A π-Halogen Bond of Dibenzofuranones with the Gatekeeper Phe113 in Human Protein Kinase CK2 Leads to Potent Tight Binding Inhibitors

 ,
,  and
and 
Abstract
1. Introduction
2. Results
2.1. Synthesis of 4a and 5
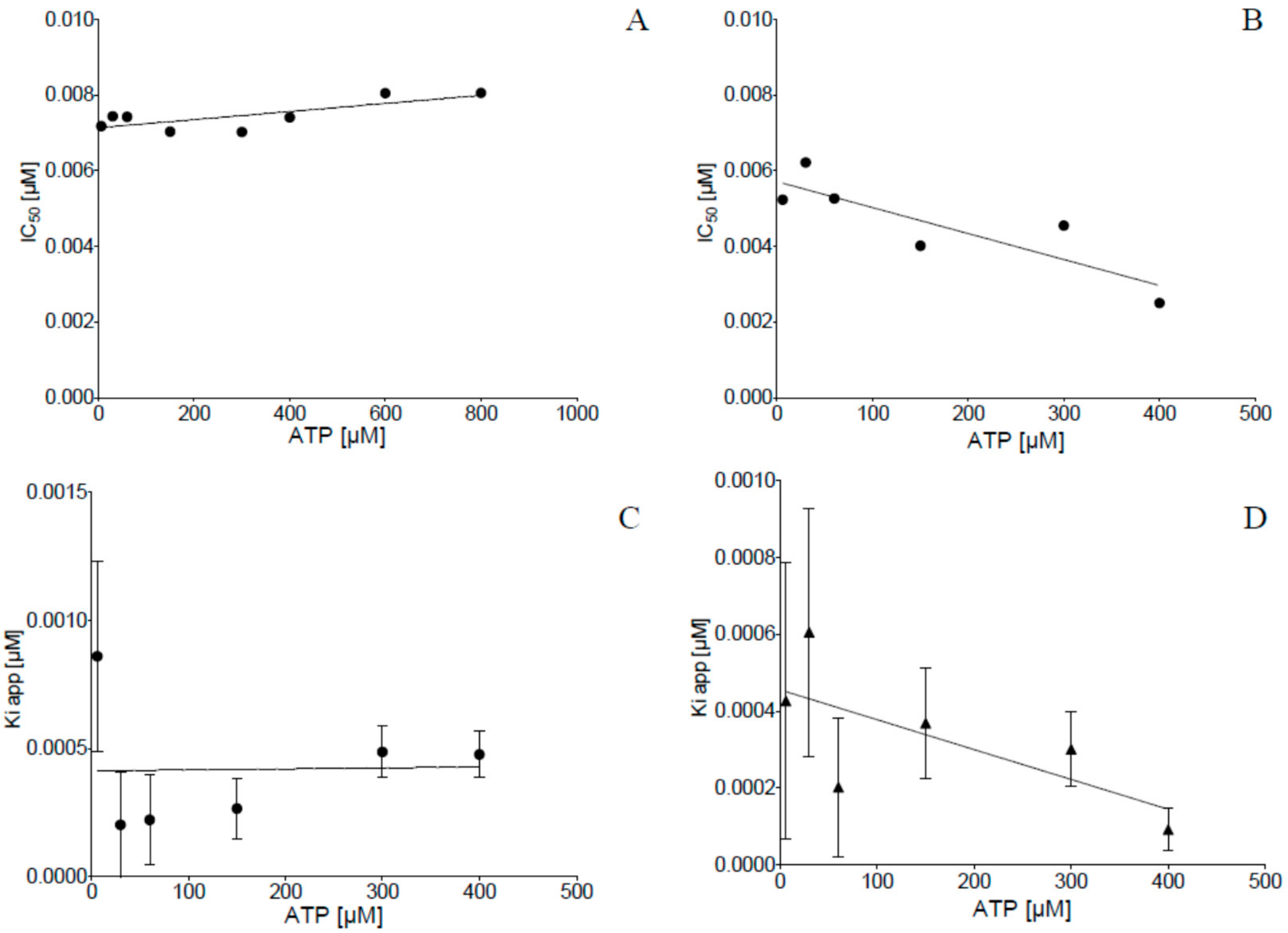
2.2. Inhibition of Human Protein Kinase CK2 In Vitro
