
Rapid Determination of Positive–Negative Bacterial Infection Based on Micro-Hyperspectral Technology
Abstract
1. Introduction
2. Materials and Methods
2.1. Micro-Hyperspectral Imaging System
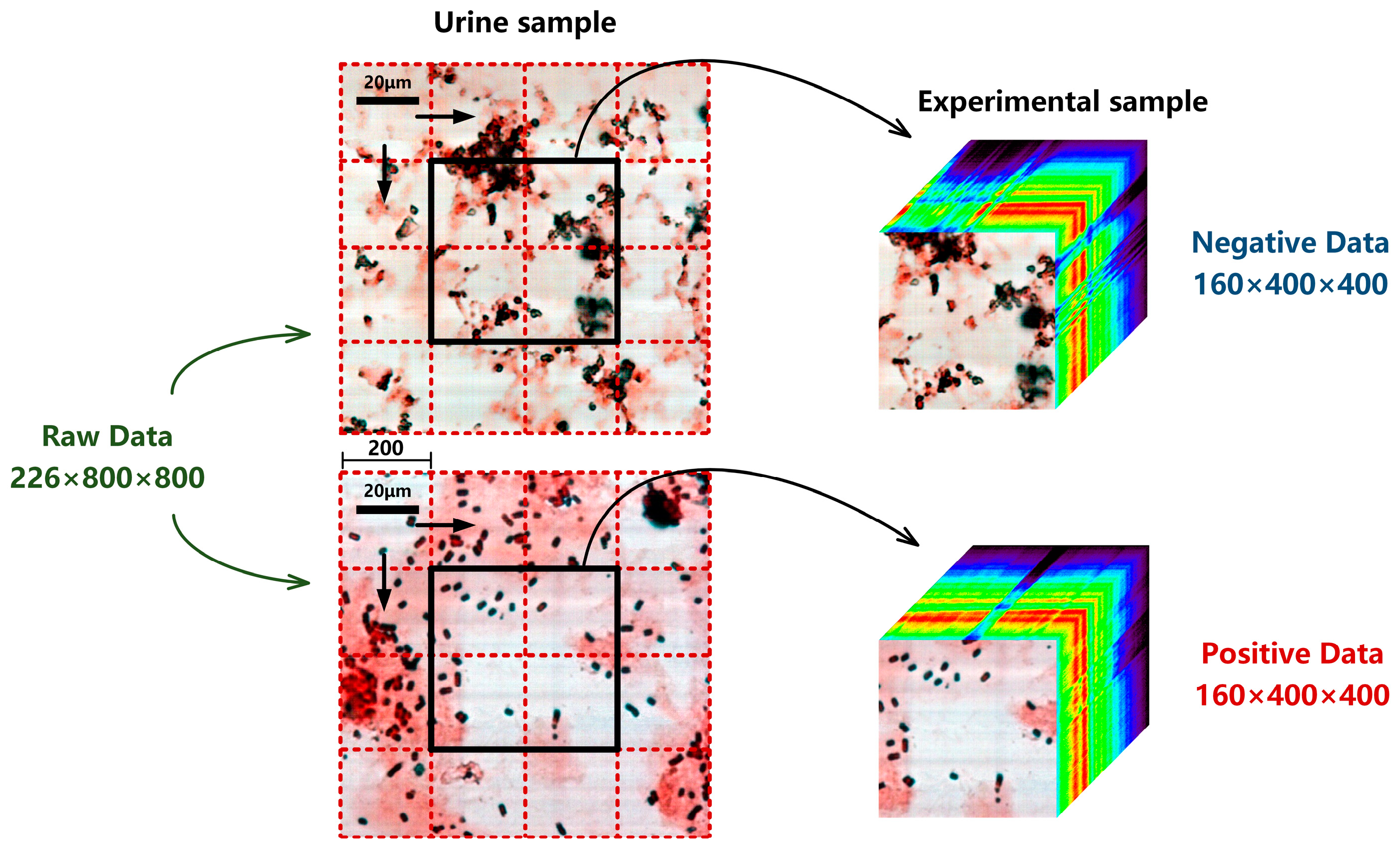
2.2. Experimental Samples Preparation
- Take a clean glass slide, disinfect it with alcohol, and rinse it with distilled water. Then, bake it with an alcohol lamp to remove wax and cool it for later use.
- Record detailed information on the urine sample and assign it a unique identifier. Pour the urine into an anticoagulant tube and balance it (so that the fluid volume in each tube is approximately the same).
- Place the urine sample in a centrifuge and spin it at a speed of 3000 r/10 min.
