
From Smartphone Lateral Flow Immunoassay Screening to Direct MS Analysis: Development and Validation of a Semi-Quantitative Direct Analysis in Real-Time Mass Spectrometric (DART-MS) Approach to the Analysis of Deoxynivalenol


Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. DART-Triple Quadrupole MS/MS Conditions
2.3. Sample Preparation
2.4. Initial In-House Method Validations
3. Results
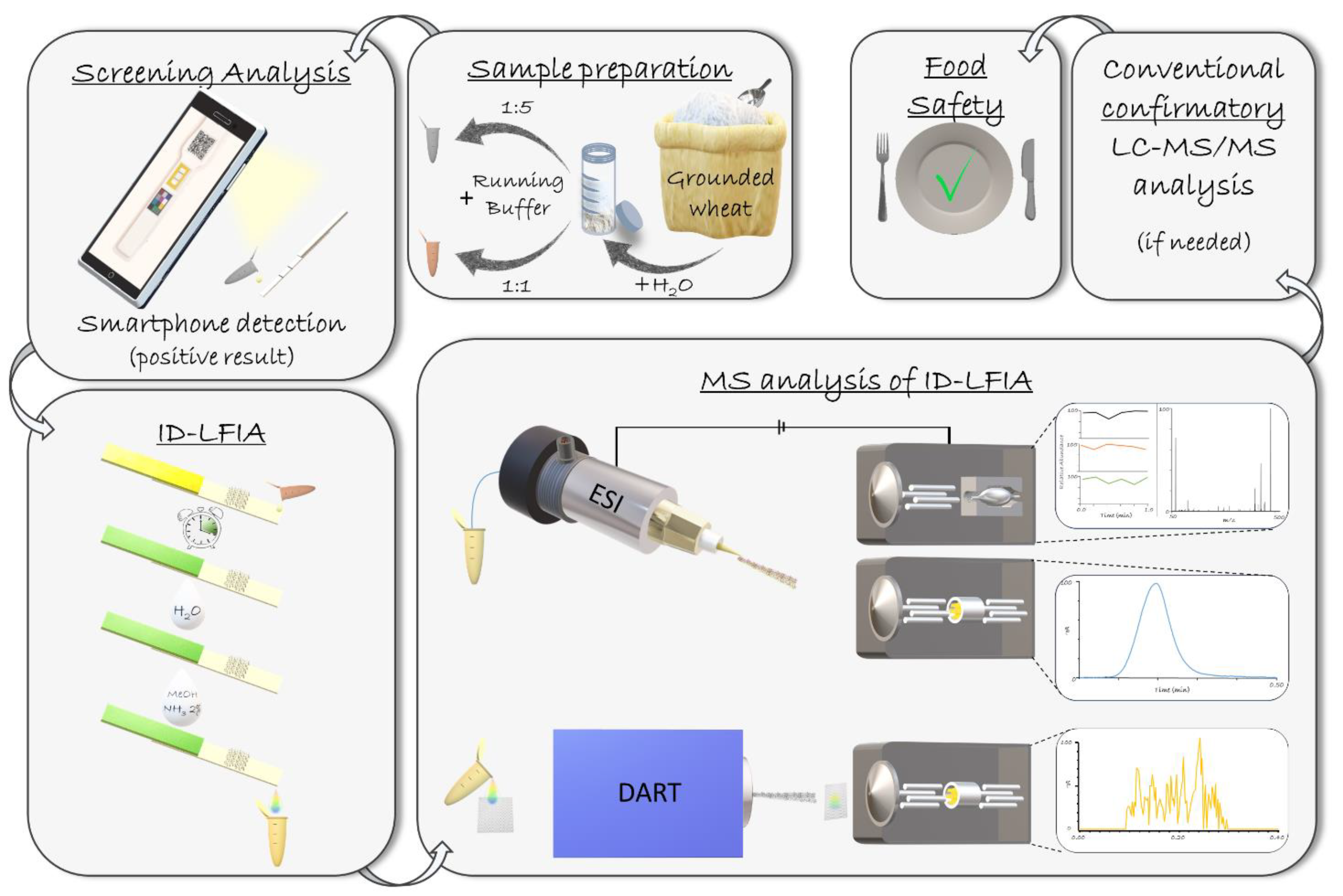
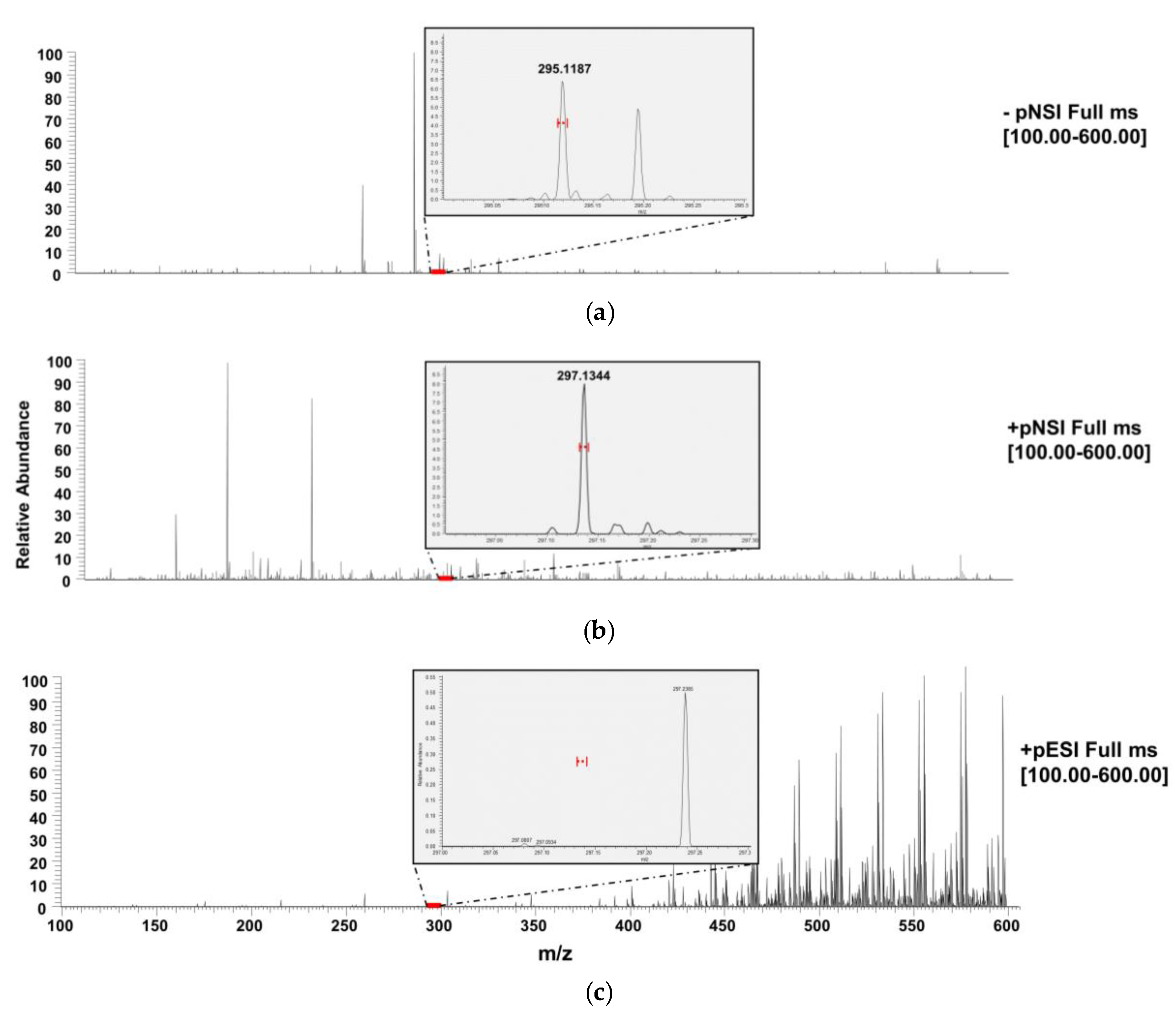
3.1. General Considerations
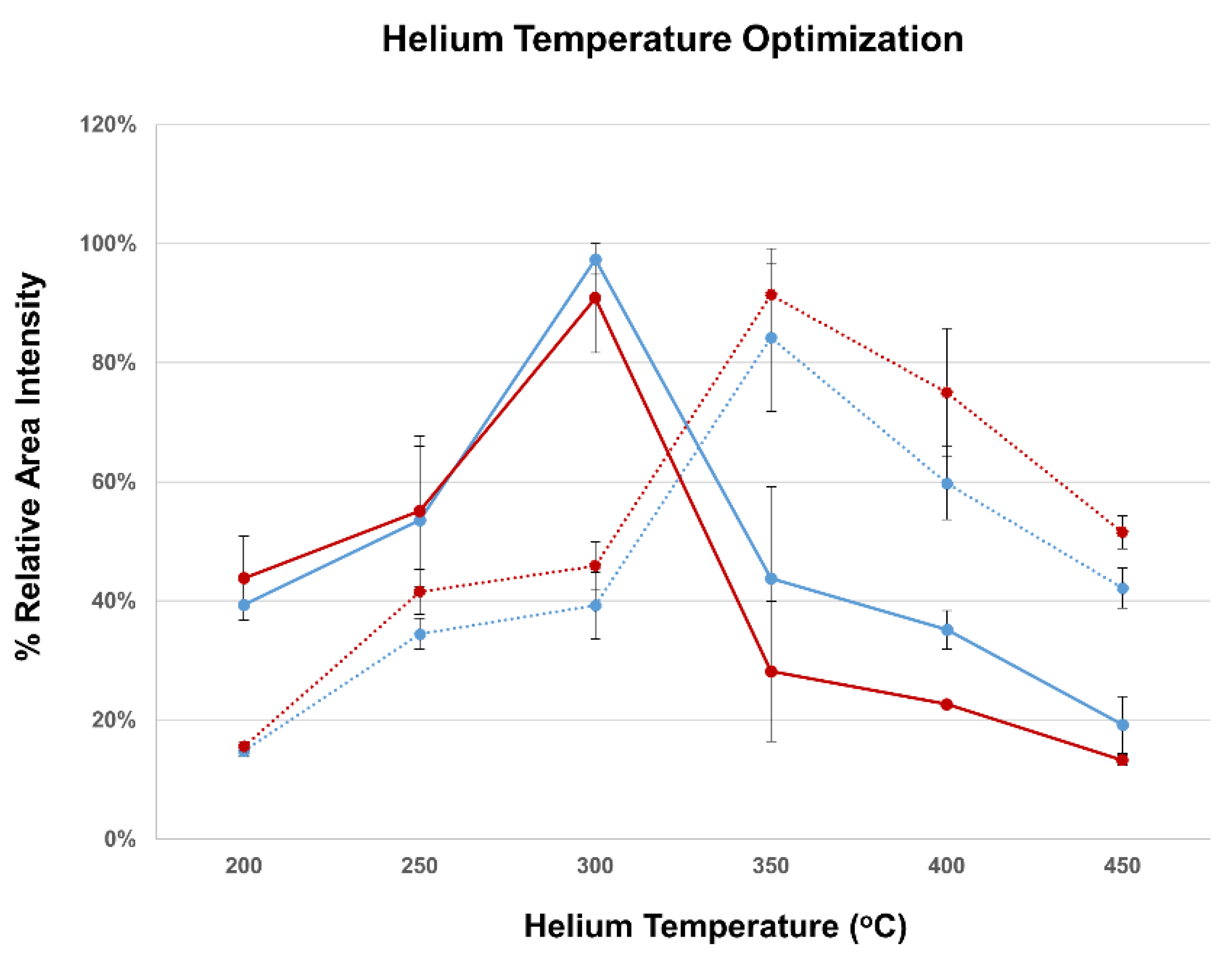
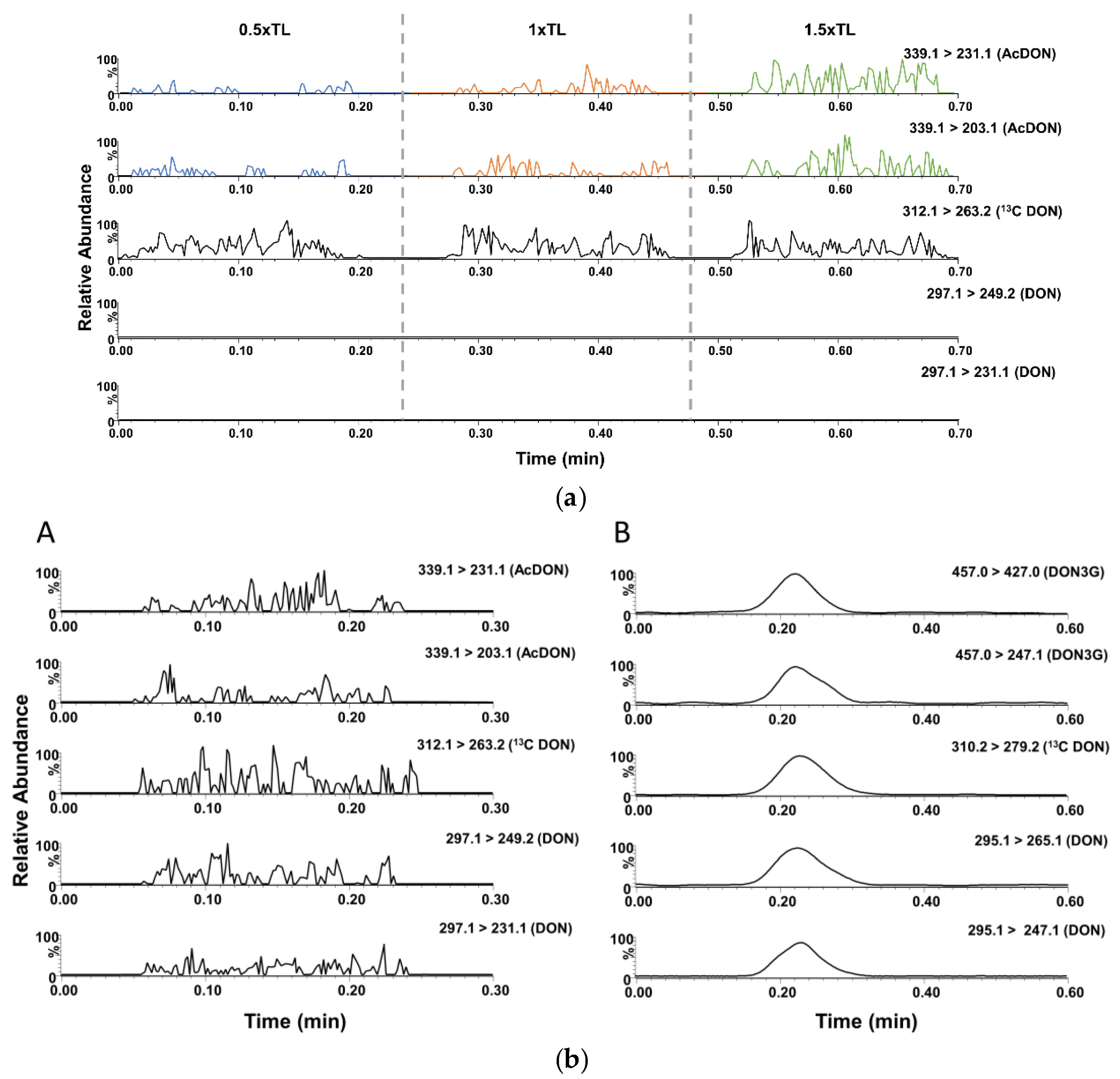
3.2. DART-QqQ-MS/MS Method Development
3.3. Initial In-House Method Validations
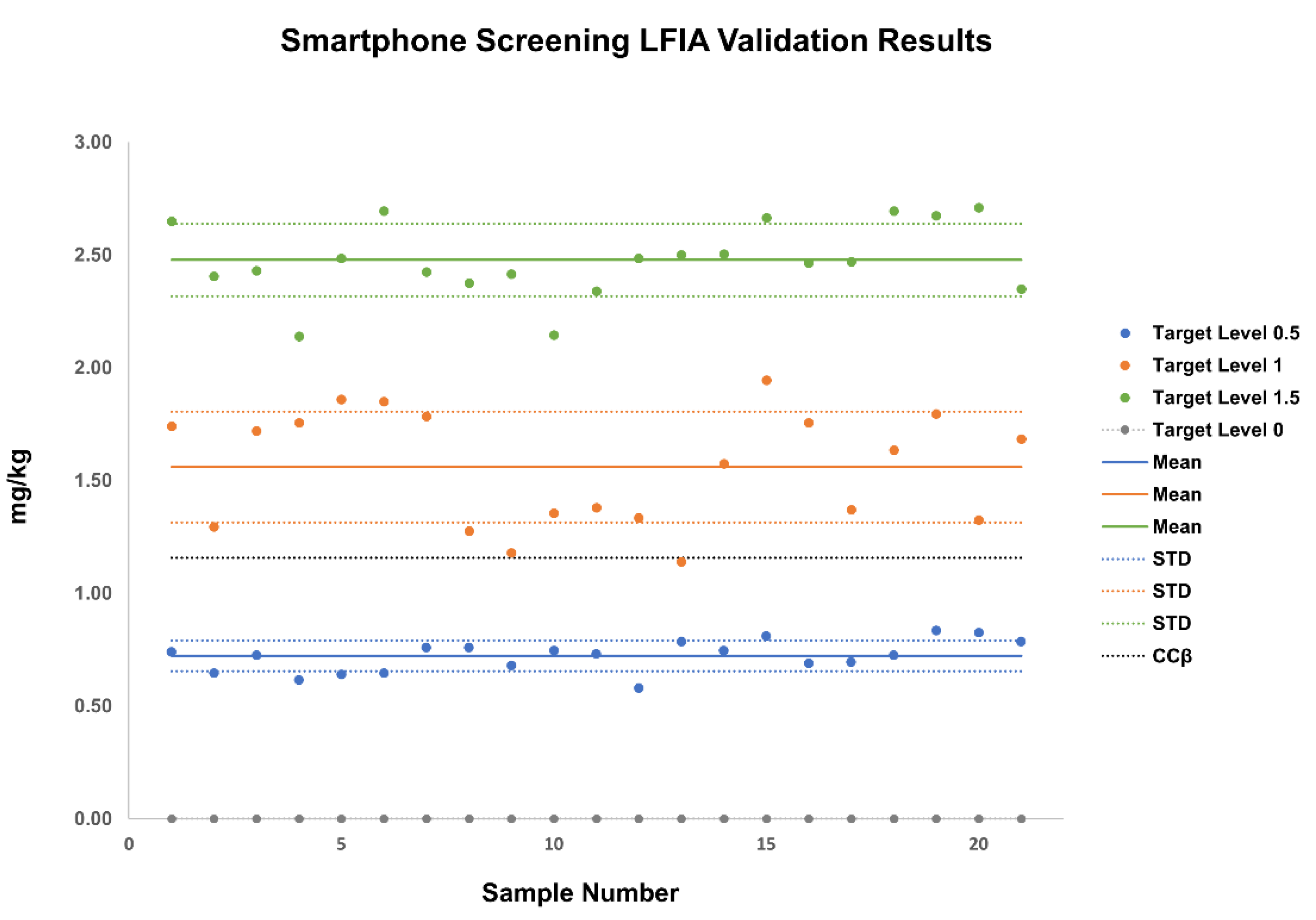
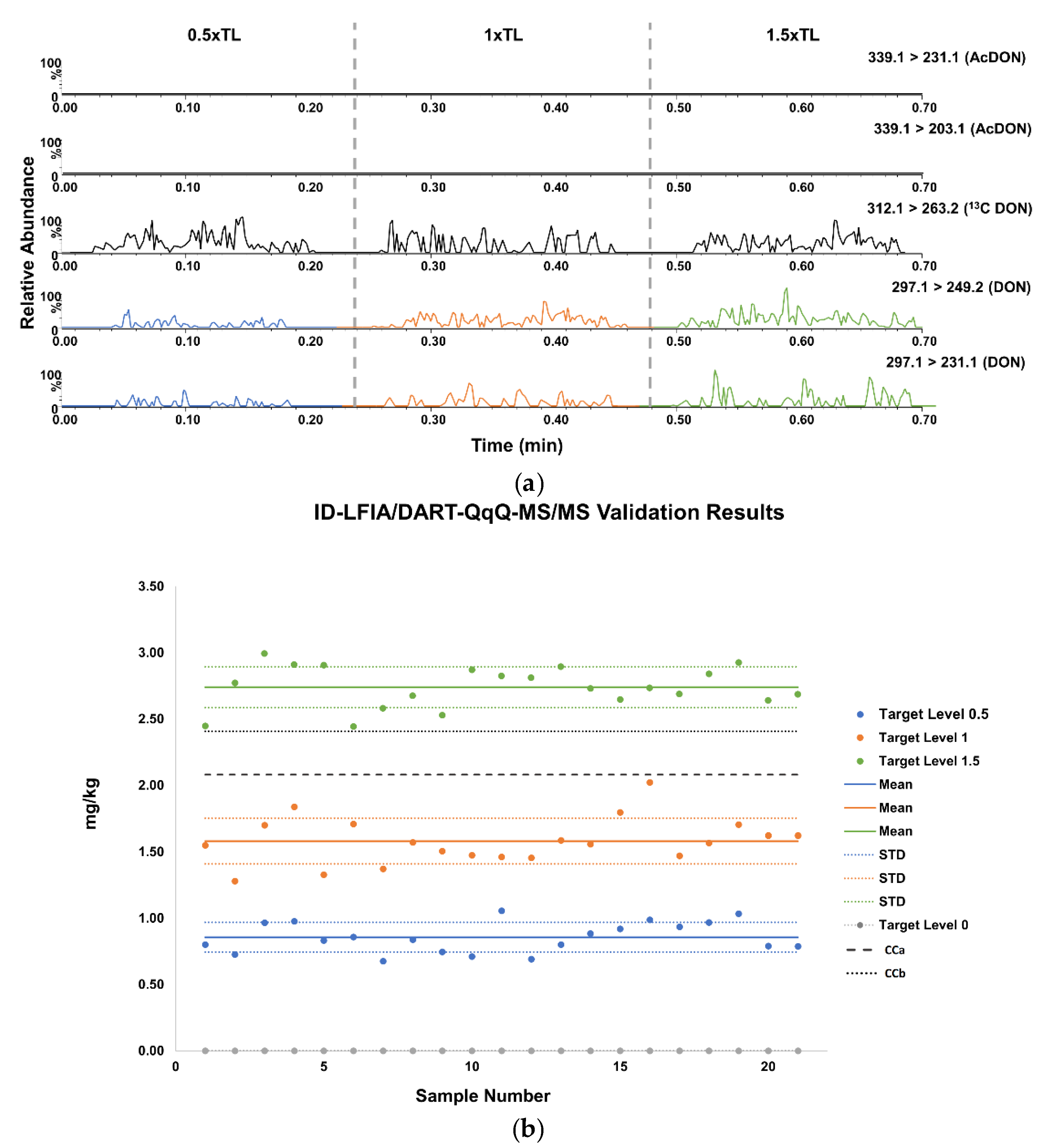
3.3.1. Validation of Screening LFIA with Smartphone Readout
3.3.2. Validation of ID-LFIA/DART-QqQ-MS/MS
3.4. Analysis of Real Samples
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- European Commission. REGULATION (EC) No 178/2002 of the European parliament and of the council of 28 January2002 laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety. Off. J. Eur. Communities 2002, 31, 1–24. [Google Scholar]
- Rodriguez, R.S.; O’Keefe, T.L.; Froehlich, C.; Lewis, R.E.; Sheldon, T.R.; Haynes, C.L. Sensing Food Contaminants: Advances in Analytical Methods and Techniques. Anal. Chem. 2021, 93, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Pedroso Pereira, L.T.; Putnik, P.; Tadashi Iwase, C.H.; de Oliveira Rocha, L. Deoxynivalenol: Insights on genetics, analytical methods and occurrence. Curr. Opin. Food Sci. 2019, 30, 85–92. [Google Scholar] [CrossRef]
- Magnoli, A.P.; Poloni, V.L.; Cavaglieri, L. Impact of mycotoxin contamination in the animal feed industry. Curr. Opin. Food Sci. 2019, 29, 99–108. [Google Scholar] [CrossRef]
- European Commission. Commission Decision 2002/657/EC of 12 August 2002 implementing Council Directive concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Communities 2002, 45, 1–39. [Google Scholar]
