Improved Syntheses of the mGlu5 Antagonists MMPEP and MTEP Using Sonogashira Cross-Coupling


 ,
,
Abstract
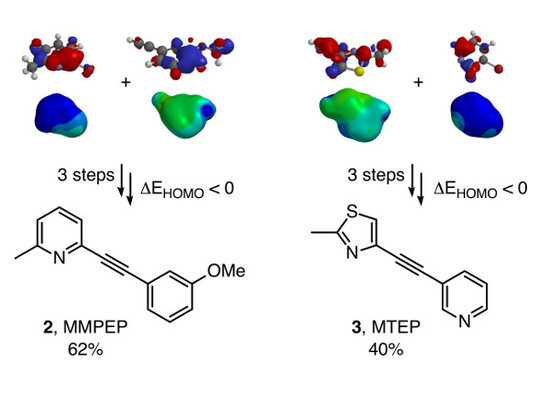
{kind=link}
{kind=link}
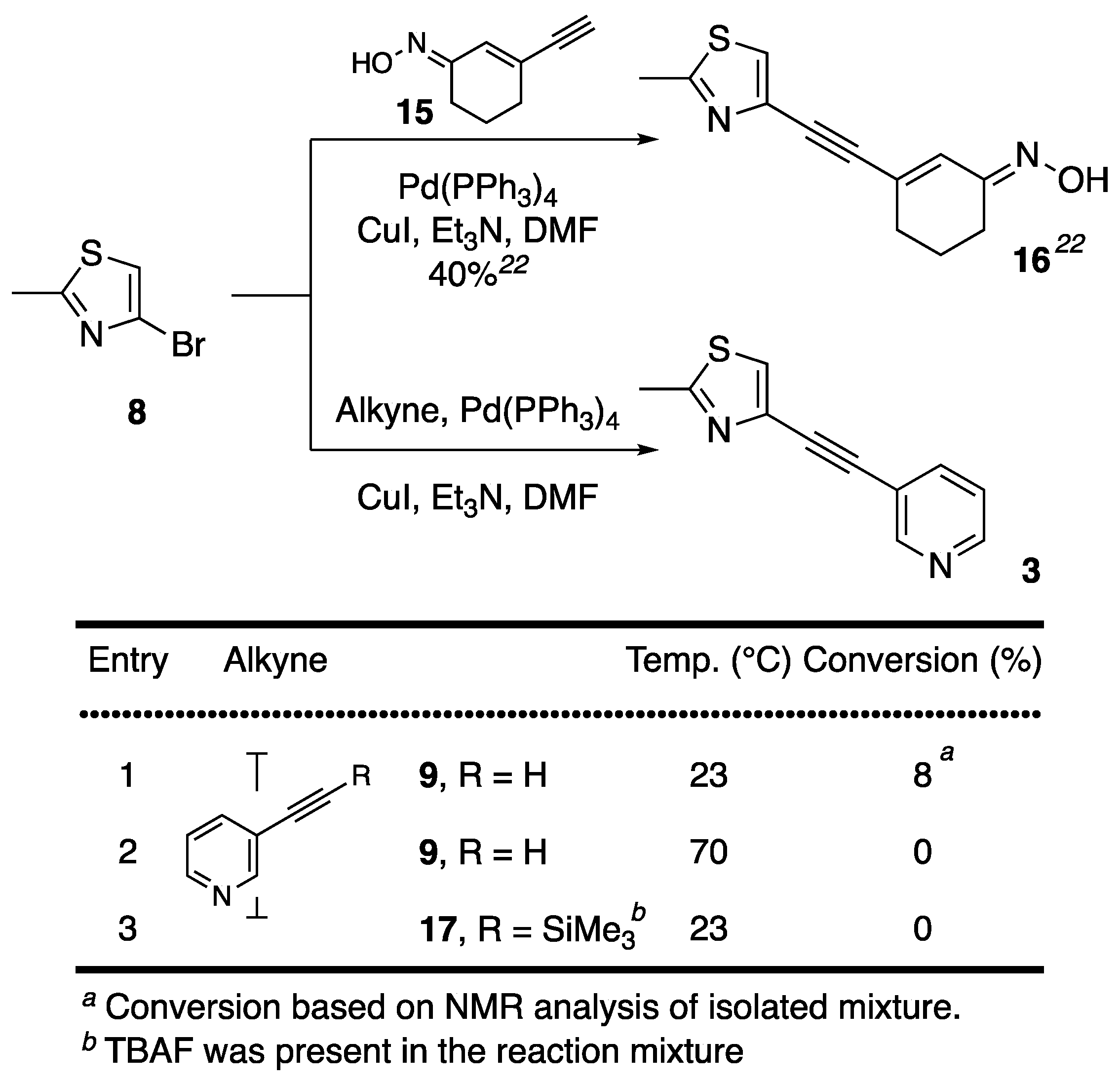{kind=link}
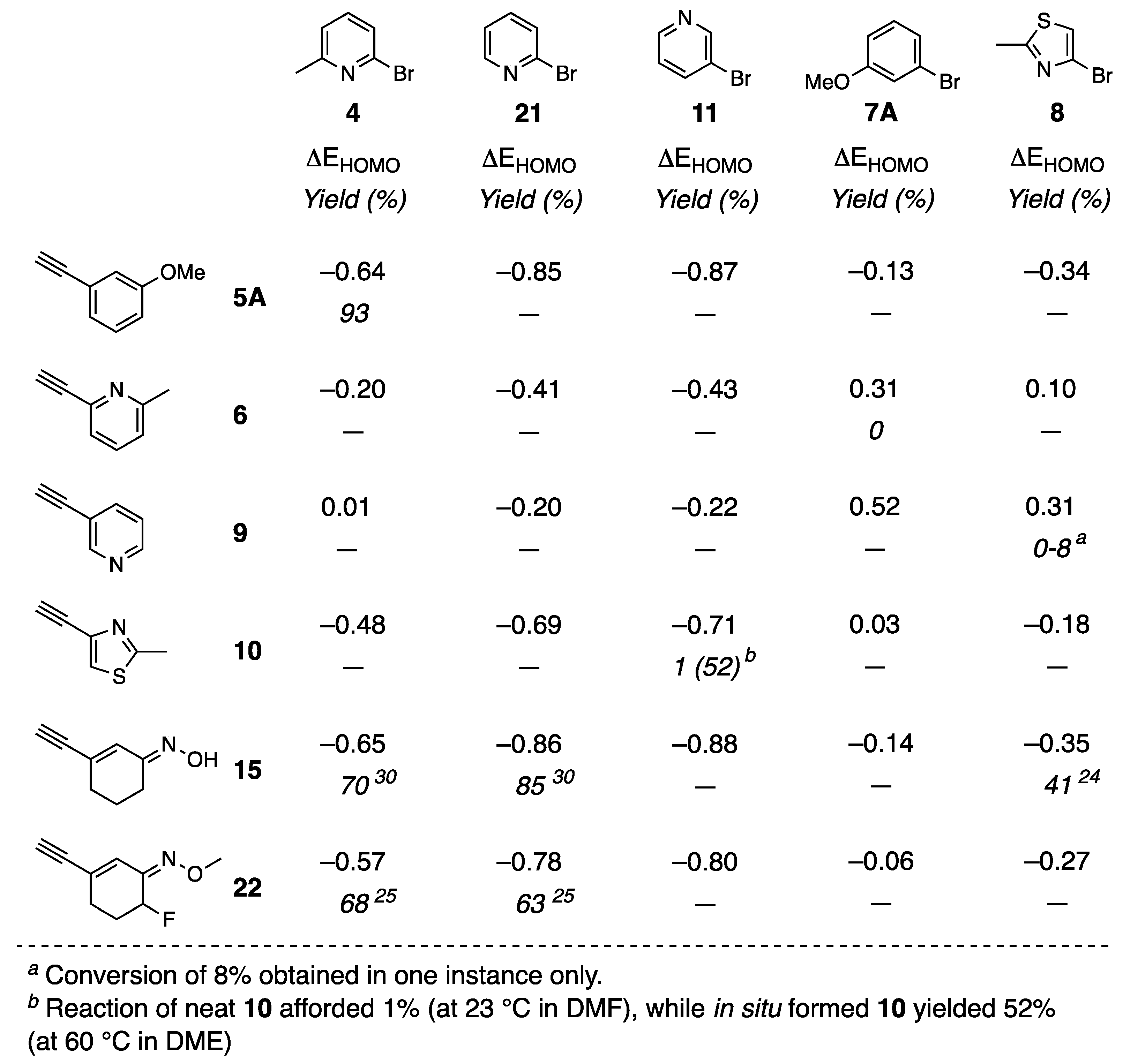{kind=link}
{kind=link}
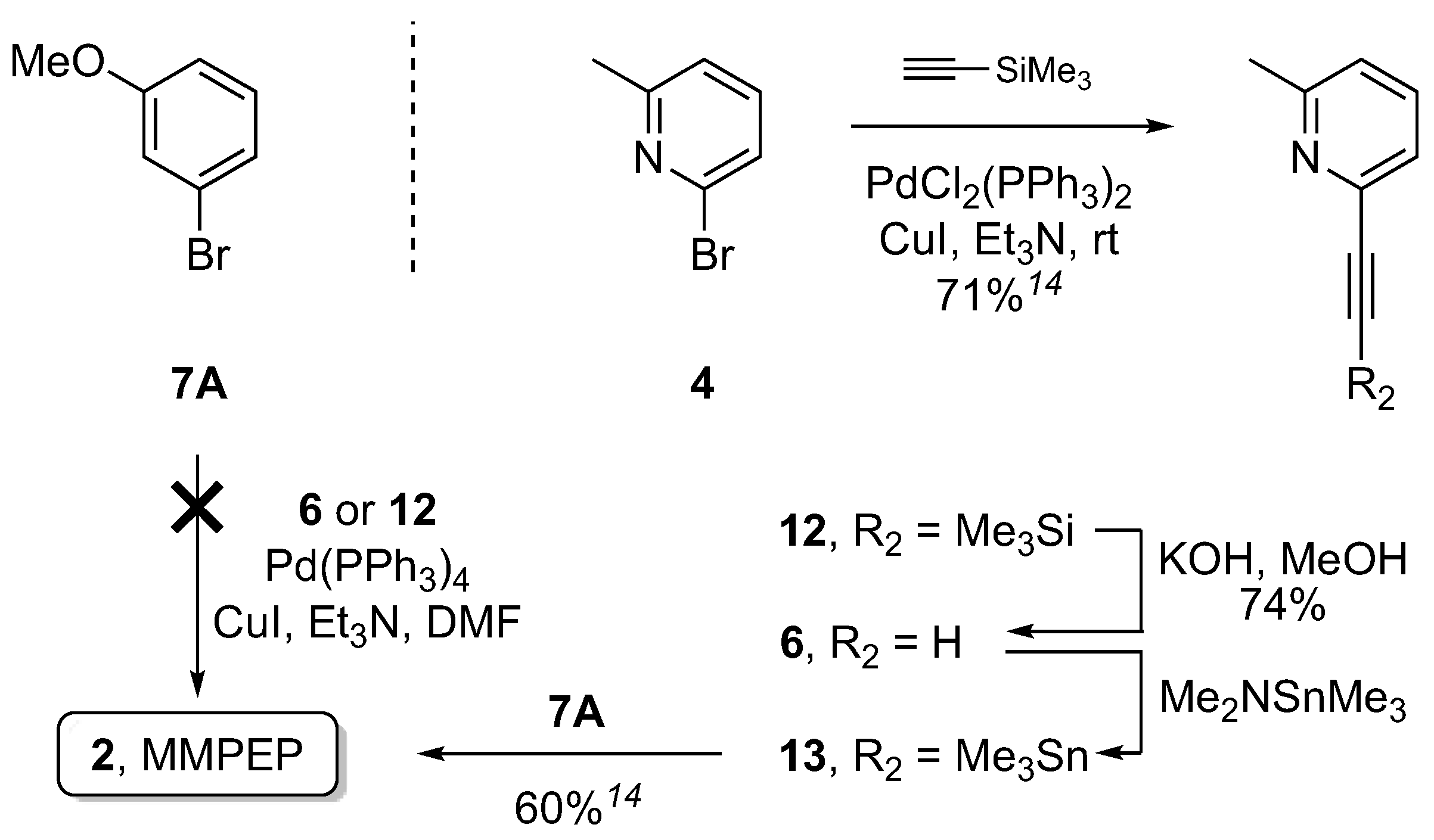{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Techniques
3.2. Syntheses
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rouse, S.T.; Marino, M.J.; Bradley, S.R.; Awad, H.; Wittmann, M.; Conn, P.J. Distribution and Roles of Metabotropic Glutamate Receptors in the Basal Ganglia Motor Circuit: Implications for Treatment of Parkinson’s Disease and Related Disorders. Pharmacol. Ther. 2000, 88, 427–435. [Google Scholar] [CrossRef]
- Daggett, L.P.; Sacaan, A.I.; Akong, M.; Rao, S.P.; Hess, S.D.; Liaw, C.; Urrutia, A.; Jachec, C.; Ellis, S.B.; Dreessen, J.; et al. Molecular and Functional Characterization of Recombinant Human Metabotropic Glutamate Receptor Subtype 5. Neuropharmacology 1995, 34, 871–886. [Google Scholar] [CrossRef]
- Tanabe, Y.; Masu, M.; Ishii, T.; Shigemoto, R.; Nakanishi, S. A Family of Metabotropic Glutamate Receptors. Neuron 1992, 8, 169–179. [Google Scholar] [CrossRef]
- Ossowska, K.; Konieczny, J.; Wardas, J.; Pietraszek, M.; Kuter, K.; Wolfarth, S.; Pilc, A. An Influence of Ligands of Metabotropic Glutamate Receptor Subtypes on Parkinsonian-like Symptoms and the Striatopallidal Pathway in Rats. Amino Acids 2007, 32, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Bruno, V.; Ksiazek, I.; Battaglia, G.; Lukic, S.; Leonhardt, T.; Sauer, D.; Gasparini, F.; Kuhn, R.; Nicoletti, F.; Flor, P. Selective Blockade of Metabotropic Glutamate Receptor Subtype 5 Is Neuroprotective. Neuropharmacology 2000, 39, 2223–2230. [Google Scholar] [CrossRef]
