The Implication of the Brain Insulin Receptor in Late Onset Alzheimer’s Disease Dementia

 , and
, and 
Abstract
1. Introduction
2. The Hippocampal Insulin Receptor Is a Key Target in Physiological Cognitive Processes and Neurodegeneration
3. Molecular Bases of Insulin Receptor Modulation
4. Relationship between Insulin Receptor Activation and TAU Phosphorylation
- Talbot and co-workers reported that LOAD patients show impaired brain insulin-signaling transduction with reduced tyrosine kinase activity of the IR [35]. IR and its receptor analogous IGF1R form heterodimers (IR/IGF1R) that modulate the selectivity and affinity for insulin and IGF1 in the activation of signaling molecules [65].
- Yarchoan and co-workers reported an increase in serine phosphorylation of IRS1 (inactivation), the phosphorylation of IRS1 on multiple serine residues can inhibit IRS1 activity, leading to insulin resistance in the hippocampus in LOAD and other tauopathies [54].
5. Role of the Glucose Transporter 4 in Cognition
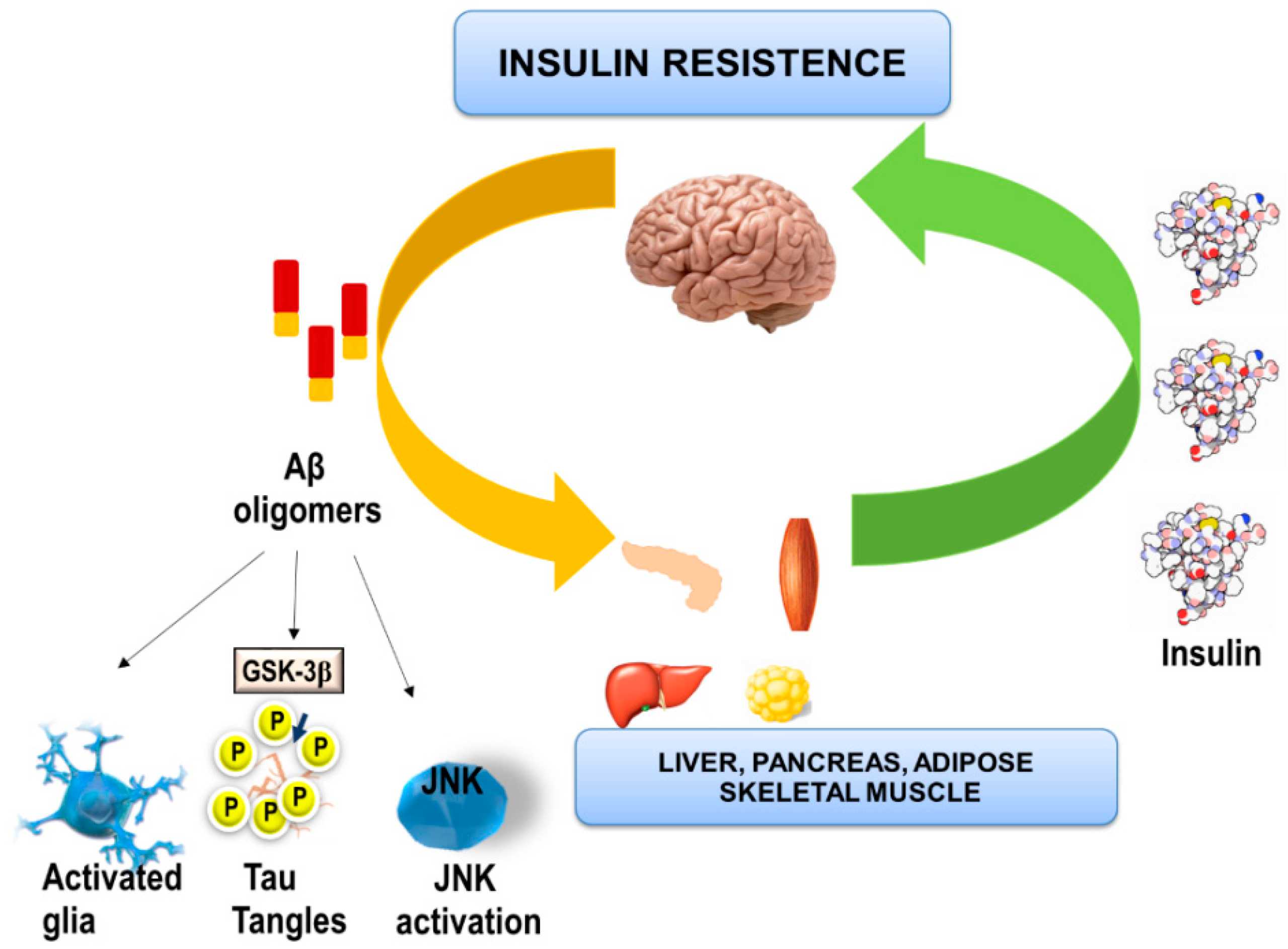
6. Effects of Aβ Oligomers on Brain Insulin and Peripheral Metabolic Tissues
7. Is BACE1 a Potential Bridge between Aβ and T2DM?
8. Potential Pharmacological Approaches for Late Onset Alzheimer’s Disease Treatment Related with the Regulation of Insulin Metabolism
8.1. Antidiabetic Drugs. Modulators of Proliferation of Activated Gamma Peroxisome Receptor. Pioglitazone
8.2. Intranasal Insulin for LOAD Treatment
8.3. Rapalogs
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2016, 12, 459–509. [Google Scholar]
- Kamat, P.K.; Kalani, A.; Rai, S.; Swarnkar, S.; Tota, S.; Nath, C.; Tyagi, N. Mechanism of Oxidative Stress and Synapse Dysfunction in the Pathogenesis of Alzheimer’s Disease: Understanding the Therapeutics Strategies. Mol. Neurobiol. 2016, 53, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkrankung der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [PubMed]
- Vishal, S.; Sourabh, A.; Harkirat, S. Alois Alzheimer (1864–1915) and the Alzheimer syndrome. J. Med. Biogr. 2011, 19, 32–33. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Spearling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Craft, S. Alzheimer disease: Insulin resistance and AD: Extending the translational path. Nat. Rev. Neurol. 2012, 8, 360–362. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, C.W.; Molinuevo, J.L.; Truyen, L.; Satlin, A.; Van der Geyten, S.; Lovestone, S. Development of interventions for the secondary prevention of Alzheimer’s dementia: The European Prevention of Alzheimer’s Dementia (EPAD) project. Lancet Psychiatry 2016, 3, 179–186. [Google Scholar] [CrossRef]
- Querfurth, H.W.; La Ferla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Mangialasche, F.; Solomon, A.; Winblad, B.; Mecocci, P.; Kivipelto, M. Alzheimer’s disease: Clinical trials and drug development. Lancet Neurol. 2010, 9, 702–716. [Google Scholar] [CrossRef]
- Selkoe, D.J. Resolving controversies on the path to Alzheimer’s therapeutics. Nat. Med. 2011, 17, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Viola, K.L.; Klein, W.L. Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015, 129, 183–206. [Google Scholar] [CrossRef] [PubMed]