- De Boer, A. Scientific assessments in European food law: Making it future-proof. Regul. Toxicol. Pharm. 2019, 108, 104437. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Shi, Z.; Liu, Q. Smartphone-based biosensors for portable food evaluation. Curr. Opin. Food Sci. 2019, 28, 74–81. [Google Scholar] [CrossRef]
- Nelis, J.L.D.; Tsagkaris, A.S.; Dillon, M.J.; Hajslova, J.; Elliott, C.T. Smartphone-based optical assays in the food safety field. Trends Anal. Chem. 2020, 129, 115934. [Google Scholar] [CrossRef]
- Cody, R.B.; Laramee, J.A.; Durst, H.D. Versatile New Ion Source for the Analysis ofMaterials in Open Air under Ambient Conditions. Anal. Chem. 2005, 77, 2297–2302. [Google Scholar] [CrossRef]
- Takats, Z.; Wiseman, J.M.; Gologan, B.; Cooks, R.G. Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science 2004, 306, 471–473. [Google Scholar] [CrossRef]
- Lu, H.; Zhang, H.; Chingin, K.; Xiong, J.; Fang, X.; Chen, H. Ambient mass spectrometry for food science and industry. Trac Trends Anal. Chem. 2018, 107, 99–115. [Google Scholar] [CrossRef]
- Zhang, X.; Ren, X.; Chingin, K. Applications of direct analysis in real time mass spectrometry (DART-MS) in food analysis: A review. Rapid Commun. Mass Spectrom. 2020, e9013. [Google Scholar] [CrossRef]
- Guo, T.; Yong, W.; Jin, Y.; Zhang, L.; Liu, J.; Wang, S.; Chen, Q.; Dong, Y.; Su, H.; Tan, T. Applications of DART-MS for food quality and safety assurance in food supply chain. Mass Spectrom. Rev. 2017, 36, 161–187. [Google Scholar] [CrossRef] [PubMed]
- Khaled, A.; Belinato, J.R.; Pawliszyn, J. Rapid and high-throughput screening of multi-residue pharmaceutical drugs in bovine tissue using solid phase microextraction and direct analysis in real time-tandem mass spectrometry (SPME-DART-MS/MS). Talanta 2020, 217, 121095. [Google Scholar] [CrossRef] [PubMed]