- Take out the centrifuged urine and use a clean sterile pipette to suck out the supernatant, leaving urine sediment at the bottom. Then, use a new pipette to suck out the urine sediment and mix it thoroughly. Smear the urine sediment on a slide and spread it quickly and evenly by a sterile loop.
- Place the prepared slide in a biosafety cabinet until it is completely dry. Then, proceed with Gram-staining in the following order: stain with crystal violet, cover with iodine, decolorize with 95% ethanol, and counterstain with safranine. Finally, rinse the slide with water and air-dry it for later use. The Gram-staining process is necessary for two reasons. First, Gram-staining is an inherent part of the current testing process, which can highlight the morphological information of bacterial targets and facilitate doctors during observation and determination. It is beneficial for our technology to adhere to the existing bacterial testing process to the maximum extent possible. Second, the bacterial profile and detailed information of the unstained sample are not clear enough without Gram-staining. It is challenging for doctors to label specific bacteria or impurities.
- Place the slide on the microscope stage and search for the field of view under a 10× objective. Convert the objective lens to a 100× objective lens and look for a field of view suspected to contain bacterial distribution. Then, perform a push scanning to capture hyperspectral images of directly smeared urine samples.
2.3. Experimental Dataset
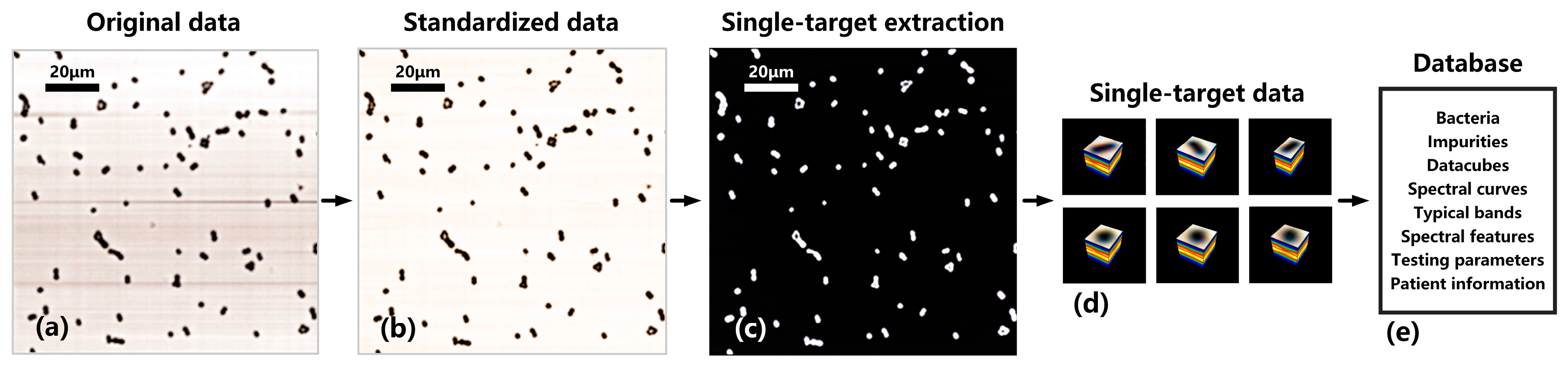
2.4. Database Standardization
- Maintain the light source intensity, focal length, and magnification constant, and collect hyperspectral image B1 of the blank sample from a blank area on the slide.
- Calculate the correction coefficient of spectral dimension:
- 3.
- Calculate the correction coefficient of spatial dimension:
- 4.