- Moser, V.A.; Pike, C.J. Obesity Accelerates Alzheimer-Related Pathology in APOE4but not APOE3Mice. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Peskind, E.; Schwartz, M.W.; Schellenberg, G.D.; Raskind, M.; Porte, D. Cerebrospinal fluid and plasma insulin levels in Alzheimer’s disease: Relationship to severity of dementia and apolipoprotein E genotype. Neurology 1998, 50, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Liu, C.C.; Van Ingelgom, A.J.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Neth, B.J.; Craft, S. Insulin Resistance and Alzheimer's Disease: Bioenergetic Linkages. Front. Aging Neurosci. 2017. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M. Insulin Resistance and Neurodegeneration: Progress towards the Development of New Therapeutics for Alzheimer’s disease. Drugs 2017, 77, 47–65. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Benedict, C. A Key Role of Insulin Receptors in Memory. Diabetes 2015, 64, 3653–3655. [Google Scholar] [CrossRef] [PubMed]
- Chneider, L.S.; Mangialasche, F.; Andreasen, N.; Feldman, H.; Giacobini, E.; Jones, R.; Mantua, V.; Mecocci, P.; Pani, L.; Winblad, B.; et al. Clinical trials and late-stage drug development for Alzheimer’s disease: An appraisal from 1984 to 2014. J. Intern. Med. 2014, 275, 251–283. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat. Rev. Drug Discov. 2006, 5, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Allgaier, M.; Allgaier, C. An update on drug treatment options of Alzheimer’s disease. Front. Biosci. 2014, 19, 1345–1354. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Frolich, L.; Blum-Degen, H.G.; Bernstein, S.; Engelsberger, J.; Humrich, S.; Laufer, D.; Muschner, A.; Thalheimer, A.; Turk, S.; Hoyer, P.; et al. Insulin and insulin receptors in the brain in aging and in sporadic Alzheimer’s disease. J. Neural Transm. 1998, 105, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Frolich, L.; Blum-Degen, D.; Riederer, P.; Hoyer, S. A disturbance of the neuronal insulin receptor signal transduction in sporadic Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1999, 893, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, S. Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur. J. Pharmacol. 2004, 490, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Maurer, K.; Hoyer, S. Alois Alzheimer revisited: Differences in origin of the disease carrying his name. J. Neural Transm. 2006, 113, 1645–1658. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, S. Neurodegeneration, Alzheimer’s disease, and beta-amyloid toxicity. Life Sci. 1994, 55, 1977–1983. [Google Scholar] [CrossRef]
- De Felice, F.G.; Lourenco, M.V.; Ferreira, S.T. How does brain insulin resistance develop in Alzheimer’s disease? Alzheimer’s Dement. 2014, 10, S26–S32. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Ferreira, S.T. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer Disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Wands, J.R. Review of insulin and insulin-like growth factor expresion, signaling, and malfunction in the central nervous system: Relevance to alzheimer’s disease. J. Alzheimer Dis. 2005, 7, 45–61. [Google Scholar] [CrossRef]