- Busman, M.; Bobell, J.R.; Maragos, C.M. Determination of the aflatoxin M1 (AFM1) from milk by direct analysis in real time—mass spectrometry (DART-MS). Food Control 2015, 47, 592–598. [Google Scholar] [CrossRef]
- Vaclavik, L.; Zachariasova, M.; Hrbek, V.; Hajslova, J. Analysis of multiple mycotoxins in cereals under ambient conditions using direct analysis in real time (DART) ionization coupled to high resolution mass spectrometry. Talanta 2010, 82, 1950–1957. [Google Scholar] [CrossRef] [PubMed]
- Geballa-Koukoula, A.; Gerssen, A.; Nielen, M.W.F. Direct analysis of lateral flow immunoassays for deoxynivalenol using electrospray ionization mass spectrometry. Anal. Bioanal. Chem. 2020, 412, 7547–7558. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Commission Regulation (EC) No 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. Off. J. Eur. Communities 2006, 364, 5–24. [Google Scholar]
- Hajslova, J.; Cajka, T.; Vaclavik, L. Challenging applications offered by direct analysis in real time (DART) in food-quality and safety analysis. Trac Trends Anal. Chem. 2011, 30, 204–218. [Google Scholar] [CrossRef]
- Ross, G.M.S.; Salentijn, G.I.; Nielen, M.W.F. A Critical Comparison between Flow-through and Lateral Flow Immunoassay Formats for Visual and Smartphone-Based Multiplex Allergen Detection. Biosensors 2019, 9, 143. [Google Scholar] [CrossRef]
- Tsagkaris, A.S.; Migliorelli, D.; Uttl, L.; Filippini, D.; Pulkrabova, J.; Hajslova, J. A microfluidic paper-based analytical device (muPAD) with smartphone readout for chlorpyrifos-oxon screening in human serum. Talanta 2021, 222, 121535. [Google Scholar] [CrossRef]
- Ross, G.M.S.; Filippini, D.; Nielen, M.W.F.; Salentijn, G.I. Interconnectable solid-liquid protein extraction unit and chip-based dilution for multiplexed consumer immunodiagnostics. Anal. Chim. Acta 2020, 1140, 190–198. [Google Scholar] [CrossRef]
