
Fluoride Anion Recognition by a Multifunctional Urea Derivative: An Experimental and Theoretical Study


 ,
,  ,
,  and
and 
Abstract
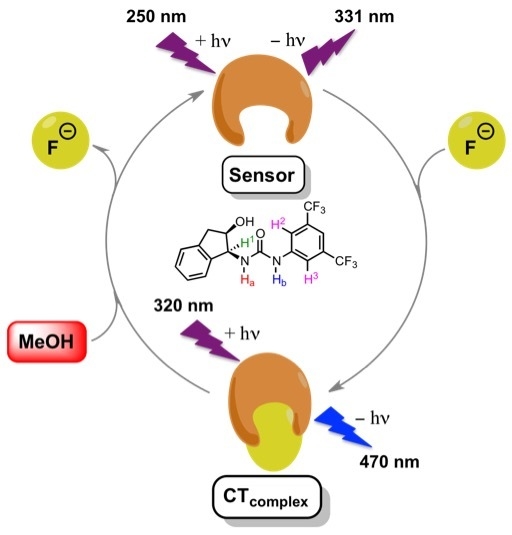
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Absorption and Fluorescence Spectroscopy
2.2.2. Theoretical Calculations
2.2.3. NMR Spectroscopy
2.2.4. Electron Microscopy
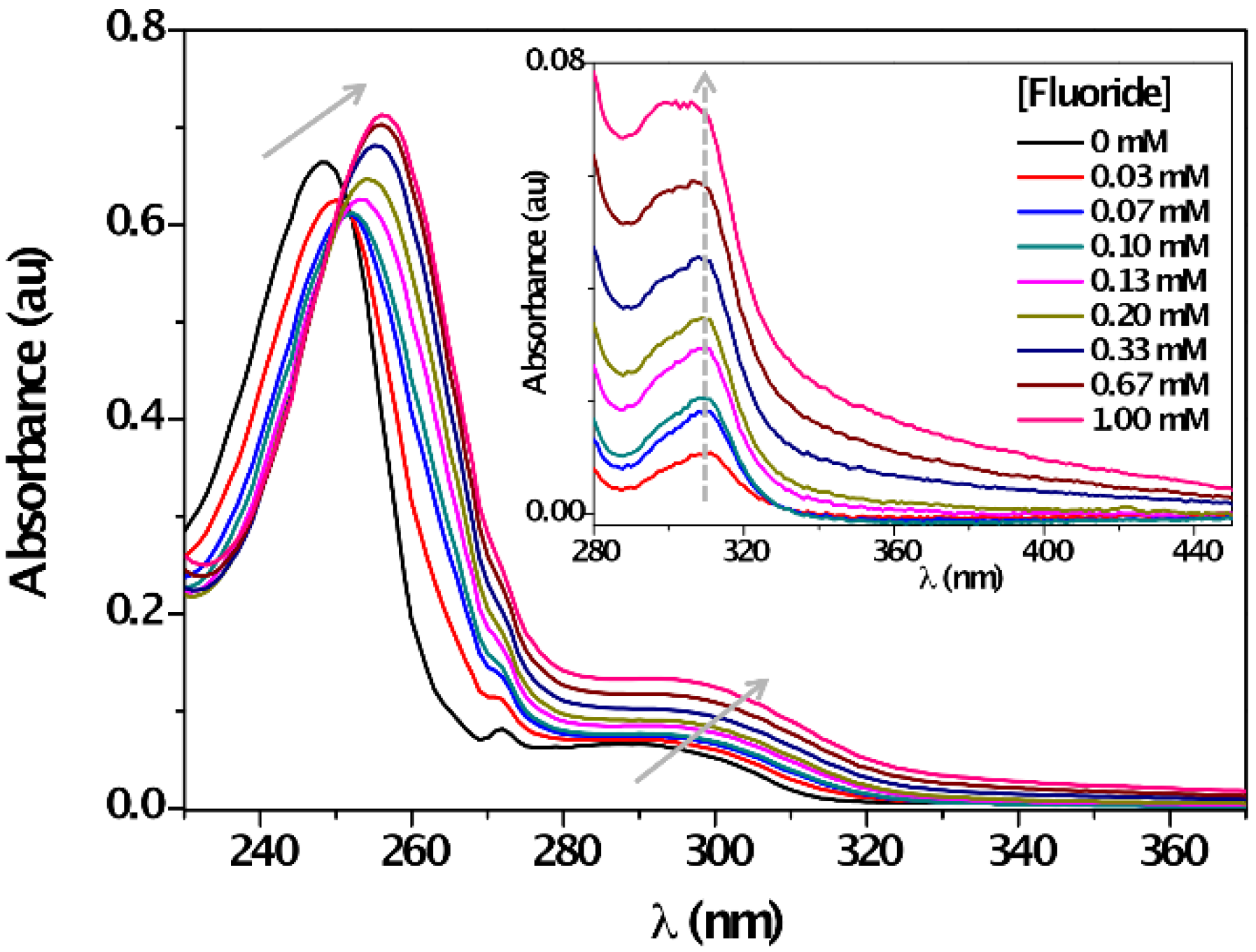
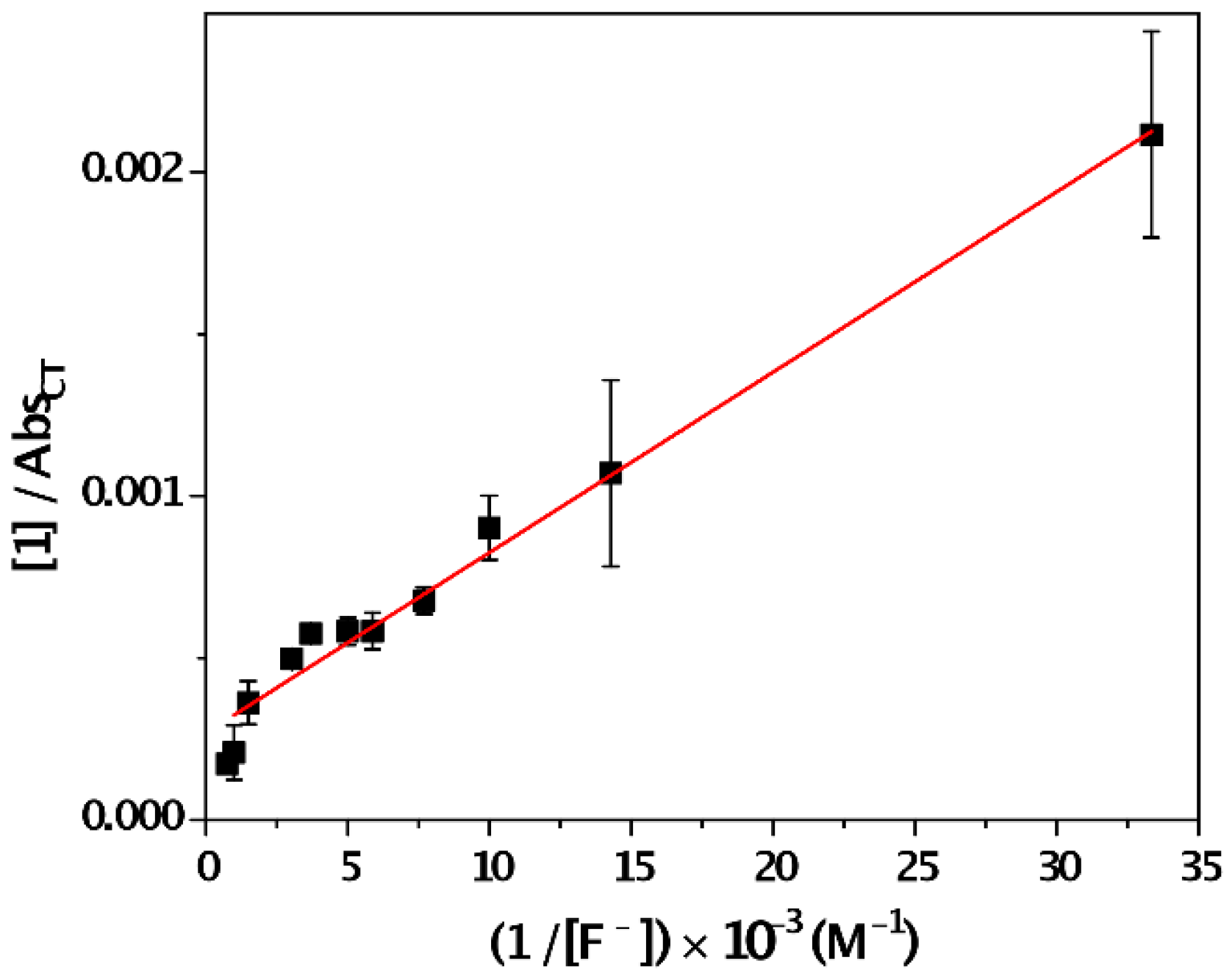
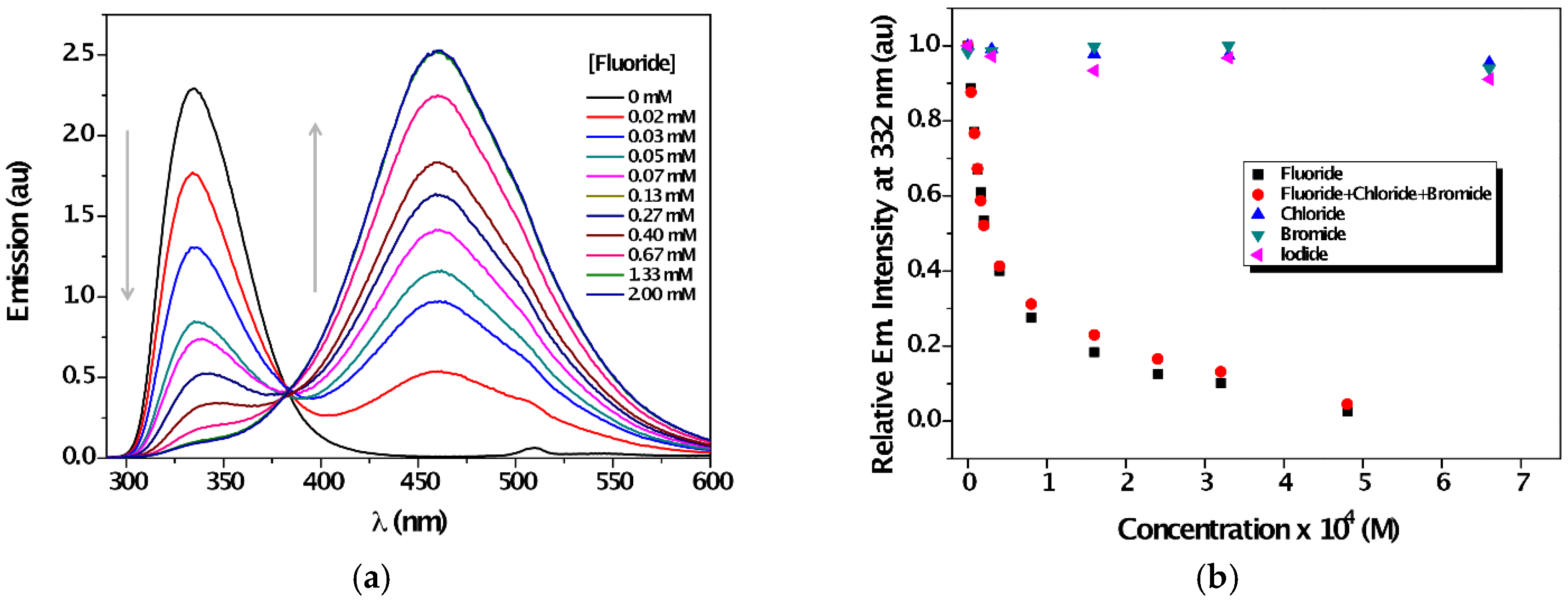
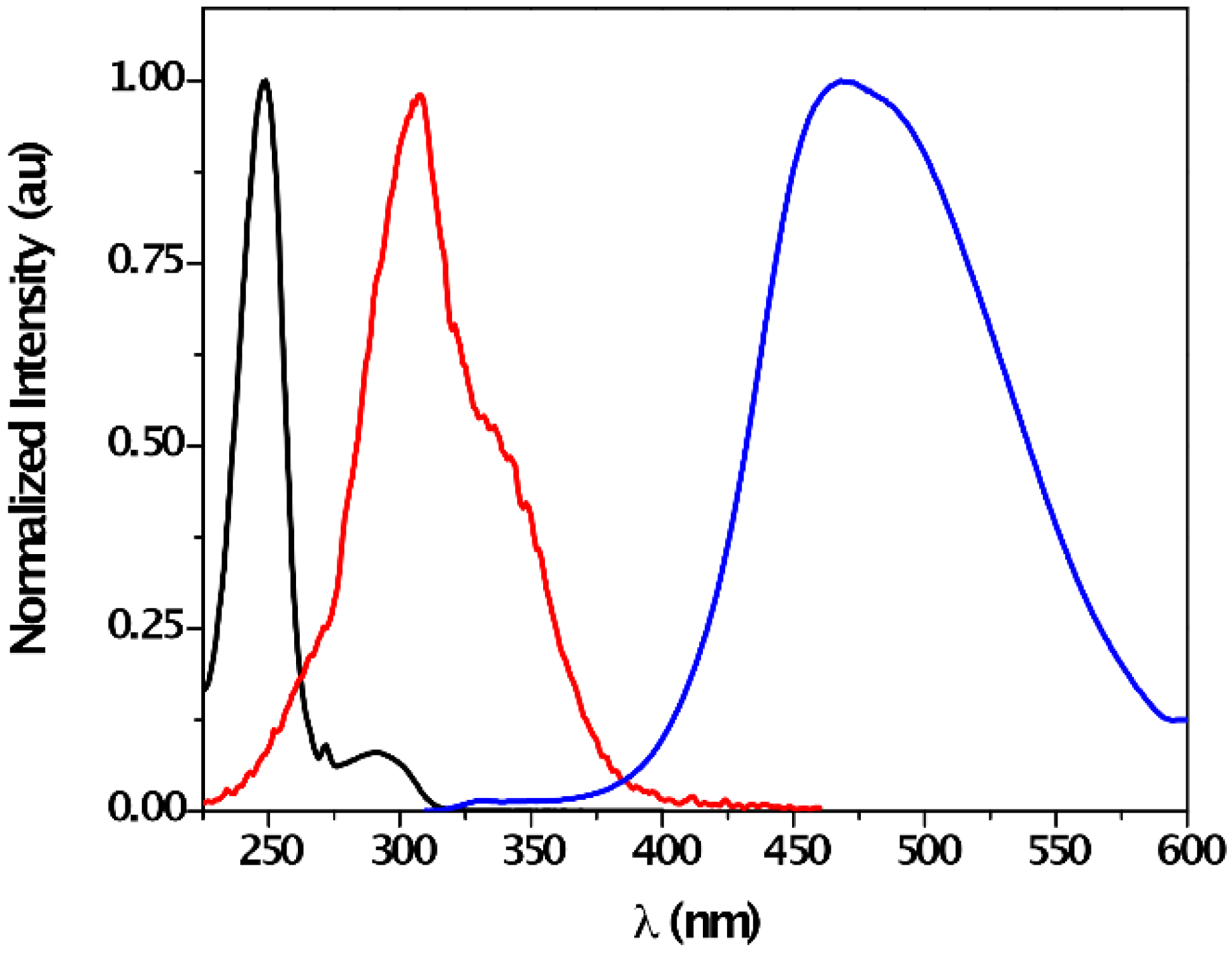
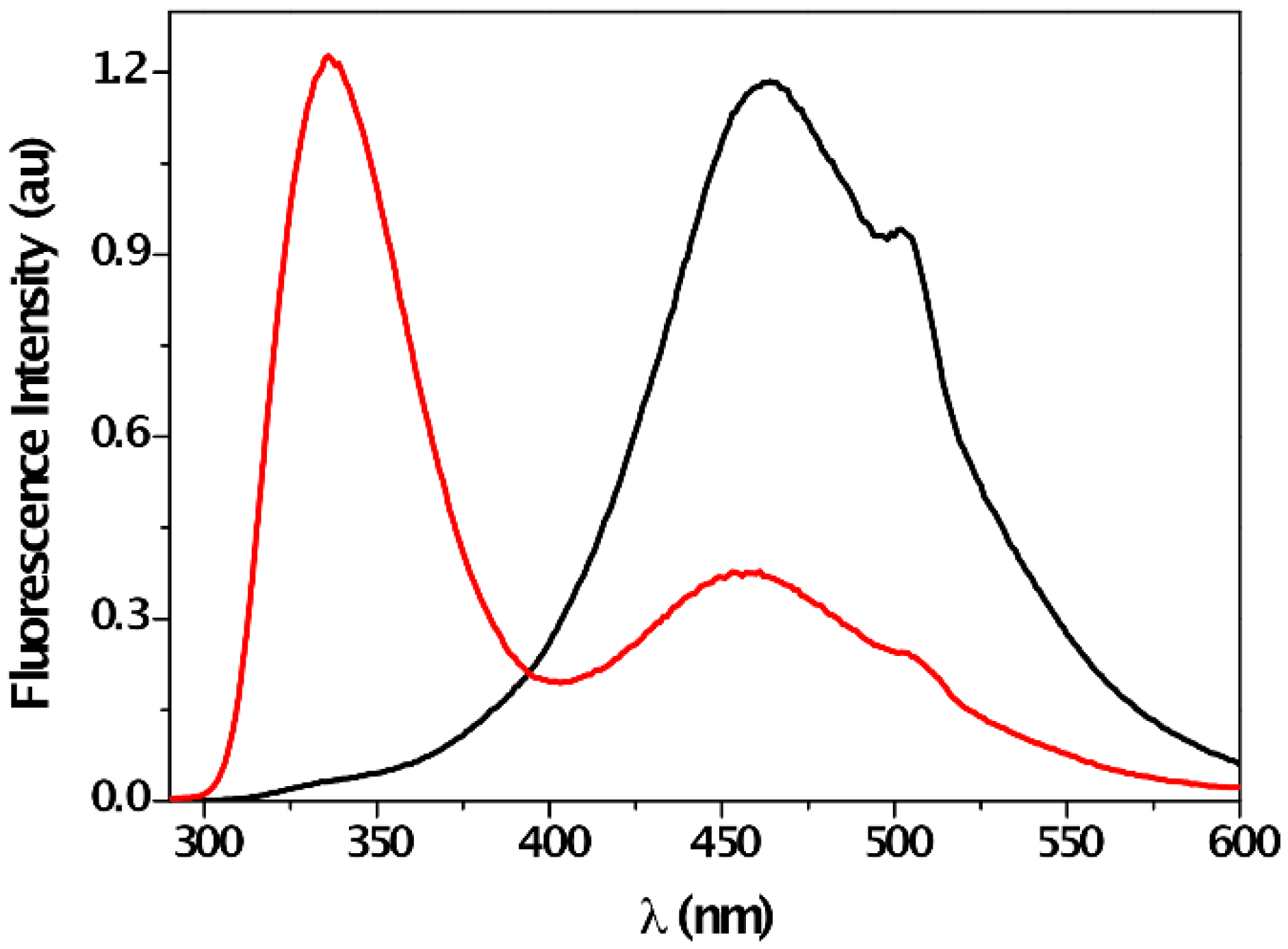
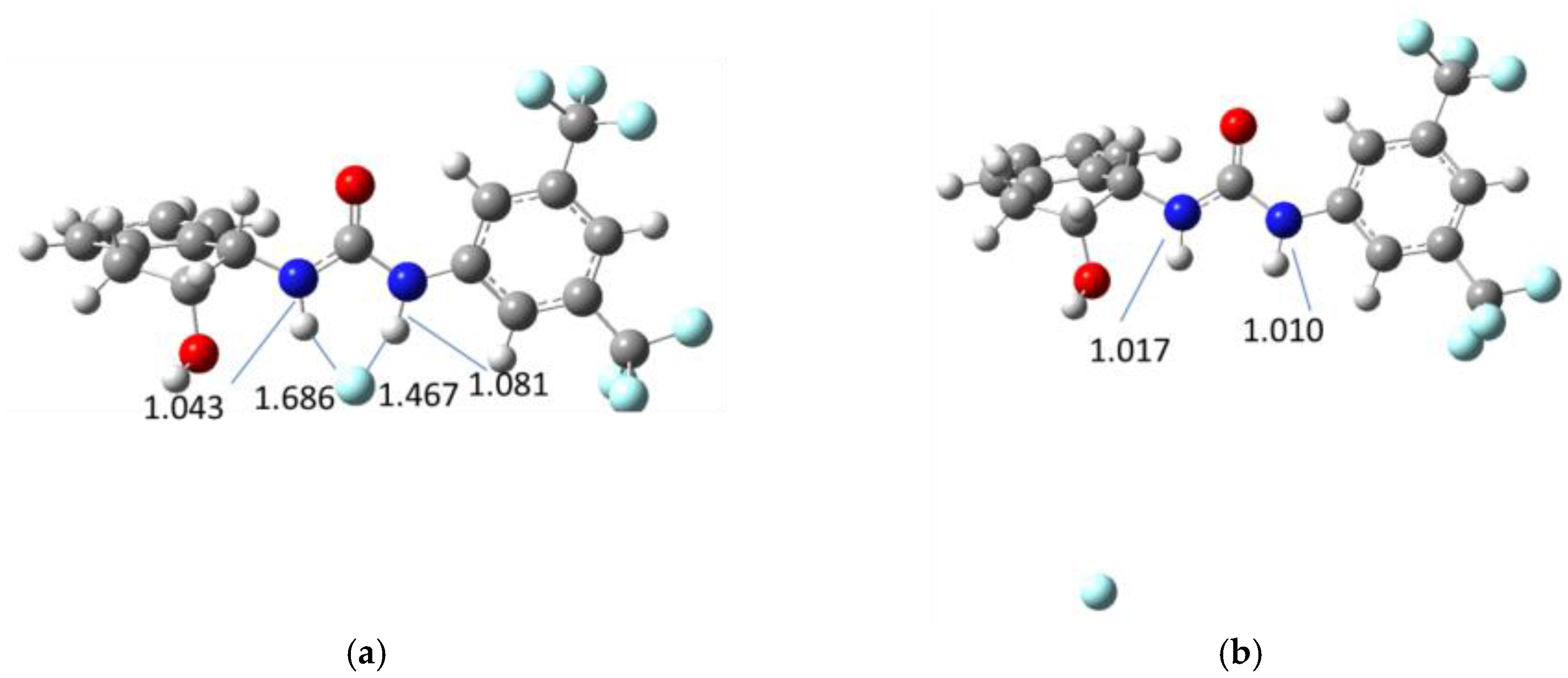
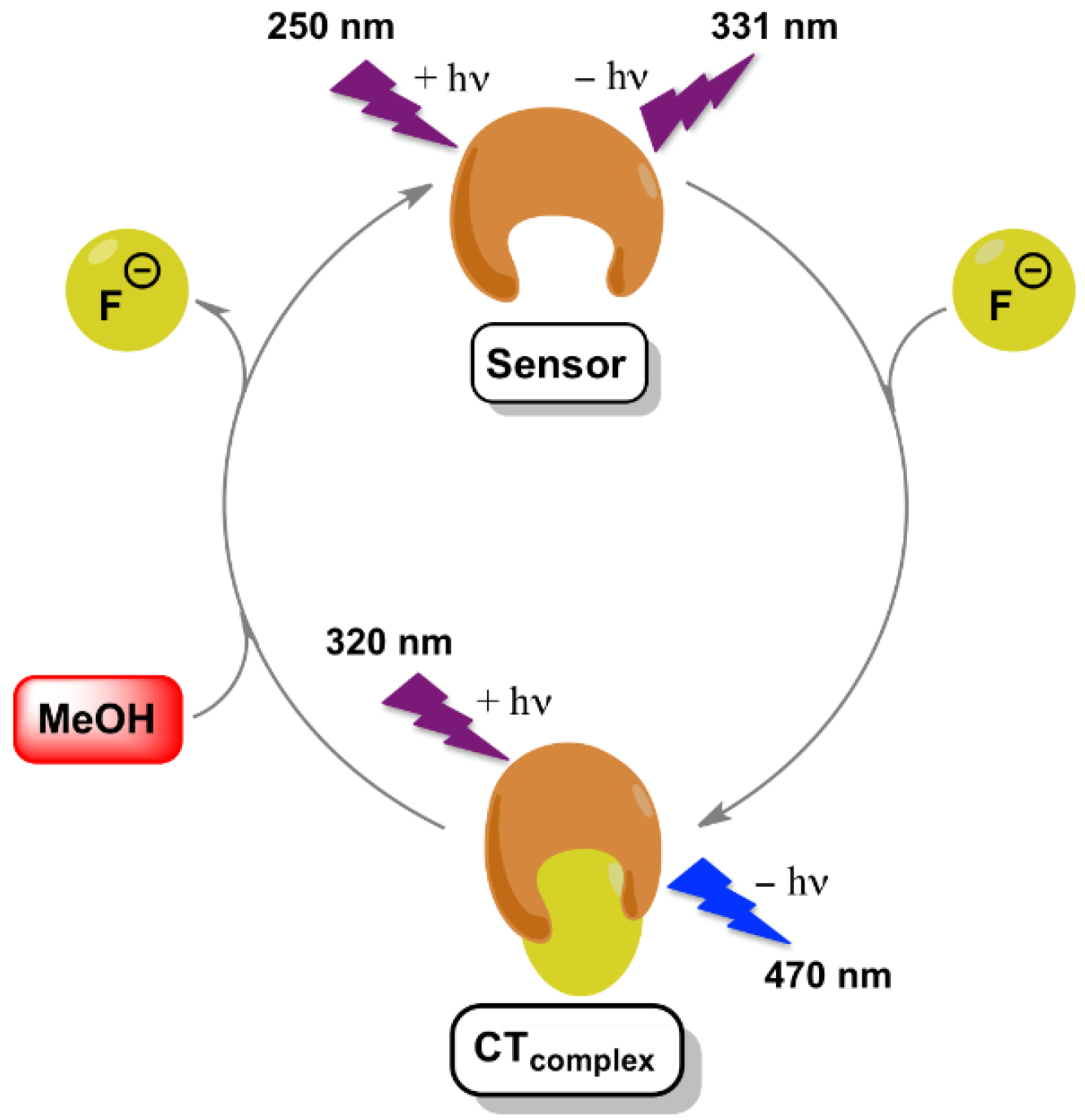
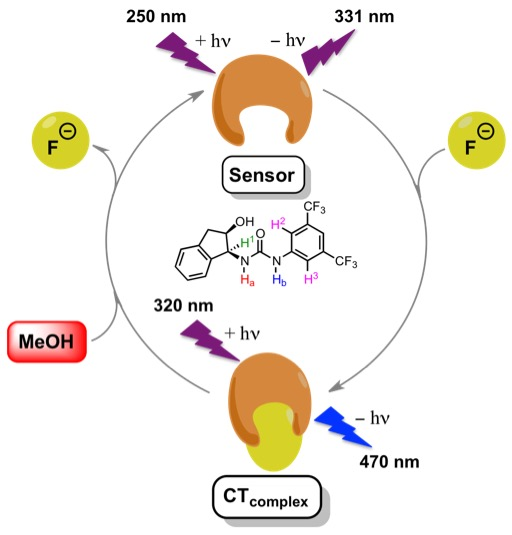
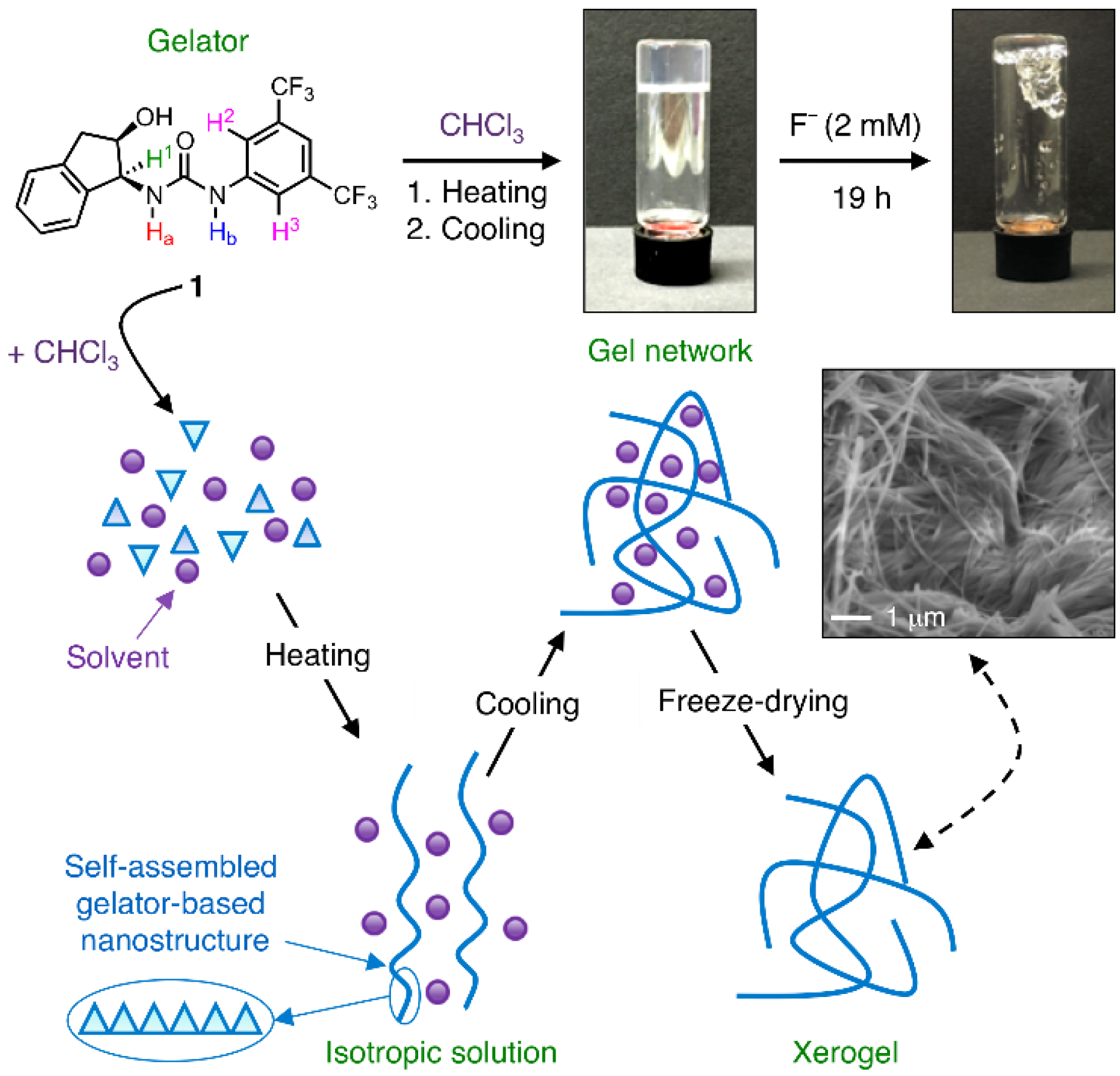
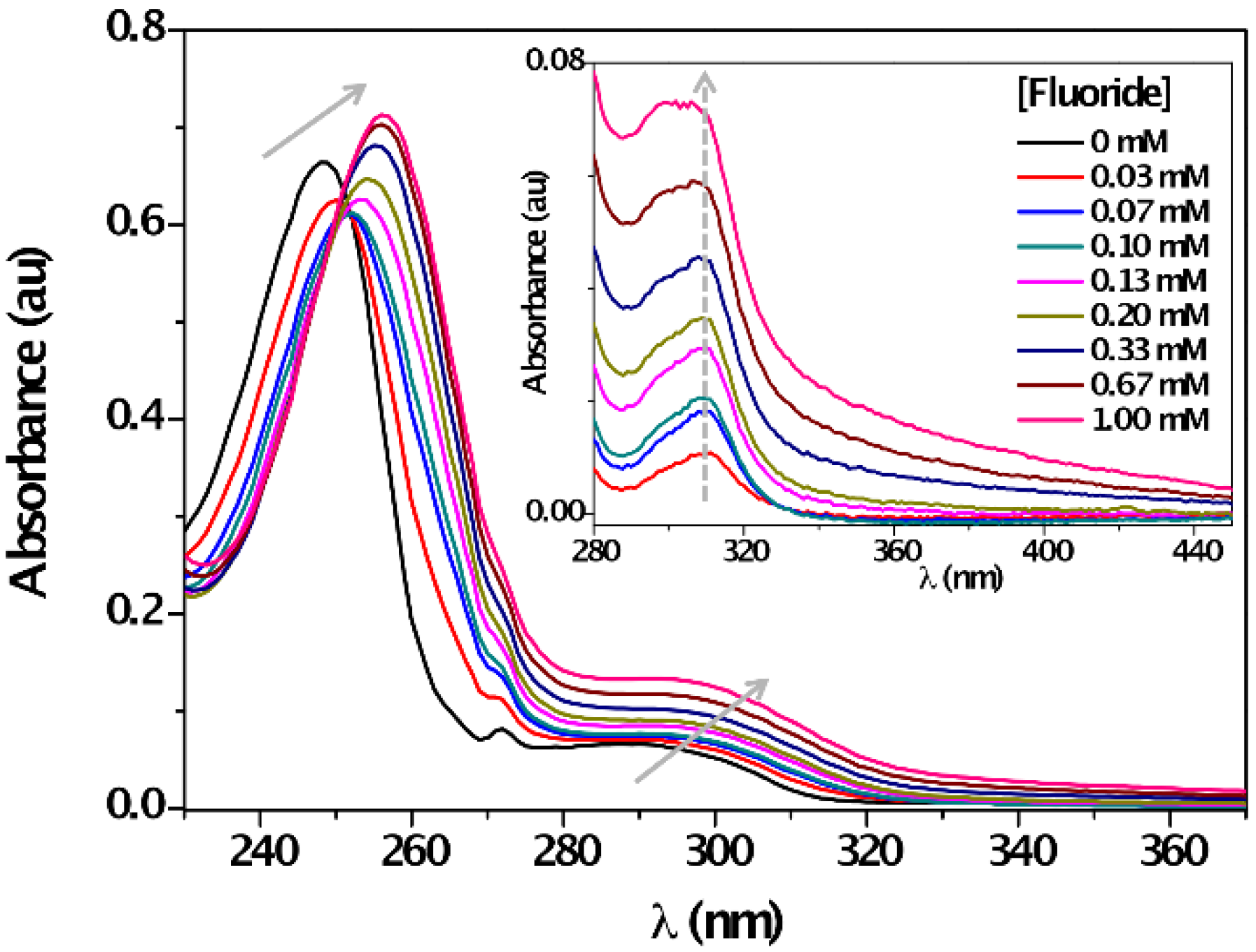
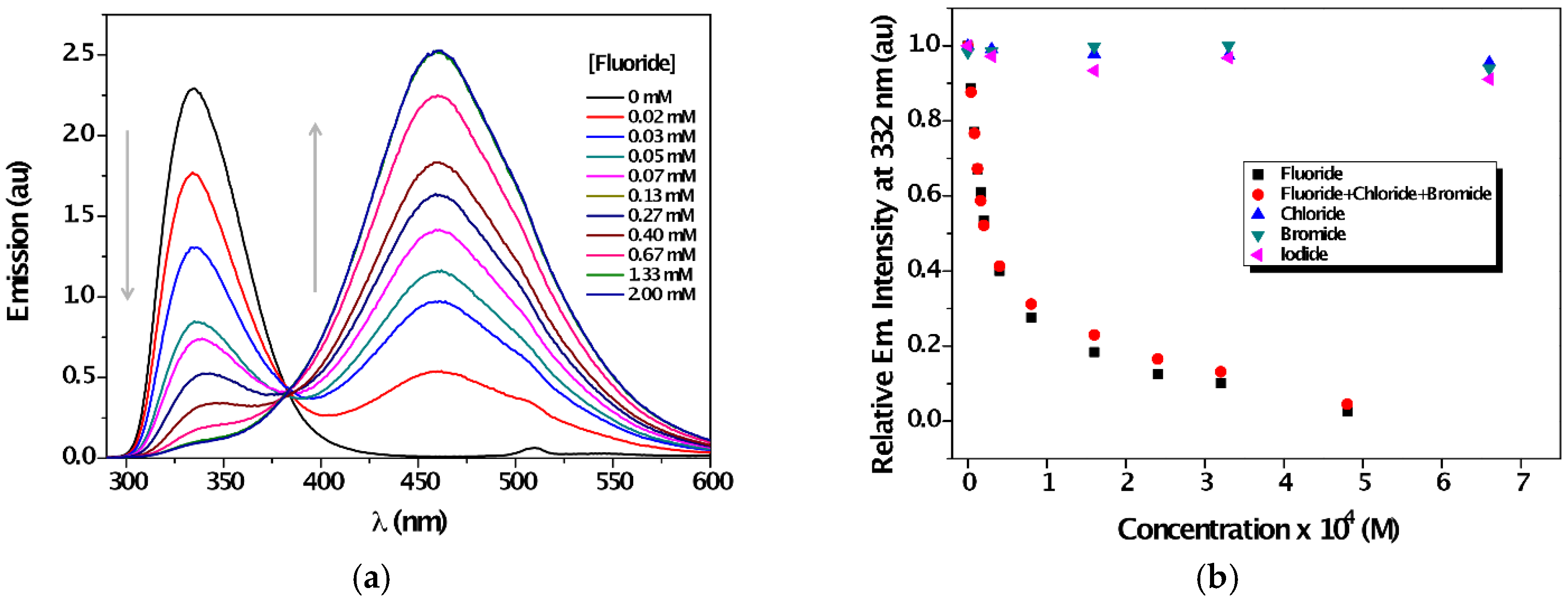
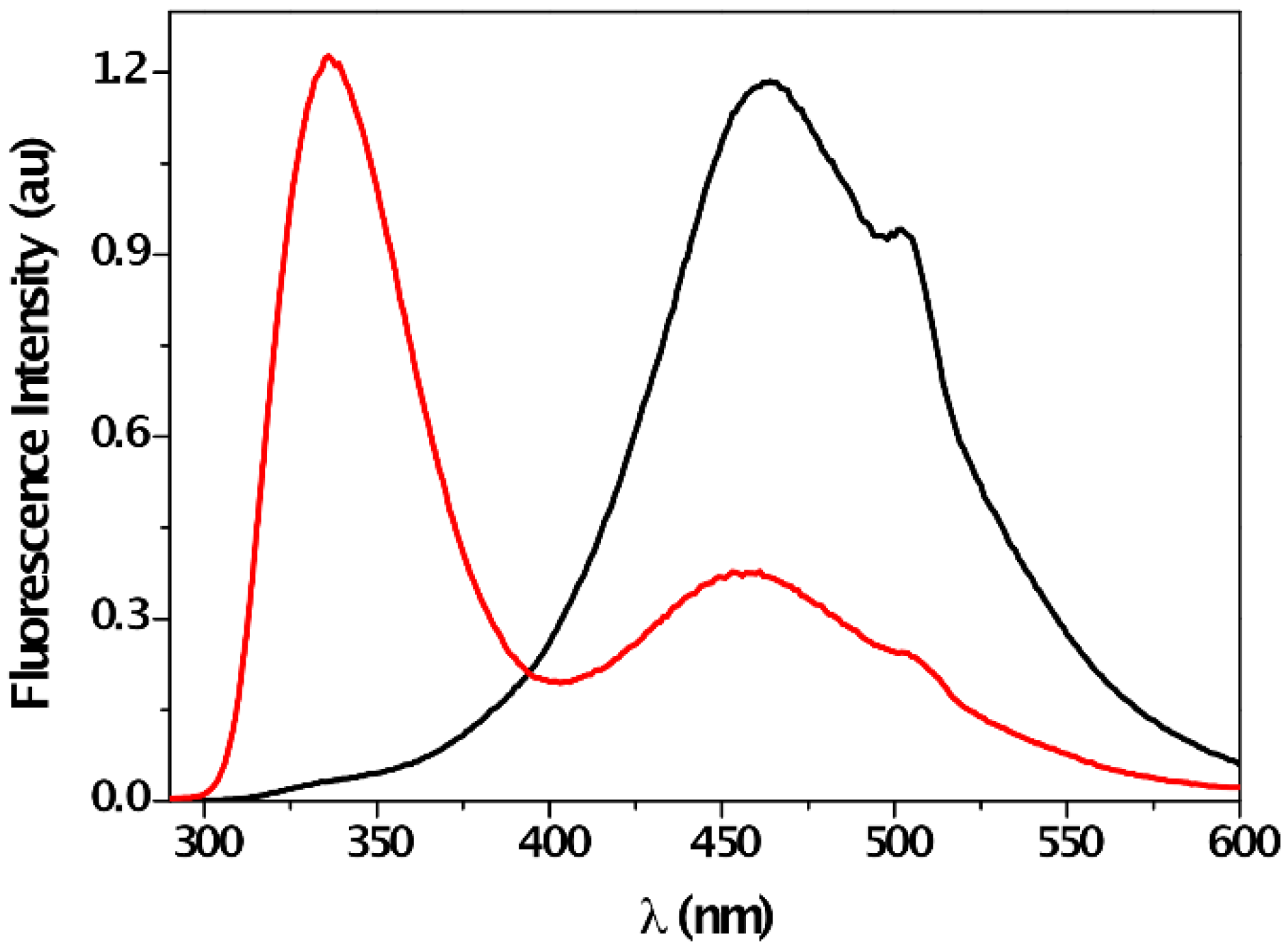
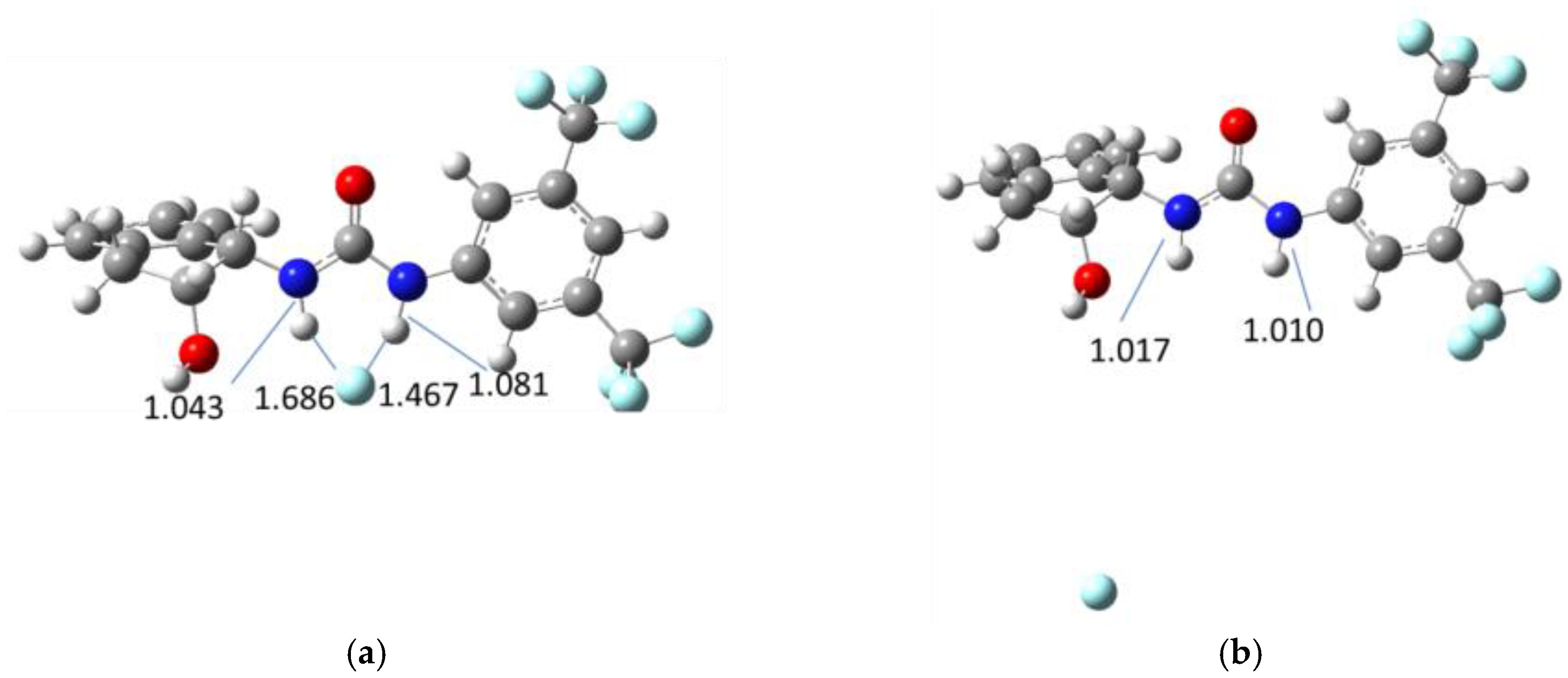
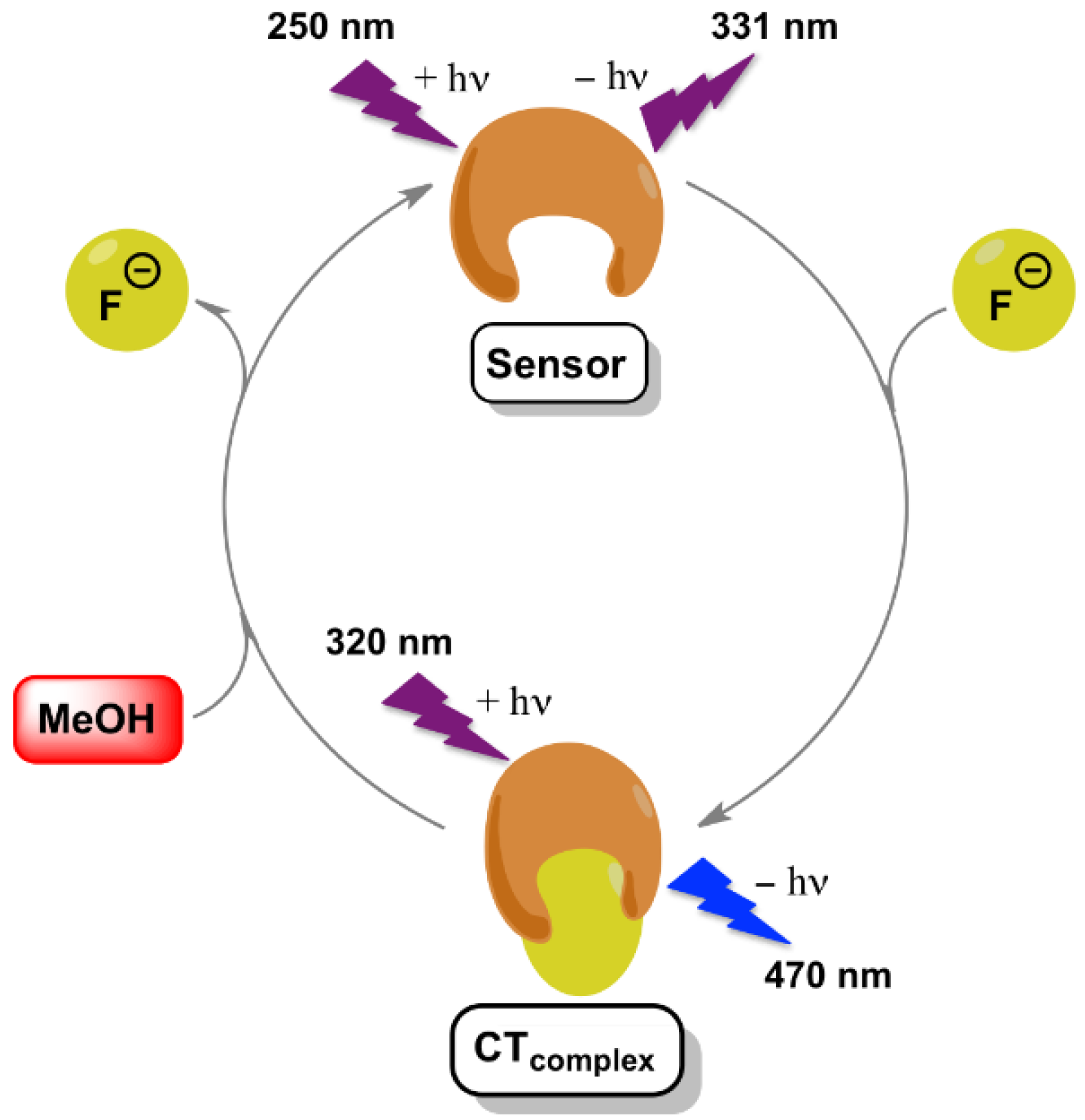
3. Results and Discussion
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CPCM | Conductor-like polarizable continuum model |
| CT | Charge-transfer |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DMSO | Dimethyl sulfoxide |
| FESEM | Field emission scanning electron microscopy |
| GIAO | Gauge-independent atomic orbital |
| ICT | Intramolecular charge transfer |
| MeCN | Acetonitrile |
| MeOH | Methanol |
| NMR | Nuclear magnetic resonance |
| PTET | Photoinduced electron transfer |
| SCRF | Self-consistent reaction field |
| UV-vis | Ultraviolet-visible |
References
- Busschaert, N.; Caltagirone, C.; Van Rossom, W.; Gale, P.A. Applications of supramolecular anion recognition. Chem. Rev. 2015, 115, 8038–8155. [Google Scholar] [CrossRef] [PubMed]
- Bregovic, V.B.; Basaric, N.; Mlinaric-Majerski, K. Anion binding with urea and thiourea derivatives. Coord. Chem. Rev. 2015, 295, 80–124. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, J.F.; Yoon, J. Fluorescence and colorimetric chemosensors for fluoride-ion detection. Chem. Rev. 2014, 114, 5511–5571. [Google Scholar] [CrossRef] [PubMed]