- De la Monte, S.M. Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 35–66. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.; Hofman, A.; Breteler, M.M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, E.M.; Witteman, J.C.; Sijbrands, E.J.; Hofman, A.; Koudstaal, P.J.; Breteler, M.M. Insulin metabolism and the risk of Alzheimer disease: The Rotterdam Study. Neurology 2010, 75, 1982–1987. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.; Kazi, H.; Han, L.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF1 resistance, IRS1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Reagan, L.P. Hippocampal insulin resistance and cognitive dysfunction. Nat. Rev. Neurosci. 2015, 16, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Porte, D., Jr. Diabetes, obesity, and the brain. Science 2005, 307, 375–379. [Google Scholar] [CrossRef] [PubMed]
- McNay, E.C.; Recknagel, A.K. Brain insulin signaling a key component of cognitive processes and a potential basis for cognitive impairment in type 2 diabetes. Neurobiol. Learn. Mem. 2011, 96, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Schiöth, H.B.; Craft, S.; Brooks, S.K.; Frey, W.H.; Benedict, C. Brain insulin signaling and Alzheimer’s disease: Current evidente and future direction. Mol. Neurobiol. 2011, 46, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Leary, J.; McNay, E.C. Intrahippocampal administration of amyloid-β(1-42) oligomers acutely impairs spatial working memory, insulin signaling, and hippocampal metabolism. J. Alzheimers Dis. 2012, 30, 413–422. [Google Scholar] [PubMed]
- Walker, J.M.; Harrison, F. Shared Neuropathological Characteristics of Obesity, Type 2 Diabetes and Alzheimer’s Disease: Impacts on Cognitive Decline. Nutrients 2015, 7, 7332–7357. [Google Scholar] [CrossRef] [PubMed]
- Grillo, C.A.; Piroli, G.G.; Lawrence, R.C.; Wrighten, S.A.; Green, A.J.; Wilson, S.P.; Sakai, R.R.; Kelly, S.J.; Wilson, M.A.; Mott, D.D.; et al. Hippocampal Insulin Resistance Impairs Spatial Learning and Synaptic Plasticity. Diabetes 2015, 64, 3927–3936. [Google Scholar] [CrossRef] [PubMed]
- Craft, S. Insulin resistance syndrome and Alzheimer’s disease: Age- and obesity-related effects on memory, amyloid, and inflammation. Neurobiol. Aging 2005, 26, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Barone, E.; Perluigi, M.; Butterfield, D.A. The Triangle of Death in Alzheimer’s Disease Brain: The Aberrant Cross-Talk Among Energy Metabolism, Mammalian Target of Rapamycin Signaling, and Protein Homeostasis Revealed by Redox Proteomics. Antioxid. Redox Signal. 2017, 26, 364–387. [Google Scholar] [CrossRef] [PubMed]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimer’s Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef]
- Nuzzo, D.; Picone, P.; Baldassano, S.; Caruana, L.; Messina, E.; Marino Gammazza, A.; Cappello, F.; Mulè, F.; Di Carlo, M. Insulin Resistance as Common Molecular Denominator Linking Obesity to Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Cholerton, B.; Baker, L.D.; Montine, T.J.; Craft, S. Type 2 Diabetes, Cognition, and Dementia in Older Adults: Toward a Precision Health Approach. Diabetes Spectr. 2016, 29, 210–219. [Google Scholar] [CrossRef] [PubMed]
- McCrimmon, R.J.; Ryan, C.M.; Frier, B.M. Diabetes and cognitive dysfunction. Lancet 2012, 379, 2291–2299. [Google Scholar] [CrossRef]
- Xu, H.; Moore, E.; Meiri, N.; Quon, M.J.; Alkon, D.L. Brain insulin receptors and spatial memory. J. Biol. Chem. 1999, 274, 34839–34842. [Google Scholar]