2.3. Selectivity
2.4. Effects of the Dibenzofuran Derivatives on the Activity of Cellular CK2
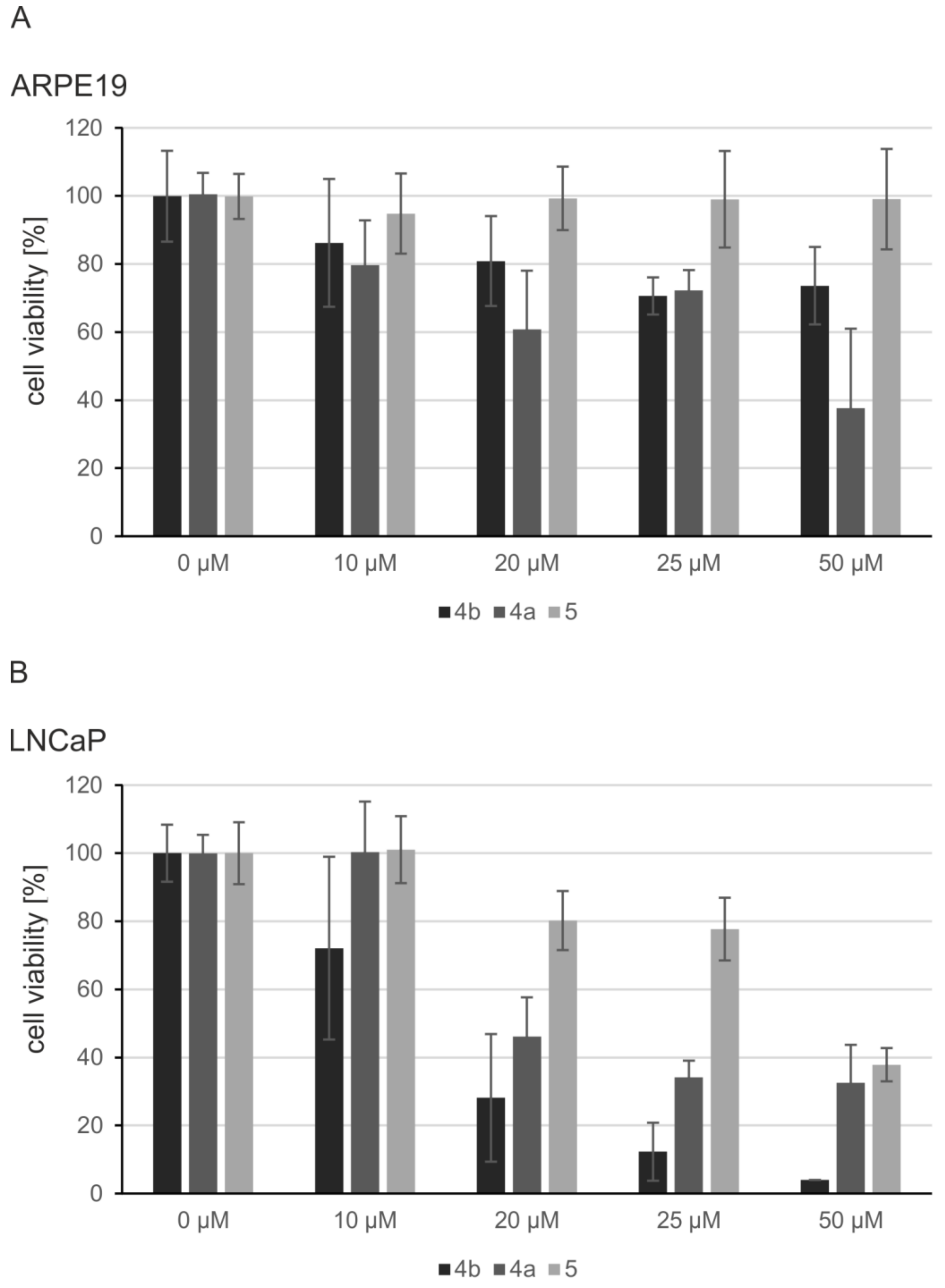
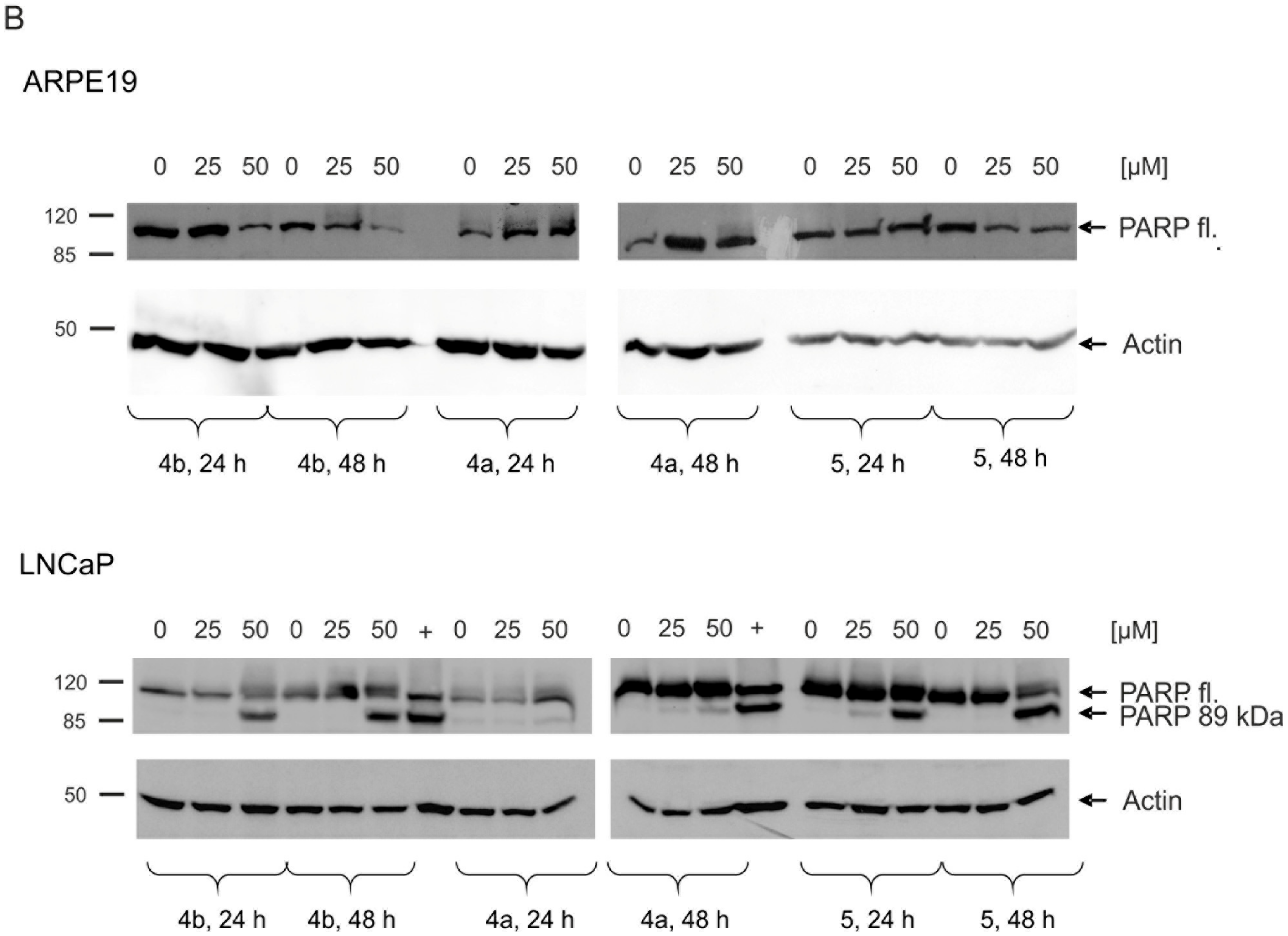
2.5. Induction of Apoptosis and Impact on Tumor Cell Viability
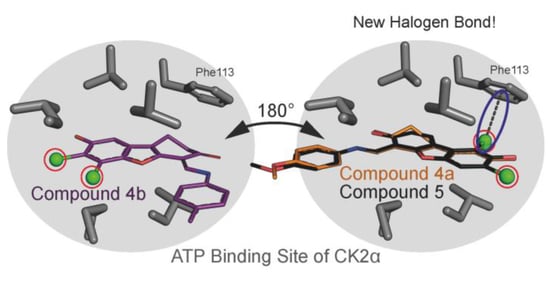