- Joint spatial and spectral dimension correction to obtain standardized hyperspectral data:
2.5. Spectral Angle Matching
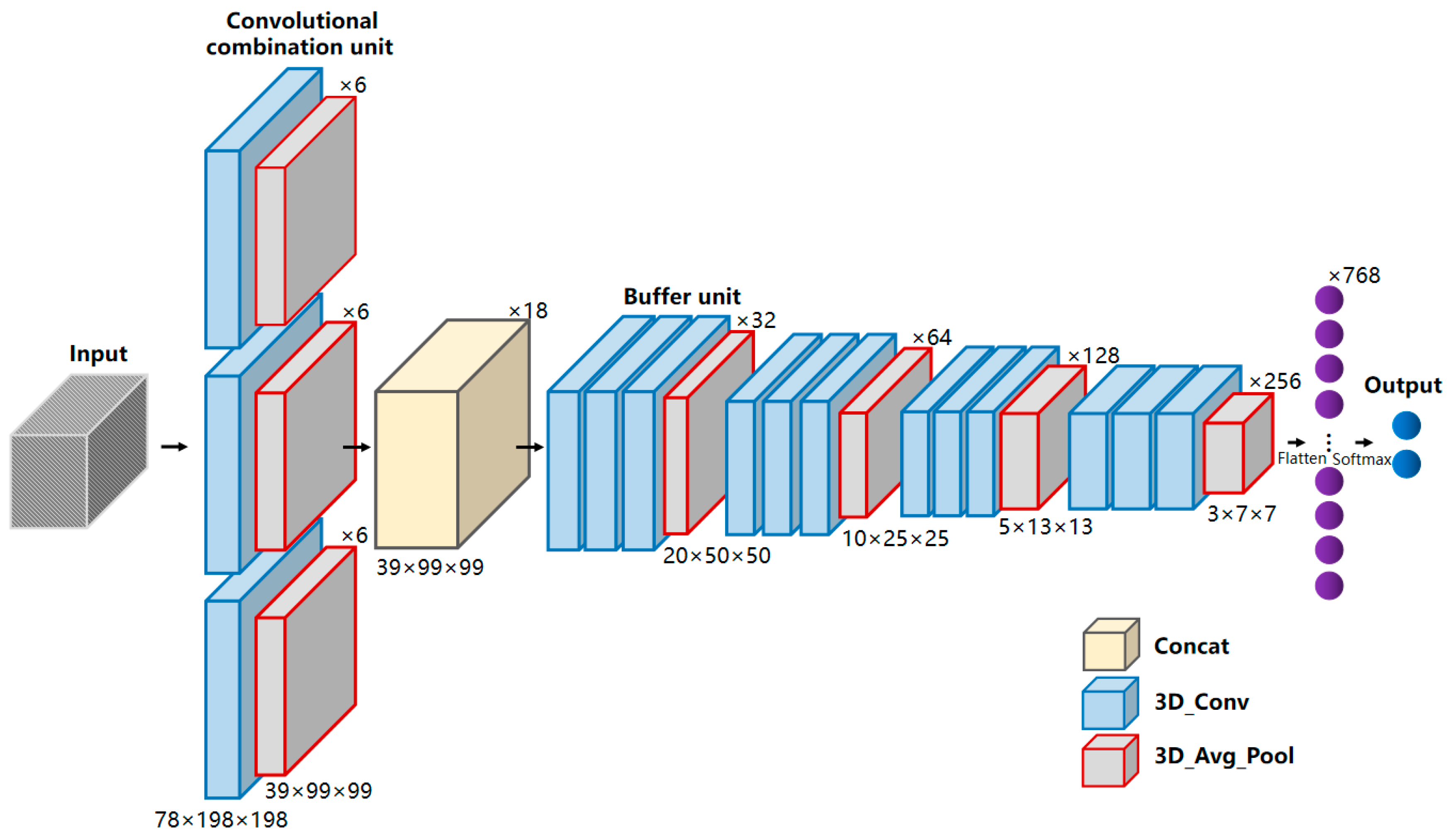
2.6. MBNet
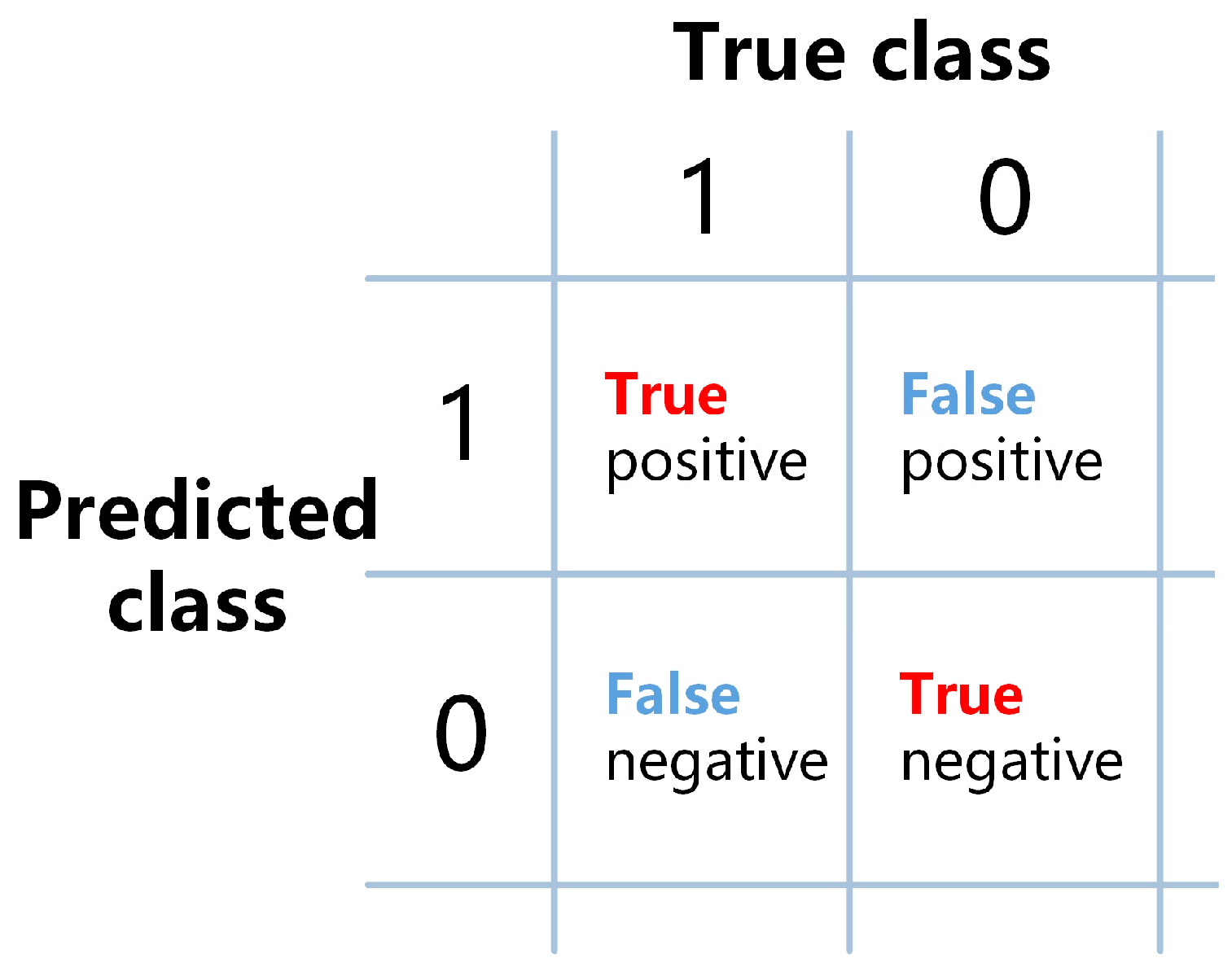
2.7. Evaluation Metrics
3. Results and Discussion
3.1. Hyperspectral Database Matching of Bacterial Sample
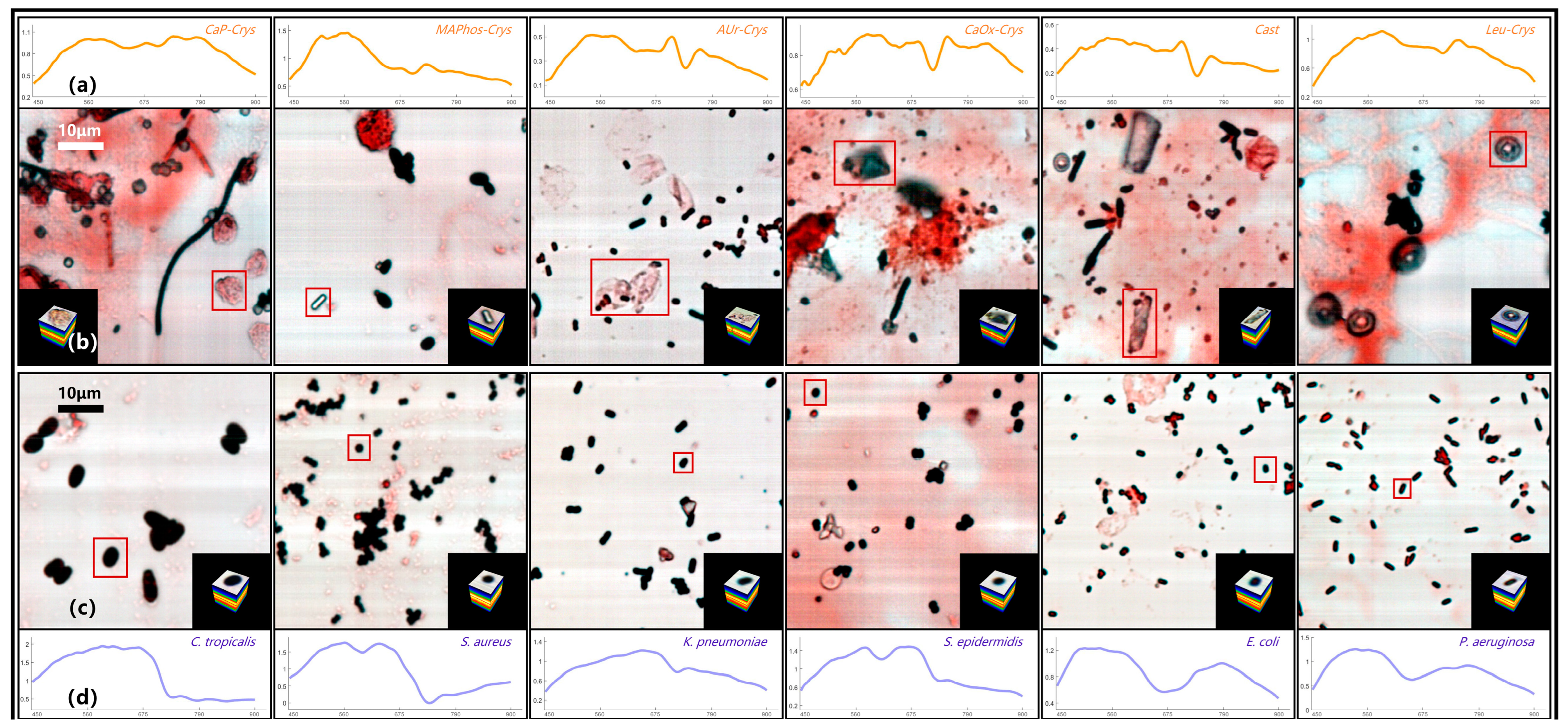
3.1.1. Hyperspectral Database of Directly Smeared Urine Sample
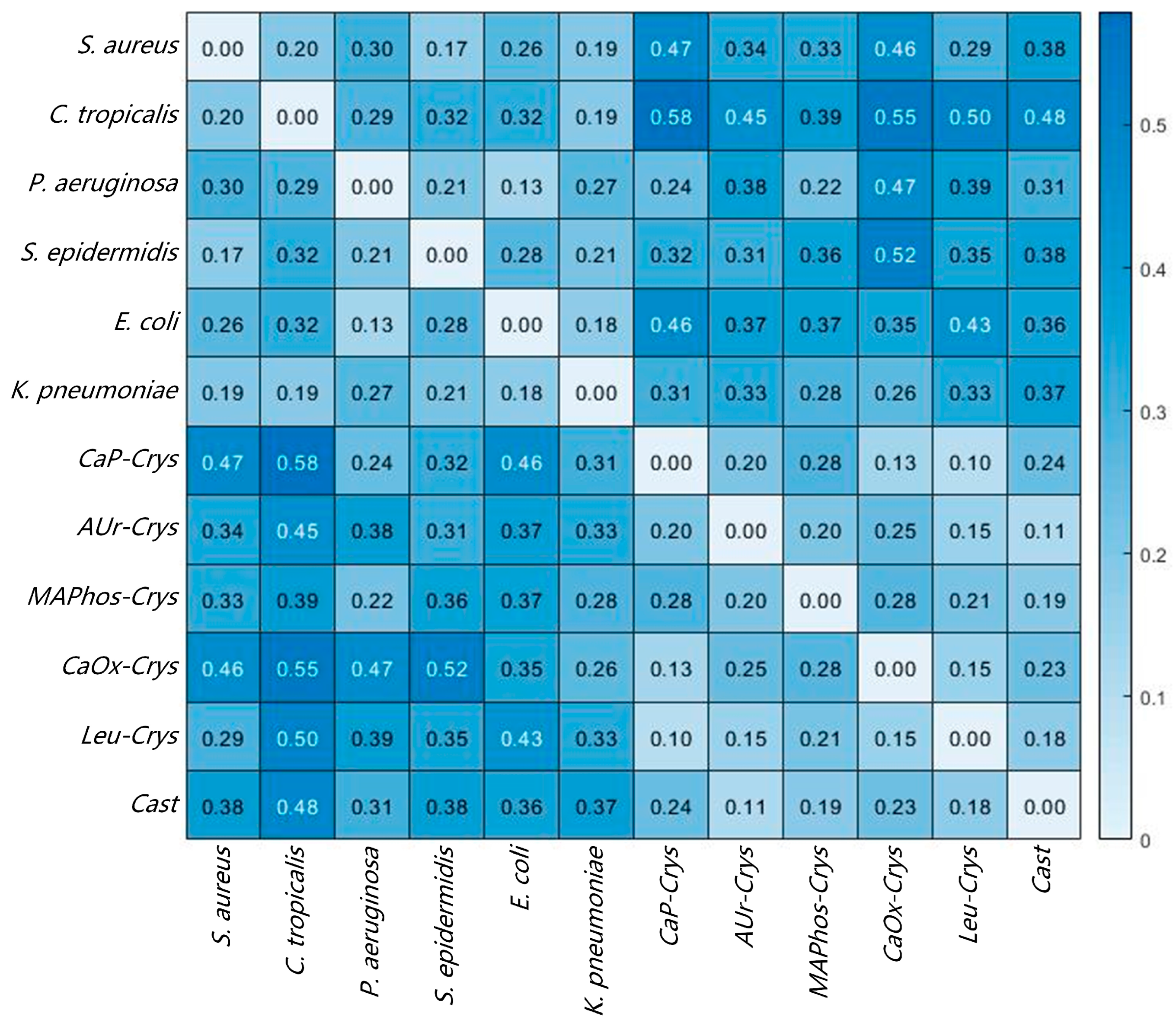
3.1.2. SAM Results of Directly Smeared Bacteria
3.2. Determination of Positive–Negative Bacterial Infection Based on MBNet
3.3. Joint Determination of Positive–Negative Bacterial Infection
- Prepare the directly smeared samples as described in Section 2.2.
- Observe the entire field of view under the microscope and locate the appropriate area.
- Collect the hyperspectral data of urine samples potentially infected with bacterial/fungal via MICROspecim.
- Standardize hyperspectral data as described in Section 2.4.
- Input data into the joint model to determine the bacterial infection (positive or negative).
- If the result is negative, issue a detection report stating “No bacteria detected in this sample.” If the result is positive, issue a detection report stating “Bacteria detected in this sample.”
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, P.; Wang, X. COVID-19: A new challenge for human beings. Cell. Mol. Immunol. 2020, 17, 555–557. [Google Scholar] [CrossRef]
- Baker, R.E.; Mahmud, A.S.; Miller, I.F.; Rajeev, M.; Rasambainarivo, F.; Rice, B.L.; Takahashi, S.; Tatem, A.J.; Wagner, C.E.; Wang, L.-F. Infectious disease in an era of global change. Nat. Rev. Microbiol. 2022, 20, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Bloom, D.E.; Cadarette, D. Infectious disease threats in the twenty-first century: Strengthening the global response. Front. Immunol. 2019, 10, 549. [Google Scholar] [CrossRef]