- Chami, B.; Steel, A.J.; De La Monte, S.M.; Sutherland, G.T. The rise and fall of insulin signaling in Alzheimer's disease. Metab Brain Dis. 2016, 31, 497–515. [Google Scholar] [CrossRef] [PubMed]
- Hokama, M.; Oka, S.; Leon, J.; Ninomiya, T.; Honda, H.; Sasaki, K.; Iwaki, T.; Ohara, T.; Sasaki, T.; LaFerla, F.M.; et al. Altered expression of diabetes-related genes in Alzheimer’s disease brains: The Hisayama study. Cereb. Cortex 2014, 24, 2476–2488. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G. Connecting type 2 diabetes to Alzheimer's disease. Expert Rev. Neurother. 2013, 13, 1297–1299. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.L.; Yang, F.; Rosario, E.R.; Ubeda, O.J.; Beech, W.; Gant, D.J.; Chen, P.P.; Hudspeth, B.; Chen, C.; Zhao, Y. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: Suppression by omega-3 fatty acids and curcumin. J. Neurosci. 2009, 29, 9078–9089. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Toledo, J.B.; Lee, E.B.; Arvanitakis, Z.; Kazi, H.; Han, L.Y.; Louneva, N.; Lee, V.M.; Kim, S.F.; Trojanowski, J.Q.; et al. Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer’s disease and tauopathies. Acta Neuropathol. 2014, 128, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Havrankova, J.; Roth, J.; Brownstein, M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 1978, 5656, 827–829. [Google Scholar] [CrossRef]
- Ribe, E.M.; Lovestone, S. Insulin signalling in Alzheimer’s disease and diabetes: From epidemiology to molecular links. J. Intern. Med. 2016, 280, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta 2017, 1863, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Diehl, T.; Mullins, R.; Kapogiannis, D. Insulin resistance in Alzheimer’s disease. Transl. Res. 2017, 183, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J. Pathol. 2011, 225, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Kappelle, L.J. Increased risk of Alzheimer’s disease in Type II diabetes: insulin resistance of the brain or insulin-induced amyloid pathology? Biochem. Soc. Trans. 2005, 33, 1041–1044. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M.; Macauley, S.L.; Holtzman, D.M. Changes in insulin and insulin signaling in Alzheimer’s disease: Cause or consequence? J. Exp. Med. 2016, 213, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Pardeshi, R.; Bolshette, N.; Gadhave, K.; Ahire, A.; Ahmed, S.; Cassano, T.; Gupta, V.B.; Lahkar, M. Insulin signaling: An opportunistic target to minify the risk of Alzheimer’s disease. Psychoneuroendocrinology 2017, 83, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Gautam, D.; Surjo, D.; Ueki, K.; Baudler, S.; Schubert, D.; Kondo, T.; Alber, J.; Galldiks, N.; Küstermann, E.; et al. Role for neuronal insulin resistance in neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2004, 101, 3100–3105. [Google Scholar] [CrossRef] [PubMed]