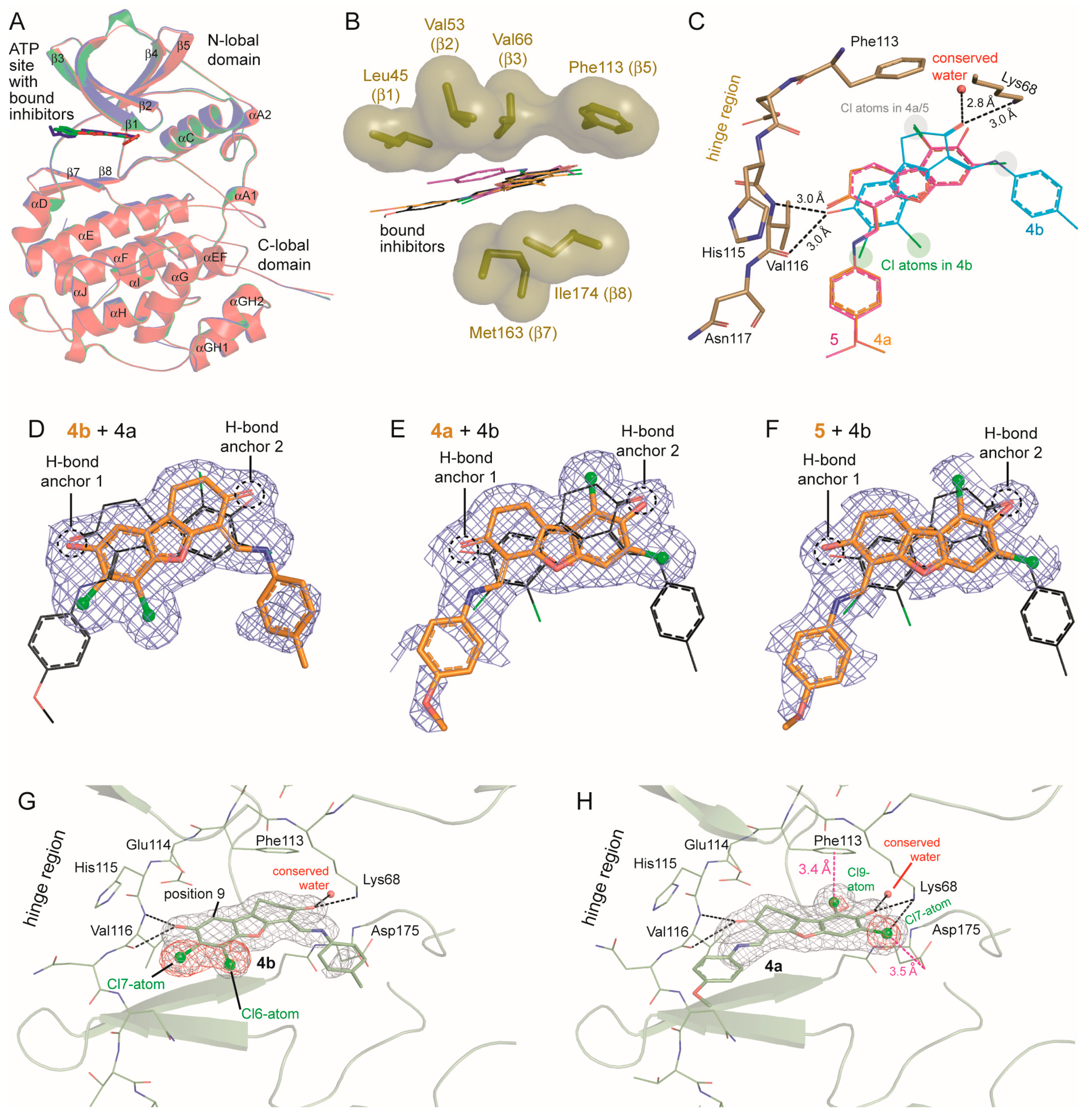
2.6. Human CK2α Crystal Structures in Complex with 4a, 5 and Their Archetype 4b
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. Chemistry
4.2.1. Synthesis of 7,9-dichloro-1,2-dihydro-8-hydroxy-4-[(4-methoxyphenylamino)methylene]dibenzo[b,d]furan-3(2H)-one (4a).