- Wang, Q.; Walsh, D.M.; Rowan, M.J.; Selkoe, D.J.; Anwyl, R. Block of Long-Term Potentiation by Naturally Secreted and Synthetic Amyloid Beta-Peptide in Hippocampal Slices Is Mediated via Activation of the Kinases c-Jun N-Terminal Kinase, Cyclin-Dependent Kinase 5, and p38 Mitogen-Activated Protein Kinase as Well as metabotropic glutamate receptor type 5. J. Neurosci. 2004, 24, 3370–3378. [Google Scholar] [CrossRef] [PubMed]
- Ritzén, A.; Mathiesen, J.M.; Thomsen, C. Molecular Pharmacology and Therapeutic Prospects of Metabotropic Glutamate Receptor Allosteric Modulators. Basic Clin. Pharmacol. Toxicol. 2005, 97, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, F.; Lingenhöhl, K.; Stoehr, N.; Flor, P.J.; Heinrich, M.; Vranesic, I.; Biollaz, M.; Allgeier, H.; Heckendorn, R.; Urwyler, S.; et al. 2-Methyl-6-(Phenylethynyl)-Pyridine (MPEP), a Potent, Selective and Systemically Active mGlu5 Receptor Antagonist. Neuropharmacology 1999, 38, 1493–1503. [Google Scholar] [CrossRef]
- Pilc, A.; Kłodzińska, A.; Brański, P.; Nowak, G.; Pałucha, A.; Szewczyk, B.; Tatarczyńska, E.; Chojnacka-Wójcik, E.; Wierońska, J. Multiple MPEP Administrations Evoke Anxiolytic- and Antidepressant-like Effects in Rats. Neuropharmacology 2002, 43, 181–187. [Google Scholar] [CrossRef]