- Chen, H.; Liu, K.; Li, Z.; Wang, P. Point of care testing for infectious diseases. Clin. Chim. Acta. 2019, 493, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ceylan Koydemir, H.; Qiu, Y.; Bai, B.; Zhang, Y.; Jin, Y.; Tok, S.; Yilmaz, E.C.; Gumustekin, E.; Rivenson, Y. Early detection and classification of live bacteria using time-lapse coherent imaging and deep learning. Light Sci. Appl. 2020, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Ombelet, S.; Barbé, B.; Affolabi, D.; Ronat, J.-B.; Lompo, P.; Lunguya, O.; Jacobs, J.; Hardy, L. Best practices of blood cultures in low-and middle-income countries. Front. Med. 2019, 6, 131. [Google Scholar] [CrossRef]
- Li, H.; Hsieh, K.; Wong, P.K.; Mach, K.E.; Liao, J.C.; Wang, T.-H. Single-cell pathogen diagnostics for combating antibiotic resistance. Nat. Rev. Methods Primers 2023, 3, 6. [Google Scholar] [CrossRef]
- Garner, C.; Brazelton de Cardenas, J.; Suganda, S.; Hayden, R. Accuracy of broad-panel PCR-based bacterial identification for blood cultures in a pediatric oncology population. Microbiol. Spectr. 2021, 9, 10–1128. [Google Scholar] [CrossRef]
- Behzadi, P.; Urbán, E.; Matuz, M.; Benkő, R.; Gajdács, M. The role of gram-negative bacteria in urinary tract infections: Current concepts and therapeutic options. Adv. Microbiol. Infect. Dis. Public Health. 2021, 15, 35–69. [Google Scholar]
- Tullus, K.; Shaikh, N. Urinary tract infections in children. Lancet 2020, 395, 1659–1668. [Google Scholar] [CrossRef]
- Paoletti, M.; Haut, J.; Plaza, J.; Plaza, A. Deep learning classifiers for hyperspectral imaging: A review. ISPRS J. Photogramm. Remote Sens. 2019, 158, 279–317. [Google Scholar] [CrossRef]