- Moragues, M.A.; Sancenon, F.; Martinez-Mañez, R. Chromogenic and fluorogenic chemosensors and reagents for anions. A comprehensive review of the year 2009. Chem. Soc. Rev. 2011, 40, 2593–2643. [Google Scholar] [CrossRef] [PubMed]
- Kubik, S. Anion recognition in water. Chem. Soc. Rev. 2010, 39, 3648–3663. [Google Scholar] [CrossRef] [PubMed]
- De Silva, A.P.; Gunaratne, H.Q.N.; Gunnlaugsson, T.; Huxley, A.J.M.; McCoy, C.P.; Rademacher, J.T.; Rice, T.E. Signaling recognition events with fluorescent sensors and switches. Chem. Rev. 1997, 97, 1515–1566. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajan, V.; Ramamurthy, P.; Thirumalai, D.; Ramakrishnan, V.T. A novel colorimetric and fluorescent chemosensor for anions involving PET and ICT pathways. Org. Lett. 2005, 7, 657–660. [Google Scholar] [CrossRef] [PubMed]
- Gunnlaugsson, T.; Davis, A.P.; Hussey, G.M.; Tierney, J.; Glynn, M. Design, synthesis and photophysical studies of simple fluorescent anion PET sensors using charge neutral thiourea receptors. Org. Biomol. Chem. 2004, 2, 1856–1863. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Yoon, J. A new fluorescent PET chemosensor for fluoride ions. Chem. Commun. 2002. [Google Scholar] [CrossRef]
- Gunnlaugsson, T.; Davis, A.P.; O’Brien, J.; Glynn, M. Fluorescent sensing of pyrophosphate and bis-carboxylates with charge neutral PET chemosensors. Org. Lett. 2002, 4, 2449–2452. [Google Scholar] [CrossRef] [PubMed]
- Gunnlaugsson, T.; Davis, A.P.; Glynn, M. Fluorescent photoinduced electron transfer (PET) sensing of anions using charge neutral chemosensors. Chem. Commun. 2001. [Google Scholar] [CrossRef]
- Nishizawa, S.; Kato, Y.; Teramae, N. Fluorescence Sensing of Anions via Intramolecular Excimer Formation in a Pyrophosphate-Induced Self-Assembly of a Pyrene-Functionalized Guanidinium Receptor. J. Am. Chem. Soc. 1999, 121, 9463–9464. [Google Scholar] [CrossRef]