- Ametamey, S.M.; Honer, M.; Schubiger, P.A. Molecular Imaging with PET. Chem. Rev. 2008, 108, 1501–1516. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Shubiger, P.A.; Ametamey, S.M. Radioligands for the PET Imaging of Metabotropic Glutamate Receptor Subtype 5 (mGluR5). Curr. Top. Med. Chem. 2010, 10, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Ametamey, S.M.; Kessler, L.J.; Honer, M.; Wyss, M.T.; Buck, A.; Hintermann, S.; Auberson, Y.P.; Gasparini, F.; Schubiger, P.A. Radiosynthesis and Preclinical Evaluation of 11C-ABP688 as a Probe for Imaging the Metabotropic Glutamate Receptor Subtype 5. J. Nucl. Med. 2006, 47, 698–705. [Google Scholar] [PubMed]
- Ametamey, S.M.; Treyer, V.; Streffer, J.; Wyss, M.T.; Schmidt, M.; Blagoev, M.; Hintermann, S.; Auberson, Y.; Gasparini, F.; Fischer, U.C.; et al. Human PET Studies of Metabotropic Glutamate Receptor Subtype 5 with 11C-ABP688. J. Nucl. Med. 2007, 48, 247–252. [Google Scholar] [PubMed]
- Alagille, D.; Baldwin, R.M.; Roth, B.L.; Wroblewski, J.T.; Grajkowska, E.; Tamagnan, G.D. Synthesis and Receptor Assay of Aromatic-Ethynyl-Aromatic Derivatives with Potent mGluR5 Antagonist Activity. Bioorg. Med. Chem. 2005, 13, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Cosford, N.D.P.; Tehrani, L.; Roppe, J.; Schweiger, E.; Smith, N.D.; Anderson, J.; Bristow, L.; Brodkin, J.; Jiang, X.; McDonald, I.; et al. 3-[(2-Methyl-1,3-Thiazol-4-Yl)ethynyl]-Pyridine: A Potent and Highly Selective Metabotropic Glutamate Subtype 5 Receptor Antagonist with Anxiolytic Activity. J. Med. Chem. 2003, 46, 204–206. [Google Scholar] [CrossRef] [PubMed]