- Stuart, M.B.; McGonigle, A.J.; Willmott, J.R. Hyperspectral imaging in environmental monitoring: A review of recent developments and technological advances in compact field deployable systems. Sensors 2019, 19, 3071. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Wang, X. Overview of hyperspectral image classification. J. Sens. 2020, 2, 4817234. [Google Scholar] [CrossRef]
- Feng, L.; Zhu, S.; Liu, F.; He, Y.; Bao, Y.; Zhang, C. Hyperspectral imaging for seed quality and safety inspection: A review. Plant Methods 2019, 15, 91. [Google Scholar] [CrossRef] [PubMed]
- Özdoğan, G.; Lin, X.; Sun, D.-W. Rapid and noninvasive sensory analyses of food products by hyperspectral imaging: Recent application developments. Trends Food Sci. Technol. 2021, 111, 151–165. [Google Scholar] [CrossRef]
- Du, J.; Tao, C.; Xue, S.; Zhang, Z. Joint Diagnostic Method of Tumor Tissue Based on Hyperspectral Spectral-Spatial Transfer Features. Diagnostics 2023, 13, 2002. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J. Hyperspectral imaging for clinical applications. BioChip J. 2022, 16, 1–12. [Google Scholar] [CrossRef]
- Zheng, L.; Wen, Y.; Ren, W.; Duan, H.; Lin, J.; Irudayaraj, J. Hyperspectral dark-field microscopy for pathogen detection based on spectral angle mapping. Sens. Actuators B Chem. 2022, 367, 132042. [Google Scholar] [CrossRef]
- Soni, A.; Dixit, Y.; Reis, M.M.; Brightwell, G. Hyperspectral imaging and machine learning in food microbiology: Developments and challenges in detection of bacterial, fungal, and viral contaminants. Compr. Rev. Food Sci. Food Saf. 2022, 21, 3717–3745. [Google Scholar] [CrossRef]
- Eady, M.; Setia, G.; Park, B. Detection of Salmonella from chicken rinsate with visible/near-infrared hyperspectral microscope imaging compared against RT-PCR. Talanta 2019, 195, 313–319. [Google Scholar] [CrossRef]