- Freude, S.; Plum, L.; Schnitker, J.; Leeser, U.; Udelhoven, M.; Krone, W.; Bruning, J.C.; Schubert, M. Peripheral hyperinsulinemia promotes tau phosphorylation in vivo. Diabetes 2005, 54, 3343–3348. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Bennet, L.; Gluckman, P.D.; Gunn, A.J. Insulin-like growth factor-1 and post-ischemic brain injury. Prog. Neurobiol. 2003, 70, 443–462. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rodriguez, P.; Sandebring-Matton, A.; Merino-Serrais, P.; Parrado-Fernandez, C.; Rabano, A.; Winblad, B.; Ávila, J.; Ferrer, I.; Cedazo-Minguez, A. Tau hyperphosphorylation induces oligomeric insulin accumulation and insulin resistance in neurons. Brain 2017, 140, 3269–3285. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Wu, Z.; Farrell, R.J.; Ryan, T.A. GLUT4 Mobilization Supports Energetic Demands of Active Synapses. Neuron 2017, 93, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Leary, J.; McNay, E.C. Novel Roles for the Insulin-Regulated Glucose Transporter-4 in Hippocampally Dependent Memory. J. Neurosci. 2016, 36, 11851–11864. [Google Scholar] [CrossRef] [PubMed]
- Mehta, V.; Parashar, A.; Sharma, A.; Singh, T.R.; Udayabanu, M. Quercetin ameliorates chronic unpredicted stress-mediated memory dysfunction in male Swiss albino mice by attenuating insulin resistance and elevating hippocampal GLUT4 levels independent of insulin receptor expression. Horm. Behav. 2017, 89, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Sato, N. Bidirectional interactions between diabetes and Alzheimer’s disease. Neurochem. Int. 2017, 108, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Lee, Y.H.; Lee, J.E. Metabolism-Centric Overview of the Pathogenesis of Alzheimer’s disease. Yonsei Med. J. 2017, 58, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Fu, B.; Lei, H.; Tang, H.; Wang, Y. Gender differences of peripheral plasma and liver metabolic profiling in APP/PS1 transgenic AD mice. Neuroscience 2016, 332, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, B.; Deng, B.; Zhang, F.; Wu, J.; Wang, Y.; Le, Y.; Zhai, Q. Amyloid-β induces hepatic insulin resistance in vivo via JAK2. Diabetes 2013, 62, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, B.; Zhang, F.; Wu, J.; Hu, Y.; Liu, Y.; Zhai, Q. Amyloid-β induces hepatic insulin resistance by activating JAK2/STAT3/SOCS-1 signaling pathway. Diabetes 2012, 61, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Forny-Germano, L.; Lyra e Silva, N.M.; Batista, A.F.; Brito-Moreira, J.; Gralle, M.; Boehnke, S.E.; Coe, B.C.; Lablans, A.; Marques, S.A.; Martinez, A.M.; et al. Alzheimer’s disease-like pathology induced by amyloid-β oligomers in nonhuman primates. J. Neurosci. 2014, 34, 13629–13643. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.R.; Lyra, E.; Silva, N.M.; Figueiredo, C.P.; Frozza, R.L.; Ledo, J.H.; Beckman, D.; Katashima, C.K.; Razolli, D.; Carvalho, B.M.; et al. Alzheimer-associated Aβ oligomers impact the central nervous system to induce peripheral metabolic deregulation. EMBO Mol. Med. 2015, 7, 190–210. [Google Scholar]
- Arrieta-Cruz, I.; Knight, C.M.; Gutiérrez-Juárez, R. Acute Exposure of the Mediobasal Hypothalamus to Amyloid-β25–35 Perturbs Hepatic Glucose Metabolism. J. Alzheimers Dis. 2015, 46, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Arrieta-Cruz, I.; Gutiérrez-Juárez, R. The Role of Insulin Resistance and Glucose Metabolism Dysregulation in the Development of Alzheimer’s Disease. Rev. Investig. Clin. 2016, 68, 53–58. [Google Scholar]
- Roher, A.E.; Esh, C.L.; Kokjohn, T.A.; Castaño, E.M.; Van Vickle, G.D.; Kalback, W.M.; Patton, R.L.; Luehrs, D.C.; Daugs, I.D.; Kuo, Y.M.; et al. Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimers Dement. 2009, 5, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Guglielmotto, M.; Monteleone, D.; Giliberto, L.; Fornaro, M.; Borghi, R.; Tamagno, E.; Tabaton, M. Amyloid-β42 activates the expression of BACE1 through the JNK pathway. J. Alzheimers Dis. 2011, 27, 871–883. [Google Scholar] [PubMed]