- McIldowie, M.J.; Gandy, M.N.; Skelton, B.W.; Brotchie, J.M.; Koutsantonis, G.A.; Spackman, M.A.; Piggott, M.J. Physical and Crystallographic Characterisation of the mGlu5 Antagonist MTEP and Its Monohydrochloride. J. Pharm. Sci. 2010, 99, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Iso, Y.; Grajkowska, E.; Wroblewski, J.T.; Davis, J.; Goeders, N.E.; Johnson, K.M.; Sanker, S.; Roth, B.L.; Tueckmantel, W.; Kozikowski, A.P. Synthesis and Structure-Activity Relationships of 3-[(2-Methyl-1,3-Thiazol-4-Yl)ethynyl]pyridine Analogues as Potent, Noncompetitive Metabotropic Glutamate Receptor Subtype 5 Antagonists; Search for Cocaine Medications. J. Med. Chem. 2006, 49, 1080–1100. [Google Scholar] [CrossRef] [PubMed]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. A Convenient Synthesis of Acetylenes: Catalytic Substitutions of Acetylenic Hydrogen with Bromoalkenes, Iodoarenes and Bromopyridines. Tetrahedron Lett. 1975, 16, 4467–4470. [Google Scholar] [CrossRef]
- Peixoto, D.; Begouin, A.; Queiroz, M.J.R.P. Synthesis of 2-(Hetero)arylthieno[2,3-b] or [3,2-b]Pyridines from 2,3-Dihalopyridines, (Hetero)arylalkynes, and Na2S. Further Functionalizations. Tetrahedron 2012, 68, 7082–7094. [Google Scholar] [CrossRef]
- Khairnar, B.J.; Dey, S.; Jain, V.K.; Bhanage, B.M. Dimethylaminoalkyl Chalcogenolate Palladium(II) Complexes as an Efficient Copper- and Phospine-free Catalyst for Sonogashira Reaction. Tetrahedron Lett. 2014, 55, 716–719. [Google Scholar] [CrossRef]
- Chinchilla, R.; Nájera, C. Recent Advances in Sonogashira Reactions. Chem. Soc. Rev. 2011, 40, 5084–5121. [Google Scholar] [CrossRef] [PubMed]
