.; Parween, S.; Kagawa, N.; Pandey, A.V. Altered CYP19A1 and CYP3A4 Activities Due to Mutations A115V, T142A, Q153R and P284L in the Human P450 Oxidoreductase. Front. Pharmacol. 2017, 8, 580. [Google Scholar] [CrossRef] [PubMed]
- Geller, D.H.; Auchus, R.J.; Miller, W.L. P450c17 mutations R347H and R358Q selectively disrupt 17,20-lyase activity by disrupting interactions with P450 oxidoreductase and cytochrome b5. Mol. Endocrinol. 1999, 13, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Flück, C.E.; Meyer-Böni, M.; Pandey, A.V.; Kempná, P.; Miller, W.L.; Schoenle, E.J.; Biason-Lauber, A. Why boys will be boys: Two pathways of fetal testicular androgen biosynthesis are needed for male sexual differentiation. Am. J. Hum. Genet. 2011, 89, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Flück, C.E.; Pandey, A.V. Steroidogenesis of the testis—New genes and pathways. Ann. Endocrinol. 2014, 75, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Biason-Lauber, A.; Miller, W.L.; Pandey, A.V.; Fluck, C.E. Of marsupials and men: “Backdoor” dihydrotestosterone synthesis in male sexual differentiation. Mol. Cell Endocrinol. 2013, 371, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Audi, L.; Fernández-Cancio, M.; Carrascosa, A.; Andaluz, P.; Torán, N.; Piró, C.; Vilaró, E.; Vicens-Calvet, E.; Gussinyé, M.; Albisu, M.A.; et al. Novel (60%) and recurrent (40%) androgen receptor gene mutations in a series of 59 patients with a 46,XY disorder of sex development. J. Clin. Endocrinol. Metab. 2010, 95, 1876–1888. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cancio, M.; Audí, L.; Andaluz, P.; Torán, N.; Piró, C.; Albisu, M.; Gussinyé, M.; Yeste, D.; Clemente, M.; Martínez-Mora, J.; et al. SRD5A2 gene mutations and polymorphisms in Spanish 46,XY patients with a disorder of sex differentiation. Int. J. Androl. 2011, 34 Pt 2, e526–e535. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Ohno, T.; Sakaki, T.; Akiyoshi-Shibata, M.; Yabusaki, Y.; Imai, T.; Kominami, S. Kinetic analysis of successive reactions catalyzed by bovine cytochrome p450(17alpha,lyase). Biochemistry 1998, 37, 2800–2806. [Google Scholar] [CrossRef] [PubMed]
- Soucy, P.; Van, L.-T. Conversion of pregnenolone to DHEA by human 17α-hydroxylase/17,20-lyase (P450c17). Eur. J. Biochem. 2000, 267, 3243–3247. [Google Scholar] [CrossRef] [PubMed]
- Brock, B.J.; Waterman, M.R. Biochemical Differences between Rat and Human Cytochrome P450c17 Support the Different Steroidogenic Needs of These Two Species. Biochemistry 1999, 38, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.V.; Kempná, P.; Hofer, G.; Mullis, P.E.; Flück, C.E. Modulation of human CYP19A1 activity by mutant NADPH P450 oxidoreductase. Mol. Endocrinol. 2007, 21, 2579–2595. [Google Scholar] [CrossRef] [PubMed]