- Nishizawa, S.; Kaneda, H.; Uchida, T.; Teramae, N. Anion sensing by a donor–spacer–acceptor system: An intra-molecular exciplex emission enhanced by hydrogen bond-mediated complexation. J. Chem. Soc. Perkin Trans. 1998, 2, 2325–2328. [Google Scholar] [CrossRef]
- Kovalchuk, A.; Bricks, J.L.; Reck, G.; Rurack, K.; Schulz, B.; Szumna, A.; Weibhoff, H. A charge transfer-type fluorescent molecular sensor that “lights up” in the visible upon hydrogen bond-assisted complexation of anions. Chem. Commun. 2004. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.-Y.; Jiang, Y.-B. p-Dimethylaminobenzamide as an ICT dual fluorescent neutral receptor for anions under proton coupled electron transfer sensing mechanism. Chem. Phys. Lett. 2002, 355, 438–444. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, L.; Wu, F.-Y.; Jiang, Y.-B. Development of fluorescent sensing of anions under excited-state intermolecular proton transfer signaling mechanism. Org. Lett. 2003, 5, 2667–2670. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Hamilton, A.D. A dual channel fluorescence chemosensor for anions involving intermolecular excited state proton transfer. Angew. Chem. Int. Ed. 2001, 40, 3912–3915. [Google Scholar] [CrossRef]
- Smith, P.J.; Reddington, M.V.; Wilcox, C.S. Ion pair binding by a urea in chloroform solution. Tetrahedron Lett. 1992, 33, 6085–6088. [Google Scholar] [CrossRef]
- Fan, E.; Van Arman, S.A.; Kincaid, S.; Hamilton, A.D. Molecular recognition: Hydrogen-bonding receptors that function in highly competitive solvents. J. Am. Chem. Soc. 1993, 115, 369–370. [Google Scholar] [CrossRef]
- Kirk, K.L. Biochemistry of the Elemental Halogens and Inorganic Halides, 1st ed.; Plenum Press: New York, NY, USA, 1991. [Google Scholar]
- Kleerekoper, M. The role of fluoride in the prevention of osteoporosis. Endocrinol. Metab. Clin. N. Am. 1998, 27, 441–452. [Google Scholar] [CrossRef]