- Steiner, D.; Sulyok, M.; Malachova, A.; Mueller, A.; Krska, R. Realizing the simultaneous liquid chromatography-tandem mass spectrometry based quantification of >1200 biotoxins, pesticides and veterinary drugs in complex feed. J. Chromatogr. A 2020, 1629, 461502. [Google Scholar] [CrossRef]
- Gross, J.H. Direct analysis in real time—A critical review on DART-MS. Anal. Bioanal. Chem. 2014, 406, 63–80. [Google Scholar] [CrossRef]
- Song, L.; Dykstra, A.B.; Yao, H.; Bartmess, J.E. Ionization mechanism of negative ion-direct analysis in real time: A comparative study with negative ion-atmospheric pressure photoionization. J. Am. Soc. Mass Spectrom. 2009, 20, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Gibson, S.; Bhandari, D.; Cook, K.; Bartmess, J. Ionization mechanism of positive-Ion direct analysis in real time: A transient microenvironment concept. Anal. Chem. 2009, 81, 10080–10088. [Google Scholar] [CrossRef]
- Juan, C.; Covarelli, L.; Beccari, G.; Colasante, V.; Mañes, J. Simultaneous analysis of twenty-six mycotoxins in durum wheat grain from Italy. Food Control 2016, 62, 322–329. [Google Scholar] [CrossRef]
- Rezazadeh, M.; Seidi, S.; Lid, M.; Pedersen-Bjergaard, S.; Yamini, Y. The modern role of smartphones in analytical chemistry. Trac Trends Anal. Chem. 2019, 118, 548–555. [Google Scholar] [CrossRef]
- Rateni, G.; Dario, P.; Cavallo, F. Smartphone-Based Food Diagnostic Technologies: A Review. Sensors 2017, 17, 1453. [Google Scholar] [CrossRef]
- Domínguez, I.; Garrido Frenich, A.; Romero-González, R. Mass spectrometry approaches to ensure food safety. Anal. Methods 2020, 12, 1148–1162. [Google Scholar] [CrossRef]
- Lattanzio, V.M.T.; Ciasca, B.; Powers, S.; von Holst, C. Validation of screening methods according to Regulation 519/2014/EU. Determination of deoxynivalenol in wheat by lateral flow immunoassay: A case study. Trac Trends Anal. Chem. 2016, 76, 137–144. [Google Scholar] [CrossRef]