- Flück, C.E.; Mullis, P.E.; Pandey, A.V. Reduction in hepatic drug metabolizing CYP3A4 activities caused by P450 oxidoreductase mutations identified in patients with disordered steroid metabolism. Biochem. Biophys. Res. Commun. 2010, 401, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Nicolo, C.; Flück, C.E.; Mullis, P.E.; Pandey, A.V. Restoration of mutant cytochrome P450 reductase activity by external flavin. Mol. Cell Endocrinol. 2010, 321, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Riddick, D.S.; Ding, X.; Wolf, C.R.; Porter, T.D.; Pandey, A.V.; Zhang, Q.; Gu, J.; Finn, R.D.; Ronseaux, S.; McLaughlin, L.A.; et al. NADPH-cytochrome P450 oxidoreductase: Roles in physiology, pharmacology, and toxicology. Drug Metab. Dispos. 2013, 41, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Zalewski, A.; Ma, N.S.; Legeza, B.; Renthal, N.; Flück, C.E.; Pandey, A.V. Vitamin D-Dependent Rickets Type 1 Caused by Mutations in CYP27B1 Affecting Protein Interactions With Adrenodoxin. J. Clin. Endocrinol. Metab. 2016, 101, 3409–3418. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.M.; Im, Sa.; Pearl, N.M.; Turcu, A.F.; Waskell, J.R.L.; Auchus, R.J. Cytochrome b5 Activates the 17,20-Lyase Activity of Human Cytochrome P450 17A1 by Increasing the Coupling of NADPH Consumption to Androgen Production. Biochemistry 2016, 55, 4356–4365. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, F.K.; Gonzalez, E.; Auchus, R.J.; Guengerich, F.P. Mechanism of 17alpha,20-Lyase and New Hydroxylation Reactions of Human Cytochrome P450 17A1: 18O Labeling And Oxygen Surrogate Evidence For A Role Of A Perferryl Oxygen. J. Biol. Chem. 2016, 291, 17143–17164. [Google Scholar] [CrossRef] [PubMed]
- Duggal, R.; Liu, Y.; Gregory, M.C.; Denisov, I.G.; Kincaid, J.R.; Sligar, S.G. Evidence that cytochrome b5 acts as a redox donor in CYP17A1 mediated androgen synthesis. Biochem. Biophys. Res. Commun. 2016, 477, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Estrada, D.F.; Skinner, A.L.; Laurence, J.S.; Scott, E.E. Human cytochrome P450 17A1 conformational selection: Modulation by ligand and cytochrome b5. J. Biol. Chem. 2014, 289, 14310–14320. [Google Scholar] [CrossRef] [PubMed]
- Pallan, P.S.; Nagy, L.D.; Lei, L.; Gonzalez, E.; Kramlinger, V.M.; Azumaya, C.M.; Wawrzak, Z.; Waterman, M.R.; Guengerich, F.P.; Egli, M. Structural and kinetic basis of steroid 17alpha,20-lyase activity in teleost fish cytochrome P450 17A1 and its absence in cytochrome P450 17A2. J. Biol. Chem. 2015, 290, 3248–3268. [Google Scholar] [CrossRef] [PubMed]
- Ramudo Cela, L.; Balea-Filgueiras, J.; Vizoso-Hermida, J.R.; Martín-Herranz, I. Study of cases of abiraterone discontinuation due to toxicity in pre-chemotherapy after 1 year's experience. J. Oncol. Pharm. Pract. 2017, 23, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Alyamani, M.; Li, J.; Rogacki, K.; Abazeed, M.; Upadhyay, S.K.; Balk, S.P.; Taplin, M.E.; Auchus, R.J.; Sharifi, N. Redirecting abiraterone metabolism to fine-tune prostate cancer anti-androgen therapy. Nature 2016, 533, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Bonomo, S.; Hansen, C.H.; Petrunak, E.M.; Scott, E.E.; Styrishave, B.; Jørgensen, F.S.; Olsen, L. Promising Tools in Prostate Cancer Research: Selective Non-Steroidal Cytochrome P450 17A1 Inhibitors. Sci. Rep. 2016, 6, 29468. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, N. Prostate cancer: CYP17A1 inhibitor failure-lessons for future drug development. Nat. Rev. Urol. 2015, 12, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Udhane, S.S.; Dick, B.; Hu, Q.; Hartmann, R.W.; Pandey, A.V. Specificity of anti-prostate cancer CYP17A1 inhibitors on androgen biosynthesis. Biochem. Biophys. Res. Commun. 2016, 477, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Monno, S.; Ogawa, H.; Date, T.; Fujioka, M.; Miller, W.L.; Kobayashi, M. Mutation of histidine 373 to leucine in cytochrome P450c17 causes 17 alpha-hydroxylase deficiency. J. Biol. Chem. 1993, 268, 25811–25817. [Google Scholar] [PubMed]
- Quattropani, C.; Vogt, B.; Odermatt, A.; Dick, B.; Frey, B.M.; Frey, F.J. Reduced activity of 11 beta-hydroxysteroid dehydrogenase in patients with cholestasis. J. Clin. Investig. 2001, 108, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Shackleton, C.H. Mass spectrometry in the diagnosis of steroid-related disorders and in hypertension research. J. Steroid Biochem. Mol. Biol. 1993, 45, 127–140. [Google Scholar] [CrossRef]
- Huang, N.; Pandey, A.V.; Agrawal, V.; Reardon, W.; Lapunzina, P.D.; Mowat, D.; Jabs, E.W.; Van Vliet, G.; Sack, J.; Flück, C.E.; et al. Diversity and function of mutations in p450 oxidoreductase in patients with Antley-Bixler syndrome and disordered steroidogenesis. Am. J. Hum. Genet. 2005, 76, 729–749. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.V.; Joshi, S.K.; Tekwani, B.L.; Chauhan, V.S. A colorimetric assay for heme in biological samples using 96-well plates. Anal. Biochem. 1999, 268, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Darden, T.; Nabuurs, S.B.; Finkelstein, A.; Vriend, G. Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins 2004, 57, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Vriend, G. WHAT IF: A molecular modeling and drug design program. J. Mol. Graph. 1990, 8, 52–56. [Google Scholar] [CrossRef]
- Canutescu, A.A.; Shelenkov, A.A.; Dunbrack, R.L. A graph-theory algorithm for rapid protein side-chain prediction. Protein Science 2003, 12, 2001–2014. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Tirado-Rives, J. Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc. Natl. Acad. Sci. USA 2005, 102, 6665–6670. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 17OH Steroids Basal | 17OH Steroids Stimulated | Cortisol Basal | Cortisol Stimulated | Activities (% of WT) | Ref | |
|---|---|---|---|---|---|---|
| 17OHase | 17,20 lyase | |||||
| Normal | Hyperresponsive | Normal | Areactive | 65 | 5 | Geller 1997 [27] |
| Elevated | Hyperresponsive | Normal | Areactive | 65 | 5 | Geller 1997 [27] |
| Slightly elevated | Not reactive | Low normal | Areactive | van den Akker 2002 [29] | ||
| Normal | Normal | Low normal | Areactive | van den Akker 2002 [29] | ||
| Normal/Elevated | Reactive | Low normal | Hyporeactive | 60 | 0 | van den Akker 2002 [29] |
| Normal/Elevated | Reactive | Low normal | Hyporeactive | 60 | 0 | van den Akker 2002 [29] |