- Liu, K.; Ke, Z.; Chen, P.; Zhu, S.; Yin, H.; Li, Z.; Chen, Z. Classification of two species of Gram-positive bacteria through hyperspectral microscopy coupled with machine learning. Biomed. Opt. Express. 2021, 12, 7906–7916. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Park, B.; Eady, M.; Ouyang, Q.; Chen, K. Single-cell classification of foodborne pathogens using hyperspectral microscope imaging coupled with deep learning frameworks. Sens. Actuators B Chem. 2020, 309, 127789. [Google Scholar] [CrossRef]
- Kang, R.; Park, B.; Ouyang, Q.; Ren, N. Rapid identification of foodborne bacteria with hyperspectral microscopic imaging and artificial intelligence classification algorithms. Food Control 2021, 130, 108379. [Google Scholar] [CrossRef]
- Park, B.; Seo, Y.; Yoon, S.-C.; Hinton, A., Jr.; Windham, W.R.; Lawrence, K.C. Hyperspectral microscope imaging methods to classify gram-positive and gram-negative foodborne pathogenic bacteria. Trans. ASABE 2015, 58, 5–16. [Google Scholar]
- Michael, M.; Phebus, R.K.; Amamcharla, J. Hyperspectral imaging of common foodborne pathogens for rapid identification and differentiation. Food Sci. Nutr. 2019, 7, 2716–2725. [Google Scholar] [CrossRef]
- Ho, C.-S.; Jean, N.; Hogan, C.A.; Blackmon, L.; Jeffrey, S.S.; Holodniy, M.; Banaei, N.; Saleh, A.A.; Ermon, S.; Dionne, J. Rapid identification of pathogenic bacteria using Raman spectroscopy and deep learning. Nat. Commun. 2019, 10, 4927. [Google Scholar] [CrossRef]
- Tao, C.; Du, J.; Tang, Y.; Wang, J.; Dong, K.; Yang, M.; Hu, B.; Zhang, Z. A Deep-Learning Based System for Rapid Genus Identification of Pathogens under Hyperspectral Microscopic Images. Cells 2022, 11, 2237. [Google Scholar] [CrossRef]