- Liu, Z.; Hua, Q.; Wang, J.; Liang, Z.; Li, J.; Wu, J.; Shen, X.; Lei, H.; Li, X. A smartphone-based dual detection mode device integrated with two lateral flow immunoassays for multiplex mycotoxins in cereals. Biosens. Bioelectron. 2020, 158, 112178. [Google Scholar] [CrossRef] [PubMed]
- Broekaert, N.; Devreese, M.; De Mil, T.; Fraeyman, S.; De Baere, S.; De Saeger, S.; De Backer, P.; Croubels, S. Development and validation of an LC-MS/MS method for the toxicokinetic study of deoxynivalenol and its acetylated derivatives in chicken and pig plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 971, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Lattanzio, V.M.; Solfrizzo, M.; De Girolamo, A.; Chulze, S.N.; Torres, A.M.; Visconti, A. LC-MS/MS characterization of the urinary excretion profile of the mycotoxin deoxynivalenol in human and rat. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 707–715. [Google Scholar] [CrossRef]
- Cunha, S.C.; Fernandes, J.O. Development and validation of a gas chromatography–mass spectrometry method for determination of deoxynivalenol and its metabolites in human urine. Food Chem. Toxicol. 2012, 50, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Olsson, J.; Börjesson, T.; Lundstedt, T.; Schnürer, J. Detection and quantification of ochratoxin A and deoxynivalenol in barley grains by GC-MS and electronic nose. Int. J. Food Microbiol. 2002, 72, 203–214. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | MRM Ion Transitions (m/z) (Positive Ion Mode) | Collision Energy (eV) |
|---|---|---|
| DON | 297.1 > 249.2 | 10 |
| 297.1 > 231.1 | 16 | |
| 13C15 DON | 312.1 > 263.2 | 10 |
| 312.1 > 245.1 | 16 | |
| AcDON | 339.1 > 231.1 | 10 |
| 339.1 > 203.1 | 16 |