- An der Heiden, M.R.; Plenio, H.; Immel, S.; Burello, E.; Rothenberg, G.; Hoefsloot, H.C.J. Insights into Sonogashira Cross-Coupling by High-Throughput Kinetics and Descriptor Modeling. Chemistry 2008, 14, 2857–2866. [Google Scholar] [CrossRef] [PubMed]
- Kessler, L.J. Development of Novel Ligands for PET Imaging of the Metabotropic Glutamate Receptor Subtype 5 (mGluR5). Ph.D. Thesis, ETH Zürich, Zürich, Switzerland, 2004. [Google Scholar] [CrossRef]
- Sephton, S.M.; Mu, L.; Müller, A.; Wanger-Baumann, C.A.; Schibli, R.; Krämer, S.D.; Ametamey, S.M. Synthesis and in Vitro/in Vivo Pharmacological Evaluation of [11C]-ThioABP, a Novel Radiotracer for Imaging mGluR5 with PET. Medchemcomm 2013, 4, 520–526. [Google Scholar] [CrossRef]
- Milicevic Sephton, S.; Mu, L.; Schweizer, W.B.; Schibli, R.; Krämer, S.D.; Ametamey, S.M. Synthesis and Evaluation of Novel α-Fluorinated (E)-3-((6-Methylpyridin-2-Yl)ethynyl)cyclohex-2-Enone-O-Methyl Oxime (ABP688) Derivatives as Metabotropic Glutamate Receptor Subtype 5 PET Radiotracers. J. Med. Chem. 2012, 55, 7154–7162. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.A.; Wirth, T. Synthesis of Indene Derivatives via Electrophilic Cyclization. Org. Lett. 2009, 11, 229–231. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Green, J.R. Benzocycloheptynedicobalt Complexes by Intramolecular Nicholas Reactions. Synlett 2005, 2005, 271–274. [Google Scholar] [CrossRef]
- Ding, C.; Babu, G.; Orita, A.; Hirate, T.; Otera, J. Synthesis and Photoluminescence Studies of Siloles with Arylene Ethynylene Strands. Synlett 2007, 2007, 2559–2563. [Google Scholar] [CrossRef]