4.2.2. Synthesis of 6,7-dichloro-1,4-dihydro-8-hydroxy-4-[(4-methylphenylamino)methylene]-dibenzo[b,d]furan-3(2H)-one (4b)
4.2.3. Synthesis of (E)-1,3-dichloro-6-[(4-methoxyphenylimino)methyl]dibenzo[b,d]furan- 2,7-diol (5)
4.3. CK2 Inhibition Assay and Kinetic Determinations
4.4. Cell Culture and Treatment of Cells with Dibenzofuran Derivatives
4.5. MTT Viability Assay
4.6. Extraction of Proteins
4.7. Determination of Cellular CK2 Activity by an In Vitro Phosphorylation Assay
4.8. Western Blot Analysis
4.9. Assay of Caspase Activity
4.10. Crystallization and Structure Determination
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Meggio, F.; Pinna, L.A. One-thousand-and-one substrates of protein kinase CK2? Faseb J. 2003, 17, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Salvi, M.; Sarno, S.; Cesaro, L.; Nakamura, H.; Pinna, L.A. Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Bba Mol. Cell Res. 2009, 1793, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Issinger, O.G. Protein kinase CK2 in human diseases. Curr. Med. Chem. 2008, 15, 1870–1886. [Google Scholar] [CrossRef] [PubMed]
- Filhol, O.; Cochet, C. Cellular functions of protein kinase CK2: A dynamic affair. Cell. Mol. Life Sci. 2009, 66, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Barz, T.; Ackermann, K.; Dubois, G.; Eils, R.; Pyerin, W. Genome-wide expression screens indicate a global role for protein kinase CK2 in chromatin remodeling. J. Cell Sci. 2003, 116, 1563–1577. [Google Scholar] [CrossRef] [PubMed]
- St-Denis, N.; Gabriel, M.; Turowec, J.P.; Gloor, G.B.; Li, S.S.C.; Gingras, A.C.; Litchfield, D.W. Systematic investigation of hierarchical phosphorylation by protein kinase CK2. J. Proteom. 2015, 118, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Franchin, C.; Borgo, C.; Cesaro, L.; Zaramella, S.; Vilardell, J.; Salvi, M.; Arrigoni, G.; Pinna, L.A. Re-evaluation of protein kinase c2 pleiotropy: New insights provided by a phosphoproteomics analysis of CK2 knockout cells. Cell. Mol. Life Sci. 2017. [CrossRef]
- Gyenis, L.; Turowec, J.P.; Bretner, M.; Litchfield, D.W. Chemical proteomics and functional proteomics strategies for protein kinase inhibitor validation and protein kinase substrate identification: Applications to protein kinase CK2. Bba Proteins Proteom. 2013, 1834, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Guerra, B.; Ermakowa, I.; Issinger, O.G. Crystal structure of human protein kinase ck2: Insights into basic properties of the CK2 holoenzyme. Embo J. 2001, 20, 5320–5331. [Google Scholar] [CrossRef] [PubMed]
- Boldyreff, B.; Meggio, F.; Pinna, L.A.; Issinger, O.G. Reconstitution of normal and hyperactivated forms of casein kinase-2 by variably mutated beta-subunits. Biochemistry 1993, 32, 12672–12677. [Google Scholar] [CrossRef] [PubMed]
- Faust, M.; Montenarh, M. Subcellular localization of protein kinase CK2—A key to its function? Cell Tissue Res. 2000, 301, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Issinger, O.G. Primary and secondary interactions between ck2 alpha and ck2 beta lead to ring-like structures in the crystals of the CK2 holoenzyme. Mol. Cell. Biochem. 2005, 274, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Raaf, J.; Issinger, O.G. Protein kinase CK2: From structures to insights. Cell. Mol. Life Sci. 2009, 66, 1800–1816. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.A.; Sarno, S.; Poletto, G.; Cozza, G.; Pinna, L.A.; Meggio, F. Autophosphorylation at the regulatory beta subunit reflects the supramolecular organization of protein kinase CK2. Mol. Cell. Biochem. 2005, 274, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. CK2: A key player in cancer biology. Cell. Mol. Life Sci. 2009, 66, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Laramas, M.; Pasquier, D.; Filhol, O.; Ringeisen, F.; Descotes, J.L.; Cochet, C. Nuclear localization of protein kinase CK2 catalytic subunit (CK2 alpha) is associated with poor prognostic factors in human prostate cancer. Eur. J. Cancer 2007, 43, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Eom, J.I.; Cheong, J.W.; Choi, A.J.; Lee, J.K.; Yang, W.I.; Min, Y.H. Protein kinase CK2 alpha as an unfavorable prognostic marker and novel therapeutic target in acute myeloid leukemia. Clin. Cancer Res. 2007, 13, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.S.; Litchfield, D.W. Too much of a good thing: The role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of ck2. Bba Proteins Proteom. 