| Normal/Elevated | Reactive | Low | Hyporeactive | 100 | 0 | Sherbet 2003 [28] |
| Normal/Elevated | Normal | Low normal | Hyporeactive | Tiosano 2008 [30] | ||
| Normal/Elevated | Normal | Low normal | Hyporeactive | Tiosano 2008 [30] | ||
| Normal/Elevated | Normal | Low normal | Hyporeactive | Tiosano 2008 [30] | ||
| Normal/Elevated | Normal | Low normal | Hyporeactive | Tiosano 2008 [30] | ||
| Normal/Elevated | Normal | Low normal | Hyporeactive | Tiosano 2008 [30] | ||
| Normal/Elevated | Normal | Low normal | Hyporeactive | Tiosano 2008 [30] | ||
| Normal/low | Hyporeactive | Low normal | Areactive | 43 | 0/0 | This report |
| Age | 3 Months | 5 Months | 20 Months | 5 Years | 6.5 Years | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Parameter | Unit | Basal | Normal Range | hCG Test (500 IU/d × 3) | Normal Range | Basal | Normal Range | ACTH-Stimulated | Basal | Basal | Normal Range |
| Sodium | mEq/L | 140 | 136–145 | - | 141 | 136–145 | - | 144 | 136–145 | ||
| Potassium | mEq/L | 4.0 | 3.5–5.1 | - | 4.6 | 3.5–5.1 | - | 4.7 | 3.5–5.1 | ||
| ACTH | pg/mL | 81 | 9–50 | 82 | 9–50 | 66 | 9–50 | - | 62 | 46 | 9–50 |
| Progesterone | ng/dL | 222 | 5–80 | - | 251 | 10–50 | 425 | 522 | 339 | 10–50 | |
| 17OH Preg | ng/dL | 132 | 60–830 | 27 | 60–830 | 13 | 10–50 | - | - | - | |
| 17OHProg | ng/dL | 300 | 40–460 | 40 | 40–460 | 29 | 19–159 | 35 | 110 | 80 | 10–470 |
| DHEA-S | µg/dL | <5 | 5–62 | <5 | 5–62 | <5 | 5–190 | - | <5 | <5 | 5–95 |
| 11-Deoxycortisol | ng/dL | 1100 | 1450 ± 790 | 200 | 1450 ± 790 | 140 | 186 ± 116 | - | 87 | - | 205 ± 108 |
| Cortisol | µg/dL | 3.7 | 4.3–22.2 | 8.6 | 4.3–22.2 | 8.9 | 4.3–22.2 | 7.8 | 7.1 | 4.1 | 4.3–22.4 |
| Δ4A | ng/dL | <30 | 63 ± 39 | <3 | 63 ± 39 | <30 | 30–330 | <30 | - | <30 | 30–330 |
| Testosterone | ng/dL | 7 | 140 ± 132 | <4 | 140 ± 132 | 28 | 15–30 | 16 | - | - | |
| LH | IU/L | - | - | 0.3 | 0.2–1.0 | - | <0.07 | - | 0.2–1.0 | ||
| FSH | IU/L | - | - | 9 | 0.4–2.0 | - | 4.3 | - | 0.4–2.0 | ||
| PRA | ng/mL/h | - | - | 1.8 | 0.6–21.3 | - | - | 0.1 | 0.3–6.4 | ||
| Aldosterone | ng/dL | - | - | 12.1 | 14–114 | 11.5 | 6.2 | 9–66 | |||
| CYP17A1 Variant | 17α-HydroxylasePROG to 17OH-PROG | 17,20 Lyase17OH-PREG to DHEA | |||
|---|---|---|---|---|---|
| Km (µM) | Vmax (min−1) | Cat Eff (%) | Km (µM) | Vmax (min−1) | |
| WT | 6.1 ± 0.7 | 0.71 | 100 | 0.92 ± 0.07 | 0.025 ± 0.004 |
| V366M | 8.4 ± 0.9 | 0.42 | 43 | - | - |
| S441P | - | - | - | - | - |
| Protein Preparation | Heme Content (nnol/nmol of Protein) |
|---|---|
| CYP17A1 WT | 0.93 |
| CYP17A1_V366M | 0.95 |
| CYP17A1-S441P | <0.05 |
| CYB5A | 1.0 |
| CYP17A1_WT | CYP17A1_V366M | |
|---|---|---|
| Binding studies | Kd (nM) | Kd (nM) |
| Binding of PROG | 163 ± 29 | 287 ± 35 |
| Binding of PREG | 62 ± 17 | 92 ± 15 |
| Binding of 17OH-PREG | 142 ± 38 | - |
| Binding of Abiraterone | 85 ± 23 | - |
| Inhibition studies | IC50 (µM) | IC50 (µM) |
| Inhibition of PROG 17α-hydroxylation by Abiraterone | 0.04 ± 0.01 | - |
| Inhibition of PROG 17α-hydroxylation by 17OH-PREG | 1.7 ± 0.2 | - |
| Inhibition of PROG 17α-hydroxylation by PREG | 0.9 ± 0.15 | 1.4 ± 0.2 |