- Zhang, S.-W.; Swager, T.M. Fluorescent detection of chemical warfare agents: Functional group specific ratiometric chemosensors. J. Am. Chem. Soc. 2003, 125, 3420–3421. [Google Scholar] [CrossRef] [PubMed]
- Sohn, H.; Létant, S.; Sailor, M.J.; Trogler, W.C. Detection of fluorophosphonate chemical warfare agents by catalytic hydrolysis with a porous silicon interferometer. J. Am. Chem. Soc. 2000, 122, 5399–5400. [Google Scholar] [CrossRef]
- Biswas, S.; Gangopadhyay, M.; Barman, S.; Sarkar, J. Simple and efficient coumarin-based colorimetric and fluorescent chemosensor for F− detection: An ON1–OFF–ON2 fluorescent assay. Sens. Actuators B Chem. 2016, 222, 823–828. [Google Scholar] [CrossRef]
- Kim, W.; Sahoo, S.K.; Kim, G.-D.; Choi, H.-J. Novel C3V-symmetric trindane based tripodal anion receptor with tris(coumarin-urea) extension for optical sensing of bioactive anions. Tetrahedron 2015, 71, 8111–8116. [Google Scholar] [CrossRef]
- Bregovic, V.B.; Halasz, I.; Basaric, N.; Mlinaric-Majerski, K. Anthracene adamantylbisurea receptors: Switching of anion binding by photocyclization. Tetrahedron 2015, 71, 9321–9327. [Google Scholar] [CrossRef]
- Duke, R.M.; Gunnlaugsson, T. 3-Urea-1,8-naphthalimides are good chemosensors: A highly selective dual colorimetric and fluorescent ICT based anion sensor for fluoride. Tetrahedron Lett. 2011, 52, 1503–1505. [Google Scholar] [CrossRef]
- Zou, Q.; Jin, J.; Xu, B.; Ding, L.; Tian, H. New photochromic chemosensors for Hg2+ and F−. Tetrahedron 2011, 67, 915–921. [Google Scholar] [CrossRef]
- Elmes, R.B.P.; Gunnlaugsson, T. Luminescence anion sensing via modulation of MLCT emission from a naphthalimide–Ru(II)–polypyridyl complex. Tetrahedron Lett. 2010, 51, 4082–4087. [Google Scholar] [CrossRef]