2008, 1784, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Di Maira, G.; Salvi, M.; Arrigoni, G.; Marin, O.; Sarno, S.; Brustolon, F.; Pinna, L.A.; Ruzzene, M. Protein kinase CK2 phosphorylates and upregulates akt/pkb. Cell Death Differ. 2005, 12, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Ruzzene, M.; Pinna, L.A. Addiction to protein kinase CK2: A common denominator of diverse cancer cells? Bba Proteins Proteom. 2010, 1804, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.S.; Turowec, J.P.; Duncan, K.E.; Vilk, G.; Wu, C.G.; Luscher, B.; Li, S.S.C.; Gloor, G.B.; Litchfield, D.W. A peptide-based target screen implicates the protein kinase CK2 in the global regulation of caspase signaling. Sci. Signal. 2011, 4, ra30. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.S.; Turowec, J.P.; Vilk, G.; Li, S.S.C.; Gloor, G.B.; Litchfield, D.W. Regulation of cell proliferation and survival: Convergence of protein kinases and caspases. Bba Proteins Proteom. 2010, 1804, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Landesman-Bollag, E.; Channavajhala, P.L.; Seldin, D.C. Murine protein kinase CK2: Gene and oncogene. Mol. Cell. Biochem. 1999, 191, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Seldin, D.C.; Leder, P. Casein kinase ii-alpha transgene-induced murine lymphoma—Relation to theileriosis in cattle. Science 1995, 267, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B. Cancer: Addiction to oncogenes—The achilles heal of cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Solimini, N.L.; Luo, J.; Elledge, S.J. Non-oncogene addiction and the stress phenotype of cancer cells. Cell 2007, 130, 986–988. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Chen, Z.; Unger, G.; Slaton, J.; Kren, B.T.; Van Waes, C.; Ahmed, K. Emergence of protein kinase CK2 as a key target in cancer therapy. Biofactors 2010, 36, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Slaton, J.W.; Unger, G.M.; Sloper, D.T.; Davis, A.T.; Ahmed, K. Induction of apoptosis by antisense CK2 in human prostate cancer xenograft model. Mol. Cancer Res. 2004, 2, 712–721. [Google Scholar] [PubMed]
- Cozza, G.; Bortolato, A.; Moro, S. How druggable is protein kinase CK2? Med. Res. Rev. 2010, 30, 419–462. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Pinna, L.A.; Moro, S. Protein kinase CK2 inhibitors: A patent review. Expert Opin. Ther. Pat. 2012, 22, 1081–1097. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Pinna, L.A.; Moro, S. Kinase CK2 inhibition: An update. Curr. Med. Chem. 2013, 20, 671–693. [Google Scholar] [CrossRef] [PubMed]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Pre-clinical characterization of cx-4945, a potent and selective small molecule inhibitor of CK2 for the treatment of cancer. Mol. Cell. Biochem. 2011, 356, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. Cx-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.H.; Song, J.S.; Kim, S.H.; Kim, J. Pharmacokinetic characterization of CK2 inhibitor cx-4945. Arch. Pharm. Res. 2013, 36, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Acero, F.R.B.; Negrin, Y.P.; Alonso, D.F.; Perea, S.E.; Gomez, D.E.; Farina, H.G. Mechanisms of cellular uptake, intracellular transportation, and degradation of cigb-300, a tat-conjugated peptide, in tumor cell lines. Mol. Pharm. 2014, 11, 1798–1807. [Google Scholar] [CrossRef] [PubMed]
- Gratz, A.; Kucklander, U.; Bollig, R.; Gotz, C.; Jose, J. Identification of novel CK2 inhibitors with a benzofuran scaffold by novel non-radiometric in vitro assays. Mol. Cell. Biochem. 2011, 356, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Gotz, C.; Gratz, A.; Kucklaender, U.; Jose, J. Tf—A novel cell-permeable and selective inhibitor of human protein kinase CK2 induces apoptosis in the prostate cancer cell line lncap. Bba Gen. Subj. 2012, 1820, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Bischoff, N.; Bdzhola, V.G.; Yarmoluk, S.M.; Issinger, O.G.; Golub, A.G.; Niefind, K. A note of caution on the role of halogen bonds for protein kinase/inhibitor recognition suggested by high- and low-salt CK2 alpha complex structures. ACS Chem. Biol. 2015, 10, 1654–1660. [Google Scholar] [CrossRef] [PubMed]
- Kucklander, U.; Toberich, H. Reaction of 2-(aminomethylene)cyclohexanone derivatives with dichloroquinones. Chemische Berichte 1983, 116, 152–158. [Google Scholar]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. Evaluation of enzyme inhibitors in drug discovery. A guide for medicinal chemists and pharmacologists. Meth. Biochem. Anal. 2005, 46, 1–265. [Google Scholar]