- Li, Y.-H.; Tan, X.; Zhang, W.; Jiao, Q.-B.; Xu, Y.-X.; Li, H.; Zou, Y.-B.; Yang, L.; Fang, Y.-P. Research and application of several key techniques in hyperspectral image preprocessing. Front. Plant Sci. 2021, 12, 627865. [Google Scholar] [CrossRef]
- Peng, J.; Sun, W.; Ma, L.; Du, Q. Discriminative transfer joint matching for domain adaptation in hyperspectral image classification. IEEE Geosci. Remote Sens. Lett. 2019, 16, 972–976. [Google Scholar] [CrossRef]
- Chang, C.-I. Hyperspectral target detection: Hypothesis testing, signal-to-noise ratio, and spectral angle theories. IEEE Trans. Geosci. Remote Sens. 2021, 60, 1–23. [Google Scholar] [CrossRef]
- Villarraga-Gómez, H.; Herazo, E.L.; Smith, S.T. X-ray computed tomography: From medical imaging to dimensional metrology. Precis. Eng. 2019, 60, 544–569. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Q.; Li, X. Exploring the relationship between 2D/3D convolution for hyperspectral image super-resolution. IEEE Trans. Geosci. Remote Sens. 2021, 59, 8693–8703. [Google Scholar] [CrossRef]
- Li, W.; Chen, H.; Liu, Q.; Liu, H.; Wang, Y.; Gui, G. Attention mechanism and depthwise separable convolution aided 3DCNN for hyperspectral remote sensing image classification. Remote Sen. 2022, 14, 2215. [Google Scholar] [CrossRef]
- Simonyan, K.; Zisserman, A. Very deep convolutional networks for large-scale image recognition. arXiv 2014, arXiv:1409.1556. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Huang, G.; Liu, Z.; Van Der Maaten, L.; Weinberger, K.Q. Densely connected convolutional networks. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Honolulu, HI, USA, 21–26 July 2017; pp. 4700–4708. [Google Scholar]
- Dosovitskiy, A.; Beyer, L.; Kolesnikov, A.; Weissenborn, D.; Zhai, X.; Unterthiner, T.; Dehghani, M.; Minderer, M.; Heigold, G.; Gelly, S. An image is worth 16x16 words: Transformers for image recognition at scale. arXiv 2020, arXiv:2010.11929. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Infection Status | Urine Sample | Experimental Sample | |