- Jia, C.; Wu, B.; Liang, J.; Huang, X.; Yang, X.-J. A colorimetric and ratiometric fluorescent chemosensor for fluoride based on proton transfer. J. Fluoresc. 2010, 20, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Veale, E.B.; Tocci, M.G.; Pfeffer, F.M.; Kruger, P.E.; Gunnlaugsson, T. Demonstration of bidirectional photoinduced electron transfer (PET) sensing in 4-amino-1,8-naphthalimide based thiourea anion sensors. Org. Biomol. Chem. 2009, 7, 3447–3454. [Google Scholar] [CrossRef] [PubMed]
- Gómez, D.E.; Fabbrizzi, L.; Liechelli, M.J. Why, on interaction of urea-based receptors with fluoride, beautiful colors develop. J. Org. Chem. 2005, 70, 5717–5720. [Google Scholar]
- Gómez, D.E.; Fabbrizzi, L.; Licchelli, M.; Monzani, E. Urea vs. thiourea in anion recognition. Org. Biomol. Chem. 2005, 3, 1495–1500. [Google Scholar]
- Liu, W.; Wang, B.; Zhang, C.; Yin, X.; Zhang, J. Theoretical study on a chemosensor for fluoride anion-based on a urea derivative. Int. J. Quantum Chem. 2014, 114, 138–144. [Google Scholar] [CrossRef]
- Pérez-Ruiz, R.; Griesbeck, A.G.; Sampedro, D. Computational study on fluoride recognition by an urea-activated phthalimide chemosensor. Tetrahedron 2012, 68, 5724–5729. [Google Scholar] [CrossRef]
- Ghosh, A.; Jose, D.A.; Das, A.; Ganguly, B. A density functional study towards substituent effects on anion sensing with urea receptors. J. Mol. Model. 2010, 16, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Zhang, J. Theoretical investigation of chemosensor for fluoride anion based on amidophthalimide derivatives. Theor. Chem. Acc. 2009, 124, 225–234. [Google Scholar] [CrossRef]
- Muhammad, S.; Liu, C.; Zhao, L.; Wu, S.; Su, Z. A theoretical investigation of intermolecular interaction of a phthalimide based “on-off” sensor with different halide ions: Tuning its efficiency and electro-optical properties. Theor. Chem. Acc. 2009, 122, 77–86. [Google Scholar] [CrossRef]
- Jose, D.A.; Singh, A.; Das, A.; Ganguly, B. A density functional study towards the preferential binding of anions to urea and thiourea. Tetrahedron Lett. 2007, 48, 3695–3698. [Google Scholar] [CrossRef]