- Guerra, B.; Hochscherf, J.; Jensen, N.B.; Issinger, O.G. Identification of a novel potent, selective and cell permeable inhibitor of protein kinase CK2 from the nih/nci diversity set library. Mol. Cell. Biochem. 2015, 406, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.A.; Kunze, K.L.; Howald, W.N.; Thummel, K.E. Effect of inhibitor depletion on inhibitory potency: Tight binding inhibition of cyp3a by clotrimazole. Drug Metab. Dispos. 1999, 27, 596–599. [Google Scholar] [PubMed]
- Copeland, R.A. The dynamics of drug-target interactions: Drug-target residence time and its impact on efficacy and safety. Expert Opin. Drug Dis. 2010, 5, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.J. Determination of accurate k-i values for tight-binding enzyme inhibitors: An in silico study of experimental error and assay design. Anal. Biochem. 2004, 327, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; Cozza, G.; Pierre, F.; Papinutto, E.; Lolli, G.; Sarno, S.; O’Brien, S.E.; Siddiqui-Jain, A.; Haddach, M.; Anderes, K.; et al. Unprecedented selectivity and structural determinants of a new class of protein kinase CK2 inhibitors in clinical trials for the treatment of cancer. Biochemistry 2011, 50, 8478–8488. [Google Scholar] [CrossRef] [PubMed]
- Bollacke, A.; Nienberg, C.; Le Borgne, M.; Jose, J. Toward selective CK2alpha and CK2alpha’ inhibitors: Development of a novel whole-cell kinase assay by autodisplay of catalytic ck2alpha’. J. Pharm. Biomed. 2016, 121, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Gratz, A.; Bollacke, A.; Stephan, S.; Nienberg, C.; Le Borgne, M.; Gotz, C.; Jose, J. Functional display of heterotetrameric human protein kinase CK2 on escherichia coli: A novel tool for drug discovery. Microb. Cell Fact. 2015, 14, 74. [Google Scholar] [CrossRef] [PubMed]
- Dunn, K.C.; AotakiKeen, A.E.; Putkey, F.R.; Hjelmeland, L.M. Arpe-19, a human retinal pigment epithelial cell line with differentiated properties. Exp. Eye Res. 1996, 62, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Fischer, M.; Schaefer, S.; Issinger, O.G. The kinase inhibitor d11 induces caspase-mediated cell death in cancer cells resistant to chemotherapeutic treatment. J. Exp. Clin. Cancer Res. 2015, 34, 125. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, S.; Svenstrup, T.; Fischer, M.; Guerra, B. D11-mediated inhibition of protein kinase CK2 impairs hif-1α-mediated signaling in human glioblastoma cells. Pharmaceuticals 2017, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, S.; Svenstrup, T.H.; Guerra, B. The small-molecule kinase inhibitor d11 counteracts 17-aag-mediated up-regulation of hsp70 in brain cancer cells. PLoS ONE 2017, 12, e017706. [Google Scholar] [CrossRef] [PubMed]
- Borgo, C.; Franchin, C.; Scalco, S.; Bosello-Travain, V.; Donella-Deana, A.; Arrigoni, G.; Salvi, M.; Pinna, L.A. Generation and quantitative proteomics analysis of CK2 alpha/alpha’((-/-)) cells. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Klopffleisch, K.; Issinger, O.G.; Niefind, K. Low-density crystal packing of human protein kinase CK2 catalytic subunit in complex with resorufin or other ligands: A tool to study the unique hinge-region plasticity of the enzyme without packing bias. Acta Crystallogr. D 2012, 68, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Raaf, J.; Klopffleisch, K.; Issinger, O.G.; Niefind, K. The catalytic subunit of human protein kinase CK2 structurally deviates from its maize homologue in complex with the nucleotide competitive inhibitor emodin. J. Mol. Biol. 2008, 377, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Raaf, J.; Issinger, O.G.; Niefind, K. Insights from soft X-rays: The chlorine and sulfur sub-structures of a CK2 alpha/drb complex. Mol. Cell. Biochem. 2008, 316, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Yde, C.W.; Ermakova, I.; Issinger, O.G.; Niefind, K. Inclining the purine base binding plane in protein kinase CK2 by exchanging the flanking side-chains generates a preference for atp as a cosubstrate. J. Mol. Biol. 2005, 347, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; Sarno, S.; De Moliner, E.; Papinutto, E.; Zanotti, G.; Pinna, L.A. The replacement of atp by the competitive inhibitor emodin induces conformational modifications in the catalytic site of protein kinase CK2. J. Biol. Chem. 2000, 275, 29618–29622. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; Mazzorana, M.; Sarno, S.; Kazimierczuk, Z.; Zanotti, G.; Pinna, L.A. Inspecting the structure-activity relationship of protein kinase CK2 inhibitors derived from tetrabromo-benzimidazole. Chem. Biol. 2005, 12, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; Mazzorana, M.; Cendron, L.; Bortolato, A.; Sarno, S.; Kazimierczuk, Z.; Zanotti, G.; Moro, S.; Pinna, L.A. The atp-binding site of protein kinase CK2 holds a positive electrostatic area and conserved water molecules. Chembiochem 2007, 8, 1804–1809. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Nakaniwa, T.; Sekiguchi, Y.; Sogabe, Y.; Sakurai, A.; Nakamura, S.; Nakanishi, I. Crystal structure of human CK2 alpha at 1.06 angstrom resolution. J. Synchrotron Radiat. 2013, 20, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.K.; Lunney, E.A. Kinase inhibition that hinges on halogen bonds. Chem. Biol. 2011, 18, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Matter, H.; Nazare, M.; Gussregen, S.; Will, D.W.; Schreuder, H.; Bauer, A.; Urmann, M.; Ritter, K.; Wagner, M.; Wehner, V. Evidence for c-cl/c-br center dot center dot center dot pi interactions as an important contribution to protein-ligand binding affinity. Angew. Chem. Int. Edit. 2009, 48, 2911–2916. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.X.; Wang, Y.; Zhu, W.L. Nonbonding interactions of organic halogens in biological systems: Implications for drug discovery and biomolecular design. Phys. Chem. Chem. Phys. 2010, 12, 4543–4551. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L. Polypharmacology in a single drug: Multitarget drugs. Curr. Med. Chem. 2013, 20, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Talevi, A. Multi-target pharmacology: Possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef] [PubMed]
- Alchab, F.; Sibille, E.; Ettouati, L.; Bana, E.; Bouaziz, Z.; Mularoni, A.; Monniot, E.; Bagrel, D.; Jose, J.; Le Borgne, M.; et al. Screening of indeno[1,2-b]indoloquinones by maldi-ms: A new set of potential cdc25 phosphatase inhibitors brought to light. J. Enzym. Inhib. Med. Chem. 2016, 31, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Gozzi, G.J.; Bouaziz, Z.; Winter, E.; Daflon-Yunes, N.; Aichele, D.; Nacereddine, A.; Marminon, C.; Valdameri, G.; Zeinyeh, W.; Bollacke, A.; et al. Converting potent indeno[1,2-b]indole inhibitors of protein kinase CK2 into selective inhibitors of the breast cancer resistance protein abcg2. J. Med. Chem. 2015, 58, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Ermakova, I.; Boldyreff, B.; Issinger, O.G.; Niefind, K. Crystal structure of a c-terminal deletion mutant of human protein kinase CK2 catalytic subunit. J. Mol. Biol. 2003, 330, 925–934. [Google Scholar] [CrossRef]
- Alchab, F.; Ettouati, L.; Bouaziz, Z.; Bollacke, A.; Delcros, J.G.; Gertzen, C.G.W.; Gohlke, H.; Pinaud, N.; Marchivie, M.; Guillon, J.; et al. Synthesis, biological evaluation and molecular modeling of substituted indeno[1,2-b]indoles as inhibitors of human protein kinase CK2. Pharmaceuticals 2015, 8, 279–302. [Google Scholar] [PubMed]
- Gratz, A.; Götz, C.; Jose, J. A ce-based assay for human protein kinase CK2 activity measurement and inhibitor screening. Electrophoresis 2010, 31, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.C.; Hessenauer, A.; Gotz, C.; Montenarh, M. Dmat, an inhibitor of protein kinase CK2 induces reactive oxygen species and DNA double strand breaks. Oncol. Rep. 2009, 21, 1593–1597. [Google Scholar] [PubMed]
- Schwind, L.; Wilhelm, N.; Kartarius, S.; Montenarh, M.; Gorjup, E.; Götz, C. Protein kinase CK2 is necessary for the adipogenic differentiation of human mesenchymal stem cells. Biochim. Biophys. Acta 2015, 1853, 2207–2216. [Google Scholar] [CrossRef] [PubMed]
- Schuster, N.; Gotz, C.; Faust, M.; Schneider, E.; Prowald, A.; Jungbluth, A.; Montenarh, M. Wild-type p53 inhibits protein kinase CK2 activity. J. Cell. Biochem. 2001, 81, 172–183. [Google Scholar] [CrossRef]
- Nastainczyk, W.; Schmidtspaniol, I.; Boldyreff, B.; Issinger, O.G. Isolation and characterization of a monoclonal anti-protein kinase CK2 beta-subunit antibody of the igg class for the direct-detection of CK2 beta-subunit in tissue-cultures of various mammalian-species and human tumors. Hybridoma 1995, 14, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Faust, M.; Schuster, N.; Montenarh, M. Specific binding of protein kinase CK2 catalytic subunits to tubulin. Febs Lett. 1999, 462, 51–56. [Google Scholar] [CrossRef]