| Day | Sample | 0× TL | 0.5× TL | 1× TL | 1.5× TL | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID-LFIA/DART-QqQ-MS/MS Response Factor Versus 13C15 DON ± SD | LFIA Smartphone Screening Result (mg/kg, Mean Response ± SD) | ID-LFIA/DART-QqQ-MS/MS | LFIA Smartphone Screening Result (mg/kg, Mean Response ± SD) | ID-LFIA/DART-QqQ-MS/MS | LFIA Smartphone Screening Result (mg/kg, Mean Response ± SD) | ID-LFIA/DART-QqQ-MS/MS | LFIA Smartphone Screening Result (mg/kg, Mean Response ± SD) | |||||
| Mean Ion Area Ratio of DON ± SD | Expected mg/kg ± SD | Mean Ion Area Ratio of DON ± SD | Expected mg/kg ± SD | Mean Ion Area Ratio of DON ± SD | Expected mg/kg ± SD | |||||||
| 1 | 1 | 0 | <0.25 | 0.30 (±0.03) | 0.80 (±0.01) | 0.74 (±0.01) | 0.27 (±0.03) | 1.55 (±0.06) | 1.74 (±0.01) | 0.29 (±0.05) | 2.40 (±0.02) | 2.65 (±0.00) |
| 2 | 0 | <0.25 | 0.30 (±0.00) | 0.73 (±0.01) | 0.65 (±0.02) | 0.32 (±0.01) | 1.28 (±0.03) | 1.30 (±0.01) | 0.30 (±0.03) | 2.77 (±0.01) | 2.41 (±0.01) | |
| 3 | 0 | <0.25 | 0.29 (±0.02) | 0.97 (±0.06) | 0.73 (±0.01) | 0.28 (±0.01) | 1.70 (±0.03) | 1.72 (±0.02) | 0.32 (±0.01) | 2.99 (±0.03) | 2.43 (±0.02) | |
| 4 | 0 | <0.25 | 0.28 (±0.04) | 0.98 (±0.01) | 0.62 (±0.01) | 0.34 (±0.00) | 1.84 (±0.04) | 1.76 (±0.01) | 0.28 (±0.00) | 2.91 (±0.11) | 2.14 (±0.03) | |
| 5 | 0 | <0.25 | 0.28 (±0.01) | 0.83 (±0.03) | 0.64 (±0.00) | 0.30 (±0.02) | 1.33 (±0.01) | 1.86 (±0.02) | 0.27 (±0.00) | 2.91 (±0.12) | 2.49 (±0.03) | |
| 6 | 0 | <0.25 | 0.32 (±0.01) | 0.86 (±0.02) | 0.65 (±0.01) | 0.23 (±0.00) | 1.71 (±0.04) | 1.85 (±0.03) | 0.32 (±0.02) | 2.42 (±0.00) | 2.70 (±003) | |
| 7 | 0 | <0.25 | 0.31 (±0.02) | 0.68 (±0.02) | 0.76 (±0.00) | 0.31 (±0.02) | 1.37 (±0.00) | 1.79 (±0.02) | 0.30 (±0.01) | 2.58 (±0.06) | 2.43 (±0.01) | |
| 2 | 8 | 0 | <0.25 | 0.34 (±0.00) | 0.84 (±0.02) | 0.76 (±0.00) | 0.31 (±0.02) | 1.57 (±0.02) | 1.28 (±0.01) | 0.31 (±0.02) | 2.98 (±0.01) | 2.38 (±0.02) |
| 9 | 0 | <0.25 | 0.34 (±0.01) | 0.75 (±0.02) | 0.68 (±0.01) | 0.25 (±0.00) | 1.51 (±0.03) | 1.18 (±0.01) | 0.29 (±0.03) | 2.53 (±0.01) | 2.42 (±0.00) | |