| CYP17A1 Protein | Binding Energy (kcal/mol) | Dissociation Constant (nM) | Contacting Residues |
|---|---|---|---|
| WT with Prog | 10.6 | 14.66 | ALA113 PHE114 ASN202 ILE205 ILE206 LEU209 ARG239 GLY297 ASP298 GLY301 ALA302 THR306 ALA367 ILE371 VAL482 VAL483 HEME |
| M366 with Prog | 9.75 | 43.71 | ALA113 PHE114 ASN202 ILE205 ILE206 LEU209 ARG239 GLY297 ASP298 GLY301 ALA302 THR306 MET366 ALA367 ILE371 VAL482 VAL483 HEME |
| WT with Preg | 10.7 | 13.29 | ALA105 ALA113 PHE114 ILE205 ILE206 LEU209 VAL236 ARG239 GLY297 ASP298 GLY301 ALA302 GLU305 THR306 VAL366 ILE371 VAL482 VAL483 HEME |
| M366 with Preg | 10.3 | 27.11 | ALA105 SER106 ALA113 PHE114 ILE205 ILE206 LEU209 VAL236 ARG239 GLY297 ASP298 GLY301 ALA302 THR306 MET366 ILE371 VAL482 VAL483 HEME |
| WT with 17OH-Preg | 11.3 | 5.22 | ALA113 PHE114 TYR201 ASN202 ILE205 ILE206 LEU209 LEU214 ARG239 GLY297 ASP298 GLY301 ALA302 THR306 VAL366 ALA367 ILE371 VAL482 VAL483 HEME |
| M366 with 17OH-Preg | 7.0 | 60.4 | ALA113 PHE114 TYR201 ASN202 ILE205 ILE206 LEU209 ARG239 GLY297 ASP298 GLY301 ALA302 GLU305 THR306 MET366 ALA367 ILE371 VAL482 VAL483 |
| WT with Abiraterone | 12.5 | 0.69 | ALA113 PHE114 TYR201 ASN202 ILE205 ILE206 LEU209 ARG239 GLY297 ASP298 GLY301 ALA302 GLU305 THR306 VAL366 ALA367 LEU370 ILE371 VAL482 VAL483 HEME |
| V366 with Abiraterone | 8.1 | 1112.9 | ALA105 SER106 ASN107 ALA113 PHE114 TYR201 ILE205 ILE206 LEU209 ARG239 THR294 GLY297 ASP298 GLY301 ALA302 THR306 MET366 ILE371 VAL482 VAL483 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Cancio, M.; Camats, N.; Flück, C.E.; Zalewski, A.; Dick, B.; Frey, B.M.; Monné, R.; Torán, N.; Audí, L.; Pandey, A.V. Mechanism of the Dual Activities of Human CYP17A1 and Binding to Anti-Prostate Cancer Drug Abiraterone Revealed by a Novel V366M Mutation Causing 17,20 Lyase Deficiency. Pharmaceuticals 2018, 11, 37. https://doi.org/10.3390/ph11020037
Fernández-Cancio M, Camats N, Flück CE, Zalewski A, Dick B, Frey BM, Monné R, Torán N, Audí L, Pandey AV. Mechanism of the Dual Activities of Human CYP17A1 and Binding to Anti-Prostate Cancer Drug Abiraterone Revealed by a Novel V366M Mutation Causing 17,20 Lyase Deficiency. Pharmaceuticals. 2018; 11(2):37. https://doi.org/10.3390/ph11020037
Chicago/Turabian StyleFernández-Cancio, Mónica, Núria Camats, Christa E. Flück, Adam Zalewski, Bernhard Dick, Brigitte M. Frey, Raquel Monné, Núria Torán, Laura Audí, and Amit V. Pandey. 2018. "Mechanism of the Dual Activities of Human CYP17A1 and Binding to Anti-Prostate Cancer Drug Abiraterone Revealed by a Novel V366M Mutation Causing 17,20 Lyase Deficiency" Pharmaceuticals 11, no. 2: 37. https://doi.org/10.3390/ph11020037
APA StyleFernández-Cancio, M., Camats, N., Flück, C. E., Zalewski, A., Dick, B., Frey, B. M., Monné, R., Torán, N., Audí, L., & Pandey, A. V. (2018). Mechanism of the Dual Activities of Human CYP17A1 and Binding to Anti-Prostate Cancer Drug Abiraterone Revealed by a Novel V366M Mutation Causing 17,20 Lyase Deficiency. Pharmaceuticals, 11(2), 37. https://doi.org/10.3390/ph11020037