- Karama, U.; Höfle, G. Synthesis of Epothilone 16,17-Alkyne Analogs by Replacement of the C13−C15(O)-Ring Segment of Natural Epothilone C. Eur. J. Org. Chem. 2003, 2003, 1042–1049. [Google Scholar] [CrossRef]
- Baumann, C.; Mu, L.; Johannsen, S.; Honer, M.; Schubiger, P.A.; Ametamey, S.M. Structure-Activity Relationships of Fluorinated (E)-3-((6-Methylpyridin-2-yl)ethynyl)cyclohex-2-enone-O-methyloxime (ABP688(=) Derivatives and the Discovery of a High Affinity Analogue as a Potential Candidate for Imaging Metabotropic Glutamate Receptors Subtype 5 (mGluR5) with Positron Emission Tomography (PET). J. Med. Chem. 2010, 53, 4009–4017. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mu, B.; Mu, L.; Schibli, R.; Ametamey, S.M.; Milicevic Sephton, S. Improved Syntheses of the mGlu5 Antagonists MMPEP and MTEP Using Sonogashira Cross-Coupling. Pharmaceuticals 2018, 11, 24. https://doi.org/10.3390/ph11010024
Mu B, Mu L, Schibli R, Ametamey SM, Milicevic Sephton S. Improved Syntheses of the mGlu5 Antagonists MMPEP and MTEP Using Sonogashira Cross-Coupling. Pharmaceuticals. 2018; 11(1):24. https://doi.org/10.3390/ph11010024
Chicago/Turabian StyleMu, Boshuai, Linjing Mu, Roger Schibli, Simon M. Ametamey, and Selena Milicevic Sephton. 2018. "Improved Syntheses of the mGlu5 Antagonists MMPEP and MTEP Using Sonogashira Cross-Coupling" Pharmaceuticals 11, no. 1: 24. https://doi.org/10.3390/ph11010024
APA StyleMu, B., Mu, L., Schibli, R., Ametamey, S. M., & Milicevic Sephton, S. (2018). Improved Syntheses of the mGlu5 Antagonists MMPEP and MTEP Using Sonogashira Cross-Coupling. Pharmaceuticals, 11(1), 24. https://doi.org/10.3390/ph11010024