- Guglielmotto, M.; Monteleone, D.; Boido, M.; Piras, A.; Giliberto, L.; Borgh, R.; Vercelli, A.; Fornaro, M.; Tabaton, M.; Tamagno, E. Aβ1–42-mediated downregulation of Uch-L1 is dependent on NF-κB activation and impaired BACE1 lysosomal degradation. Aging Cell 2012, 11, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Guglielmotto, M.; Monteleone, D.; Piras, A.; Valsecchi, V.; Tropiano, M.; Ariano, S.; Fornaro, M.; Vercelli, A.; Puyal, J.; Arancio, O.; et al. Aβ1–42 monomers or oligomers have different effects on autophagy and apoptosis. Autophagy 2014, 10, 1827–1843. [Google Scholar] [CrossRef] [PubMed]
- Plucińska, K.; Crouch, B.; Koss, D.; Robinson, L.; Siebrecht, M.; Riedel, G.; Platt, B. Knock-in of human BACE1 cleaves murine APP and reiterates Alzheimer-like phenotypes. J. Neurosci. 2014, 34, 10710–10728. [Google Scholar] [CrossRef] [PubMed]
- Plucińska, K.; Dekeryte, R.; Koss, D.; Shearer, K.; Mody, N.; Whitfield, P.D.; Doherty, M.K.L.; Mingarelli, M.; Welch, A.; Riedel, G.; et al. Neuronal human BACE1 knockin induces systemic diabetes in mice. Diabetologia 2016, 59, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Meakin, P.J.; Harper, A.J.; Hamilton, D.L.; Gallagher, J.; McNeilly, A.D.; Burgess, L.A.; Vaanholt, L.M.; Bannon, K.A.; Latcham, J.; Hussain, I.; et al. Reduction in BACE1 decreases body weight, protects against diet-induced obesity and enhances insulin sensitivity in mice. Biochem. J. 2012, 441, 285–296. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M. Type 3 diabetes is sporadic Alzheimer’s disease: Mini-review. Eur. Neuropsychopharmacol. 2014, 24, 1954–1960. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.; Correia, S.; Santos, R.X.; Carvalho, C.; Santos, M.S.; Oliveira, C.R.; Perry, G.; Smith, M.A.; Zhu, X.; Moreira, P.I. Insulin is a two-edged knife on the brain. J. Alzheimers Dis. 2009, 18, 483–507. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Tong, M.; Lester-Coll, N.; Plater, M., Jr.; Wands, J.R. Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: Relevance to Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 10, 89–109. [Google Scholar] [CrossRef]
- Ott, V.; Benedict, C.; Schultes, B.; Born, J.; Hallschmid, M. Intranasal administration of insulin to the brain impacts cognitive function and peripheral metabolism. Diabetes Obes. Metab. 2012, 14, 214–221. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M. Early intranasal insulin therapy halts progression of neurodegeneration: Progress in Alzheimer’s disease therapeutics. Aging Health 2012, 8, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Scarpini, E. Pioglitazone for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2017, 26, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.B.; Zhang, Y.; Chen, C.; Li, Y.Q.; Ma, C.; Wang, Z.J. Pioglitazone inhibits advanced glycation end product-induced matrix metalloproteinases and apoptosis by suppressing the activation of MAPK and NF-κB. Apoptosis 2016, 21, 1082–1093. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Hanyu, H.; Hirao, K.; Kanetaka, H.; Sakurai, H.; Iwamoto, T. Efficacy of PPAR-gamma agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging 2011, 32, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Hölscher, C. Drugs developed for treatment of diabetes show protective effects in Alzheimer’s and Parkinson’s diseases. Sheng Li Xue Bao 2014, 66, 497–510. [Google Scholar] [PubMed]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed]