- Turner, D.R.; Paterson, M.J.; Steed, J.W. A conformationally flexible, urea-based tripodal anion receptor: Solid-state, solution, and theoretical studies. J. Org. Chem. 2006, 71, 1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Cho, E.J.; Mukamel, S.; Nam, K.C. Efficient fluoride-selective fluorescent host: Experiment and theory. J. Org. Chem. 2004, 69, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Herrera, R.P.; Sgarzani, V.; Bernardi, L.; Ricci, A. Catalytic enantioselective friedel–crafts alkylation of indoles with nitroalkenes by using a simple thiourea organocatalyst. Angew. Chem. Int. Ed. 2005, 44, 6576–6579. [Google Scholar] [CrossRef] [PubMed]
- Dessole, G.; Herrera, R.P.; Ricci, A. H-bonding organocatalysed friedel-crafts alkylation of aromatic and heteroaromatic systems with nitroolefins. Synlett 2004. [Google Scholar] [CrossRef]
- Schön, E.-M.; Marqués-López, E.; Herrera, R.P.; Alemán, C.; Díaz, D.D. Exploiting Molecular Self-Assembly: From Urea-Based Organocatalysts to Multifunctional Supramolecular Gels. Chem. Eur. J. 2014, 20, 10720–10731. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.M. Nanoscale Multifunctional Materials: Science and Applications; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. Influence of polarization functions on MO hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 6, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Wolinski, K.; Hilton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Kim, B.; Kim, Y.-H.; Kim, Y.; Kang, J.; Lee, W. An anion sensing photonic gel by hydrogen bonding of anions to the N-allyl-N′-ethyl urea receptor. J. Mater. Chem. A 2014, 2, 5682–5687. [Google Scholar] [CrossRef]
- Lin, Q.; Zhu, X.; Fu, Y.-P.; Zhang, Y.-M.; Fang, R.; Yang, L.-Z.; Wei, T.-B. Rationally designed anion-responsive-organogels: Sensing F− via reversible color changes in gel–gel states with specific selectivity. Soft Matter 2014, 10, 5715–5723. [Google Scholar] [CrossRef] [PubMed]