| 10 | 0 | <0.25 | 0.26 (±0.01) | 0.71 (±0.03) | 0.75 (±0.01) | 0.27 (±0.02) | 1.47 (±0.06) | 1.36 (±0.01) | 0.29 (±0.02) | 2.87 (±0.07) | 2.15 (±0.02) | |
| 11 | 0 | <0.25 | 0.31 (±0.02) | 1.06 (±0.01) | 0.73 (±0.00) | 0.28 (±0.05) | 1.46 (±0.02) | 1.38 (±0.00) | 0.28 (±0.04) | 2.82 (±0.00) | 2.34 (±0.07) | |
| 12 | 0 | <0.25 | 0.31 (±0.01) | 0.69 (±0.00) | 0.58 (±0.01) | 0.28 (±0.01) | 1.46 (±0.00) | 1.34 (±0.01) | 0.33 (±0.02) | 2.81 (±0.01) | 2.49 (±0.05) | |
| 13 | 0 | <0.25 | 0.30 (±0.04) | 0.80 (±0.01) | 0.79 (±0.01) | 0.30 (±0.02) | 1.59 (±0.01) | 1.14 (±0.01) | 0.31 (±0.04) | 2.90 (±0.05) | 2.50 (±0.00) | |
| 14 | 0 | <0.25 | 0.26 (±0.01) | 0.88 (±0.00) | 0.75 (±0.01) | 0.24 (±0.00) | 1.56 (±0.00) | 1.58 (±0.03) | 0.27 (±0.01) | 2.73 (±0.00) | 2.51 (±0.00) | |
| 3 | 15 | 0 | <0.25 | 0.31 (±0.03) | 0.92 (±0.02) | 0.81 (±0.00) | 0.26 (±0.03) | 1.80 (±0.07) | 1.95 (±0.02) | 0.29 (±0.02) | 2.65 (±0.01) | 2.67 (±0.02) |
| 16 | 0 | <0.25 | 0.33 (±0.02) | 0.99 (±0.03) | 0.69 (±0.01) | 0.25 (±0.02) | 2.12 (±0.02) | 1.76 (±0.02) | 0.29 (±0.05) | 2.73 (±0.03) | 2.47 (±0.03) | |
| 17 | 0 | <0.25 | 0.33 (±0.01) | 0.94 (±0.01) | 0.70 (±0.02) | 0.31 (±0.03) | 1.47 (±0.03) | 1.37 (±0.01) | 0.29 (±0.01) | 2.69 (±0.04) | 2.47 (±0.01) | |
| 18 | 0 | <0.25 | 0.30 (±0.04) | 0.97 (±0.03) | 0.73 (±0.05) | 0.25 (±0.00) | 1.57 (±0.01) | 1.64 (±0.02) | 0.26 (±0.01) | 2.84 (±0.07) | 2.70 (±0.03) | |
| 19 | 0 | <0.25 | 0.27 (±0.02) | 1.03 (±0.01) | 0.84 (±0.04) | 0.30 (±0.03) | 1.70 (±0.02) | 1.80 (±0.01) | 0.31 (±0.01) | 2.92 (±0.07) | 2.68 (±0.01) | |
| 20 | 0 | <0.25 | 0.29 (±0.00) | 0.79 (±0.02) | 0.83 (±0.06) | 0.26 (±0.02) | 1.62 (±0.01) | 1.33 (±0.01) | 0.34 (±0.00) | 2.64 (±0.01) | 2.71 (±0.03) | |
| 21 | 0 | <0.25 | 0.32 (±0.00) | 0.79 (±0.03) | 0.79 (±0.01) | 0.25 (±0.00) | 1.62 (±0.06) | 1.69 (±0.05) | 0.32 (±0.01) | 2.69 (±0.05) | 2.35 (±0.25) | |
| TL | ID-LFIA/DART-QqQ-MS/MS | LFIA Smartphone Screening Result (mg/kg, Mean Response ± SD) | ||||