- Pimplikar, S.W. Neuroinflammation in Alzheimer’s disease: From pathogenesis to a therapeutic target. J. Clin. Immunol. 2014, 34, S64–S69. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.C.; Yu, J.T.; Jiang, T.; Tan, L. Autophagy modulation for Alzheimer’s disease therapy. Mol. Neurobiol. 2013, 48, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Tan, M.S.; Wang, H.F.; Cao, L.; Zhang, Q.Q.; Shi, J.Q.; Gao, L.; Qin, H.; et al. Temsirolimus promotes autophagic clearance of amyloid-β and provides protective effects in cellular and animal models of Alzheimer’s disease. Pharmacol. Res. 2014, 81, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Tramutola, A.; Lanzillotta, C.; Di Domenico, T. Targeting mTOR to reduce Alzheimer-related cognitive decline: From current hits to future therapies. Expert Rev. Neurother. 2017, 17, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Hanyu, H.; Sato, T.; Kiuchi, A.; Sakurai, H.; Iwamoto, T. Pioglitazone improved cognition in a pilot study on patients with Alzheimer’s disease and mild cognitive impairment with diabetes mellitus. J. Am. Geriatr. Soc. 2009, 57, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Geldmacher, D.S.; Fritsch, T.; McClendon, M.J.; Landreth, G. A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with Alzheimer disease. Arch. Neurol. 2011, 68, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Martos, C.M.; Atkinson, R.A.K.; Chuah, M.I.; King, A.E.; Vickers, J.C. Combination treatment with leptin and pioglitazone in a mouse model of Alzheimer’s disease. Alzheimers Dement. 2016, 3, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, L.; Gouras, G.K.; Wang, R.; Gross, R.S.; Beal, M.F.; Greengard, P.; Xu, H. Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J. Neurosci. 2001, 21, 2561–2570. [Google Scholar] [PubMed]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; de la Monte, S.M. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J. Alzheimers Dis. 2005, 8, 247–268. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Farr, S.A.; Salameh, T.S.; Niehoff, M.L.; Rhea, E.M.; Morley, J.E.; Hanson, A.J.; Hansen, K.M.; Craft, S. Triglycerides cross the blood-brain barrier and induce central leptin and insulin receptor resistance. Int. J. Obes. 2017. [Google Scholar] [CrossRef] [PubMed]
- Baura, G.D.; Foster, D.M.; Porte, D.; Kahn, S.E.; Bergman, R.N.; Cobelli, C.; Schwartz, M.W. Saturable transport of insulin from plasma into the central-nervous-system of dogs in-vivo—A mechanism for regulated insulin delivery to the brain. J. Clin. Investig. 1993, 92, 1824–1830. [Google Scholar] [CrossRef] [PubMed]