- Kabsch, W. Xds. Acta Crystallogr. D 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D 2013, 69, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the ccp4 suite and current developments. Acta Crystallogr. D 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Mccoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Yde, C.W.; Ermakova, I.; Issinger, O.G. Evolved to be active: Sulfate ions define substrate recognition sites of CK2 alpha and emphasise its exceptional role within the cmgc family of eukaryotic protein kinases. J. Mol. Biol. 2007, 370, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. Phenix: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Schuttelkopf, A.W.; van Aalten, D.M.F. Prodrg: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 4a | 5 | 4b |
|---|---|---|
| Aurora A | Aurora A | Aurora A |
| SKG1 | SKG1 | SKG1 |
| CAMKK2 | CAMKK2 | |
| DYRKB1 | DYRKB1 | |
| FLT3 (D835Y) | FLT3 (D835Y) | |
| FLT3 | FLT3 | |
| FLT4/VEGFR3 | FLT4/VEGFR3 | |
| KDR/VEGFR2 | KDR/VEGFR2 | |
| PIM1 | PIM1 | |
| LCK (68%) * | LCK | |
| PKA | ||
| C-Met | ||
| PKD/PRKD2 |
| Compound | logP | TPSA (A2) |
|---|---|---|
| 4b | 4.95 | 62.47 |
| 4a | 4.55 | 71.70 |
| 5 | 6.02 | 75.19 |
| Complex | hsCK2α1–335/4b | hsCK2α1–335/4a | hsCK2α1–335/5 | ||
|---|---|---|---|---|---|
| X-ray diffraction data collection | |||||
| Wavelength [Å] | 1.0 | 2.0 | 1.0 | 2.0 | 1.0 |
| Synchrotron (beamline) | SLS (PX-III) | ||||
| Space group | P21 | ||||
| Lattice constants | |||||
| a, b, c [Å] | 57.44, 45.71, 63.19 | 57.43, 45.38, 63.33 | 57.48, 45.51, 63.49 | 57.67, 45.50, 63.32 | 57.88, 45.67, 63.75 |
| α, β, γ [°] | 90, 110.94, 90 | 90, 110.81, 90 | 90, 110.97, 90 | 90, 110.81, 90 | 90, 111.14, 90 |
| Protomers per asymmetric unit | 1 | 1 | 1 | 1 | 1 |
| Resolution [Å] (highest res. shell) | 34.79–1.79 (1.85–1.79)1 | 36.01–2.90 (3.00–2.90) | 34.16–1.84 (1.91–1.84) | 36.10–2.99 (3.10–2.99) | 36.22–1.64 (1.70–1.64) |
| Rsym [%] | 5.4 (51.3) | 3.3 (6.1) | 5.5 (70.6) | 9.2 (28.6) | 3.5 (61.5) |
| CC1/2 | 0.998 (0.818) | 0.999 (0.998) | 0.999 (0.705) | 0.996 (0.958) | 0.999 (0.738) |
| Signal-to-noise ratio (I/σI) | 12.86 (1.74) | 43.5 (26.8) | 14.02 (1.55) | 16.00 (5.20) | 18.71 (1.70) |
| No. of unique reflections | 27,735 (2290) | 6609 (610) | 25,781 (2428) | 6058 (525) | 37,646 (3505) |
| Completeness [%] | 95.24 (79.14) | 95.3 (88.2) | 96.10 (90.42) | 94.4 (84.2) | 98.85 (93.39) |
| Multiplicity | 3.5 (3.1) | 6.5 (6.8) | 3.5 (3.2) | 6.5 (6.1) | 3.3 (3.0) |
| Wilson B-factor [Å2] | 25.37 | 29.55 | 28.71 | 48.50 | 23.85 |
| Structure refinement with high-resolution data sets | |||||
| No. of reflections for Rwork/Rfree | 26,336/1389 | 14,456/1317 | 36,809/1826 | ||
| Rwork/Rfree [%] | 16.35/19.92 | 16.70/20.05 | 16.38/18.43 | ||
| Mean coordinate error [Å] | 0.180 | 0.190 | 0.180 | ||
| No. of non-H-atoms | 3094 | 3055 | 3147 | ||
| Protein | 2818 | 2803 | 2841 | ||
| Ligands/ions | 58 | 77 | 57 | ||
| Water | 218 | 175 | 249 | ||
| Mean B-factors [Å2] | 33.67 | 39.42 | 33.60 | ||
| Protein | 32.91 | 38.67 | 32.74 | ||
| Ligands/ions | 47.47 | 59.56 | 48.47 | ||
| Water | 39.83 | 42.46 | 40.07 | ||
| RMS deviations | |||||
| Bond lengths [Å] | 0.002 | 0.003 | 0.004 | ||
| Bond angles (°) | 0.55 | 0.53 | 0.65 | ||
| Ramachandran plot | |||||
| favored [%] | 97.25 | 97.86 | 97.55 | ||
| allowed [%] | 2.45 | 2.14 | 2.45 | ||
| outliers [%] | 0.31 | 0.00 | 0.00 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schnitzler, A.; Gratz, A.; Bollacke, A.; Weyrich, M.; Kuckländer, U.; Wünsch, B.; Götz, C.; Niefind, K.; Jose, J. A π-Halogen Bond of Dibenzofuranones with the Gatekeeper Phe113 in Human Protein Kinase CK2 Leads to Potent Tight Binding Inhibitors. Pharmaceuticals 2018, 11, 23. https://doi.org/10.3390/ph11010023
Schnitzler A, Gratz A, Bollacke A, Weyrich M, Kuckländer U, Wünsch B, Götz C, Niefind K, Jose J. A π-Halogen Bond of Dibenzofuranones with the Gatekeeper Phe113 in Human Protein Kinase CK2 Leads to Potent Tight Binding Inhibitors. Pharmaceuticals. 2018; 11(1):23. https://doi.org/10.3390/ph11010023
Chicago/Turabian StyleSchnitzler, Alexander, Andreas Gratz, Andre Bollacke, Michael Weyrich, Uwe Kuckländer, Bernhard Wünsch, Claudia Götz, Karsten Niefind, and Joachim Jose. 2018. "A π-Halogen Bond of Dibenzofuranones with the Gatekeeper Phe113 in Human Protein Kinase CK2 Leads to Potent Tight Binding Inhibitors" Pharmaceuticals 11, no. 1: 23. https://doi.org/10.3390/ph11010023
APA StyleSchnitzler, A., Gratz, A., Bollacke, A., Weyrich, M., Kuckländer, U., Wünsch, B., Götz, C., Niefind, K., & Jose, J. (2018). A π-Halogen Bond of Dibenzofuranones with the Gatekeeper Phe113 in Human Protein Kinase CK2 Leads to Potent Tight Binding Inhibitors. Pharmaceuticals, 11(1), 23. https://doi.org/10.3390/ph11010023