- Rajamalli, P.; Prasad, E. Non-amphiphilic pyrene cored poly(aryl ether) dendron based gels: Tunable morphology, unusual solvent effects on the emission and fluoride ion detection by the self-assembled superstructures. Soft Matter 2012, 8, 8896–8903. [Google Scholar] [CrossRef]
- Liu, J.-W.; Yang, Y.; Chen, C.-F.; Ma, J.-T. Novel anion-tuning supramolecular gels with dual-channel response: Reversible sol–gel transition and color changes. Langmuir 2010, 26, 9040–9044. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.; Kuang, G.; Jia, X.; Gao, M.; Li, Y.; Wei, Y. Glycine-glutamic-acid-based organogelators and their fluoride anion responsive properties. J. Mater. Chem. 2009, 19, 5648–5654. [Google Scholar] [CrossRef]
- Shen, J.-S.; Li, D.-H.; Cai, Q.-G.; Jiang, Y.-B. Highly selective iodide-responsive gel–sol state transition in supramolecular hydrogels. J. Mater. Chem. 2009, 19, 6219–6224. [Google Scholar] [CrossRef]
- Maeda, H. Anion-responsive supramolecular gels. Chem. Eur. J. 2008, 14, 11274–11282. [Google Scholar] [CrossRef] [PubMed]
- Griesbeck, A.G.; Hanft, S.; Miara, Y.D. Colorimetric detection of achiral anions and chiral carboxylates by a chiral thiourea–phthalimide dyad. Photochem. Photobiol. Sci. 2010, 9, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Ruiz, R.; Díaz, Y.; Goldfuss, B.; Hertel, D.; Meerholz, K.; Griesbeck, A.G. Fluoride recognition by a chiral urea receptor linked to a phthalimide chromophore. Org. Biomol. Chem. 2009, 7, 3499–3504. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Wang, C.; Smith, M.D.; Pellechia, P.J.; Shimizu, L.S. Alkali metal ions as probes of structure and recognition properties of macrocyclic pyridyl urea hosts. J. Org. Chem. 2010, 75, 5453–5460. [Google Scholar] [CrossRef] [PubMed]
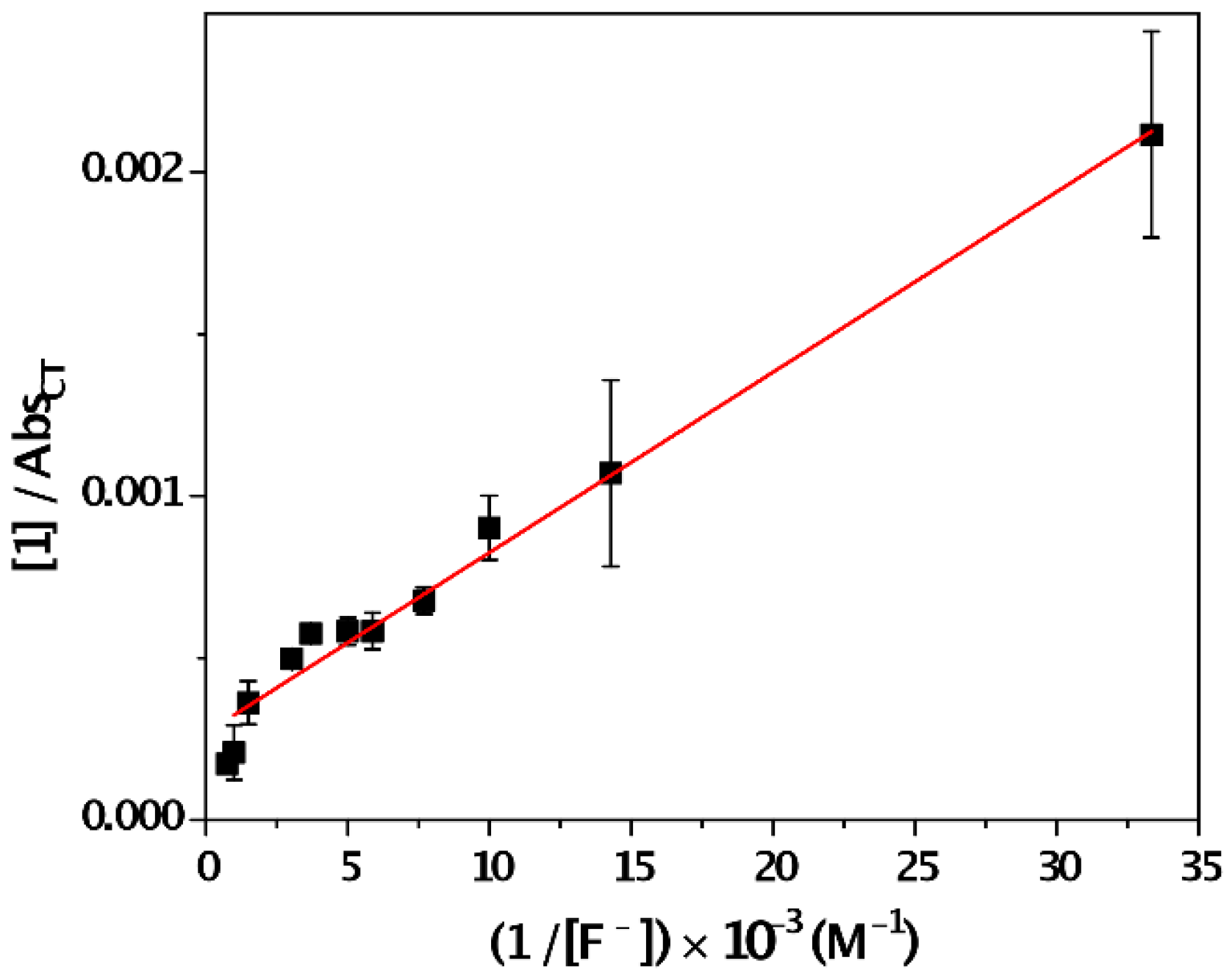
- Benesi, H.G.; Hildebrand, J.H. A spectrophotometric investigation of the interaction of iodine with aromatic hydrocarbons. J. Am. Chem. Soc. 1949, 71, 2703–2707. [Google Scholar] [CrossRef]