- Plum, L.; Schubert, M.; Brüning, J.C. The role of insulin receptor signaling in the brain. Trends Endocrinol. Metab. 2005, 16, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimers Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, L.; Maute, L.; Guschmann, M. Temsirolimus for advanced renal cellcarcinoma. Expert Rev. Anticancer Ther. 2014, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Kwitkowski, V.E.; Prowell, T.M.; Ibrahim, A.; Farrell, A.T.; Justice, R.; Mitchell, S.S.; Sridhara, R.; Pazdur, R. FDA approval summary: Temsirolimus as treatment foradvanced renal cell carcinoma. Oncologist 2010, 15, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.J. A systemic view of Alzheimer disease—Insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Reference | Physiological Alterations | Pathological Effects |
|---|---|---|
| Biessels and Reagan, 2015 [36] | Down regulation in neurogenesis were associated with reductions in dendritic spine density in CA1 pyramidal neurons. | Learning and memory loss. |
| Hoyer, S., 2004 [26] | Decline in ATP levels (mitochondrial alteration). PKB activity inhibition GSK3 activity increase. | Amount in TAU phosphorylation. Oxidative stress increases |
| De Felice, F.G., 2014 [29] | Neuroinflammation and TNFα increase associated with neuronal ER stress and JNK activation | Brain IR down regulation and synaptic alteration. |
| Grillo, C.A., 2015 [42] | Hippocampal-specific insulin resistance using a lentiviral vector expressing an IR antisense sequence | Down regulation of GluN2B and GluA1 phosphorylation at synapses. Memory failure independent of peripheral metabolic alterations. |
| Hoyer, S., 1994 [28] | Insulin modulates levels of acetylcholine and norepinephrine neurotransmitters, | Cognition loss |
| Frolich, L.D., 1999 [25] | Formation and deposition of advanced glycation end products (AGEs) | Up-regulate APP via oxidative stress and Aβ production enhancement |
| De Felice and Ferreira, 2014 [30] | mTOR dysregulation | Learning and memory deficits, cell cycle reentry |
| Craft, S. 2012 [6] | Insulin resistance increases vascular dysfunction | Vascular dementia |
| Craft, S. 2005 [43] | Insulin resistance inhibits IDE activity | Aβ levels Increase |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Folch, J.; Ettcheto, M.; Busquets, O.; Sánchez-López, E.; Castro-Torres, R.D.; Verdaguer, E.; Manzine, P.R.; Poor, S.R.; García, M.L.; Olloquequi, J.; et al. The Implication of the Brain Insulin Receptor in Late Onset Alzheimer’s Disease Dementia. Pharmaceuticals 2018, 11, 11. https://doi.org/10.3390/ph11010011
Folch J, Ettcheto M, Busquets O, Sánchez-López E, Castro-Torres RD, Verdaguer E, Manzine PR, Poor SR, García ML, Olloquequi J, et al. The Implication of the Brain Insulin Receptor in Late Onset Alzheimer’s Disease Dementia. Pharmaceuticals. 2018; 11(1):11. https://doi.org/10.3390/ph11010011
Chicago/Turabian StyleFolch, Jaume, Miren Ettcheto, Oriol Busquets, Elena Sánchez-López, Rubén D. Castro-Torres, Ester Verdaguer, Patricia R. Manzine, Saghar Rabiei Poor, María Luisa García, Jordi Olloquequi, and et al. 2018. "The Implication of the Brain Insulin Receptor in Late Onset Alzheimer’s Disease Dementia" Pharmaceuticals 11, no. 1: 11. https://doi.org/10.3390/ph11010011
APA StyleFolch, J., Ettcheto, M., Busquets, O., Sánchez-López, E., Castro-Torres, R. D., Verdaguer, E., Manzine, P. R., Poor, S. R., García, M. L., Olloquequi, J., Beas-Zarate, C., Auladell, C., & Camins, A. (2018). The Implication of the Brain Insulin Receptor in Late Onset Alzheimer’s Disease Dementia. Pharmaceuticals, 11(1), 11. https://doi.org/10.3390/ph11010011