|---|---|---|---|---|---|---|
| Mean Ion Area Ratio of DON ± SD | Response Factor ± SD | Mean Ion Area Ratio of AcDON ± SD | Response Factor ± SD | |||
| AcDON-spiked wheat | 0.5 | - | - | 0.73 (±0.06) | 0.09 (±0.01) | 0.64 (±0.01) |
| 1 | - | - | 0.76 (±0.03) | 0.16 (±0.02) | 1.87 (±0.04) | |
| 1.5 | - | - | 0.65 (±0.02) | 0.31 (±0.01) | 2.62 (±0.03) | |
| Contaminated corn starch | 0.32 (±0.09) | 0.82 (±0.08) | 0.71 (±0.08) | 0.45 (±0.05) | 12.76 (±0.07) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geballa-Koukoula, A.; Gerssen, A.; Nielen, M.W.F. From Smartphone Lateral Flow Immunoassay Screening to Direct MS Analysis: Development and Validation of a Semi-Quantitative Direct Analysis in Real-Time Mass Spectrometric (DART-MS) Approach to the Analysis of Deoxynivalenol. Sensors 2021, 21, 1861. https://doi.org/10.3390/s21051861
Geballa-Koukoula A, Gerssen A, Nielen MWF. From Smartphone Lateral Flow Immunoassay Screening to Direct MS Analysis: Development and Validation of a Semi-Quantitative Direct Analysis in Real-Time Mass Spectrometric (DART-MS) Approach to the Analysis of Deoxynivalenol. Sensors. 2021; 21(5):1861. https://doi.org/10.3390/s21051861
Chicago/Turabian StyleGeballa-Koukoula, Ariadni, Arjen Gerssen, and Michel W. F. Nielen. 2021. "From Smartphone Lateral Flow Immunoassay Screening to Direct MS Analysis: Development and Validation of a Semi-Quantitative Direct Analysis in Real-Time Mass Spectrometric (DART-MS) Approach to the Analysis of Deoxynivalenol" Sensors 21, no. 5: 1861. https://doi.org/10.3390/s21051861
APA StyleGeballa-Koukoula, A., Gerssen, A., & Nielen, M. W. F. (2021). From Smartphone Lateral Flow Immunoassay Screening to Direct MS Analysis: Development and Validation of a Semi-Quantitative Direct Analysis in Real-Time Mass Spectrometric (DART-MS) Approach to the Analysis of Deoxynivalenol. Sensors, 21(5), 1861. https://doi.org/10.3390/s21051861