|---|---|---|---|
| Negative | 2864 | 25,776 | |
| Positive | E. coli | 1442 | 12,978 |
| K. pneumoniae | 510 | 4590 | |
| A. baumannii | 236 | 2124 | |
| P. mirabilis | 365 | 3285 | |
| E. faecalis | 720 | 6480 | |
| S. epidermidis | 322 | 2898 | |
| P. aeruginosa | 315 | 2835 | |
| S. aureus | 296 | 2664 | |
| C. albicans | 460 | 4140 | |
| C. tropicalis | 594 | 5346 | |
| Total | 8124 | 73,116 | |
| Model | ACC/% | PPV/% | NPV/% |
|---|---|---|---|
| MBNet-0 h | 95.50 | 97.18 | 92.54 |
| VGGNet | 91.54 | 93.00 | 88.77 |
| ResNet | 91.30 | 92.61 | 88.79 |
| DenseNet | 92.71 | 94.12 | 90.08 |
| ViT | 94.74 | 96.16 | 92.18 |
| MBNet-3 h | 95.62 | 97.13 | 92.93 |
| Joint Model | 97.29 | 97.17 | 97.60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, J.; Tao, C.; Qi, M.; Hu, B.; Zhang, Z. Rapid Determination of Positive–Negative Bacterial Infection Based on Micro-Hyperspectral Technology. Sensors 2024, 24, 507. https://doi.org/10.3390/s24020507
Du J, Tao C, Qi M, Hu B, Zhang Z. Rapid Determination of Positive–Negative Bacterial Infection Based on Micro-Hyperspectral Technology. Sensors. 2024; 24(2):507. https://doi.org/10.3390/s24020507
Chicago/Turabian StyleDu, Jian, Chenglong Tao, Meijie Qi, Bingliang Hu, and Zhoufeng Zhang. 2024. "Rapid Determination of Positive–Negative Bacterial Infection Based on Micro-Hyperspectral Technology" Sensors 24, no. 2: 507. https://doi.org/10.3390/s24020507
APA StyleDu, J., Tao, C., Qi, M., Hu, B., & Zhang, Z. (2024). Rapid Determination of Positive–Negative Bacterial Infection Based on Micro-Hyperspectral Technology. Sensors, 24(2), 507. https://doi.org/10.3390/s24020507