- Endermitte, E.; Saava, A.; Karro, E. Exposure to High Fluoride Drinking Water and Risk of Dental Fluorosis in Estonia. Int. J. Environ. Res. Public Health 2009, 6, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.J.; Evans, L.S.; Gale, P.A.; Hursthouse, M.B.; Light, M.E. ‘Twisted’ isophthalamide analogues. Chem. Commun. 2005. [Google Scholar] [CrossRef] [PubMed]
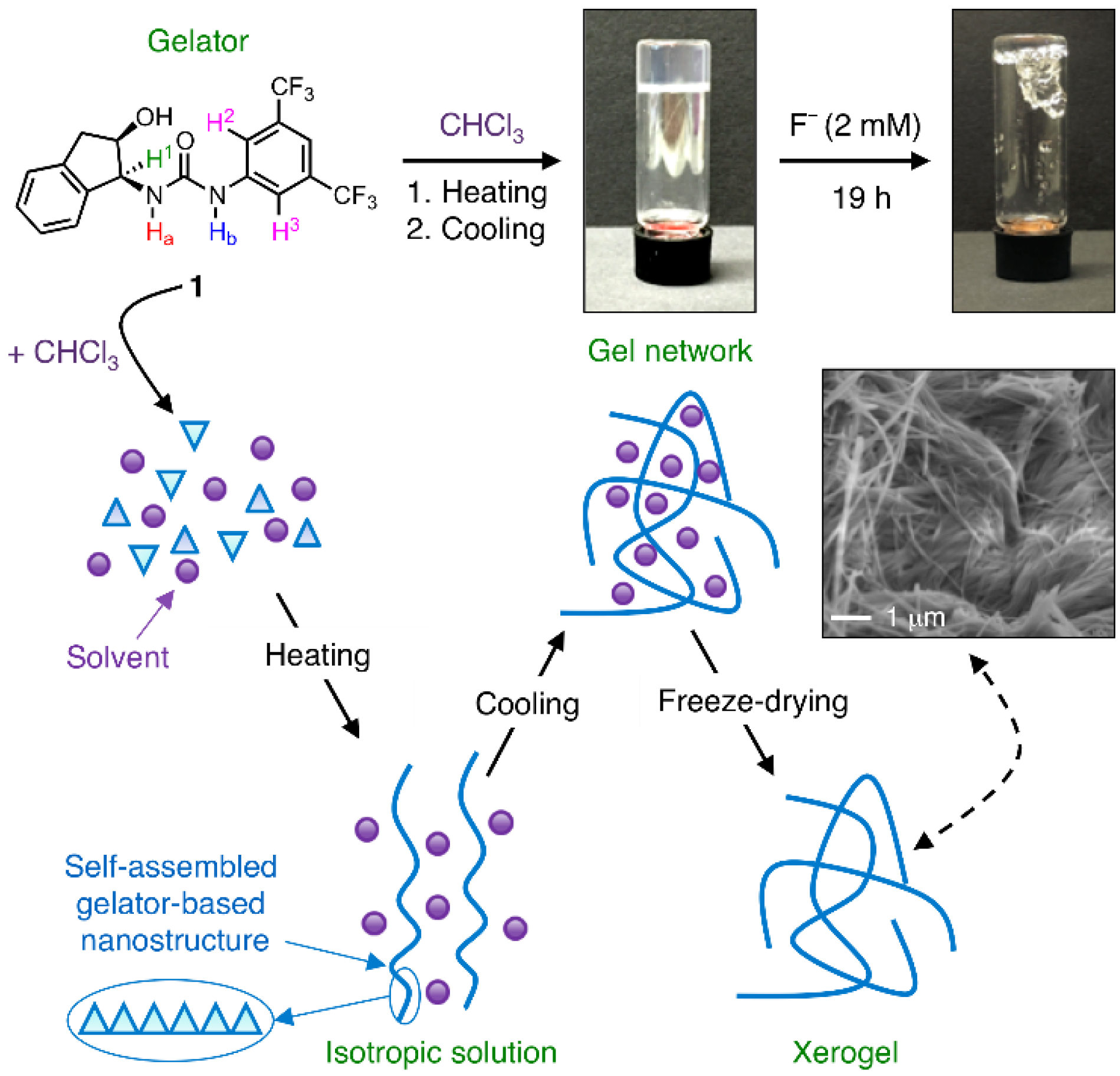

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
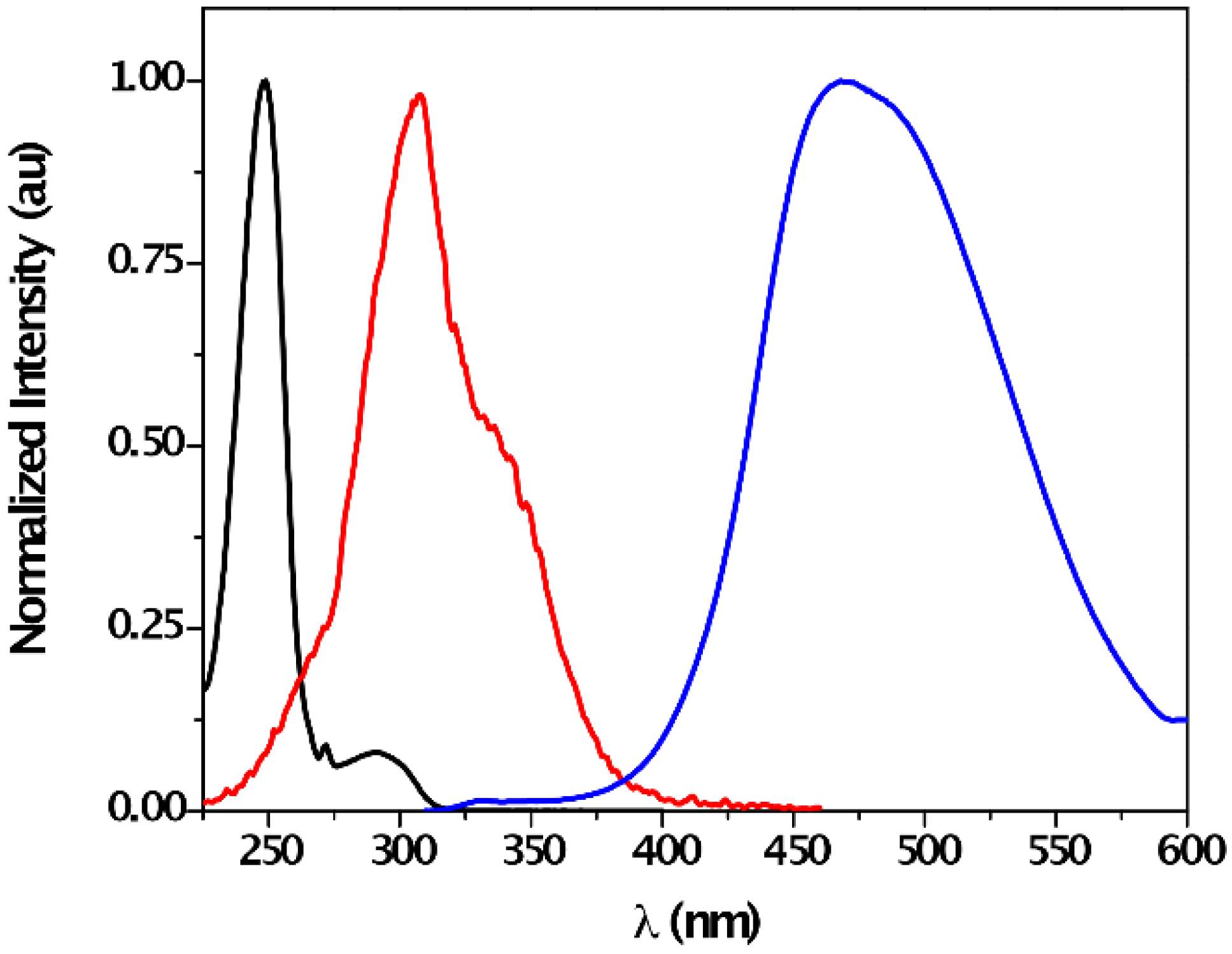
| Solvent | λabs (nm) (ππ*/nπ*) | log ε (M−1·cm−1) (ππ*/nπ*) | λem (nm) | Stokes (cm−1) | Singlet Energy (eV) |
|---|---|---|---|---|---|
| CHCl3 | 243/287 | 4.39/3.34 | 324 | 3149 | 4.07 |
| DMSO | -/297 | -/3.50 | 345 | 4237 | 3.87 |
| MeCN | 249/291 | 4.38/2.80 | 332 | 3324 | 3.98 |
| MeOH | 249/291 | 4.42/3.09 | 335 | 3640 | 3.99 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schiller, J.; Pérez-Ruiz, R.; Sampedro, D.; Marqués-López, E.; Herrera, R.P.; Díaz Díaz, D. Fluoride Anion Recognition by a Multifunctional Urea Derivative: An Experimental and Theoretical Study. Sensors 2016, 16, 658. https://doi.org/10.3390/s16050658
Schiller J, Pérez-Ruiz R, Sampedro D, Marqués-López E, Herrera RP, Díaz Díaz D. Fluoride Anion Recognition by a Multifunctional Urea Derivative: An Experimental and Theoretical Study. Sensors. 2016; 16(5):658. https://doi.org/10.3390/s16050658
Chicago/Turabian StyleSchiller, Jana, Raúl Pérez-Ruiz, Diego Sampedro, Eugenia Marqués-López, Raquel P. Herrera, and David Díaz Díaz. 2016. "Fluoride Anion Recognition by a Multifunctional Urea Derivative: An Experimental and Theoretical Study" Sensors 16, no. 5: 658. https://doi.org/10.3390/s16050658
APA StyleSchiller, J., Pérez-Ruiz, R., Sampedro, D., Marqués-López, E., Herrera, R. P., & Díaz Díaz, D. (2016). Fluoride Anion Recognition by a Multifunctional Urea Derivative: An Experimental and Theoretical Study. Sensors, 16(5), 658. https://doi.org/10.3390/s16050658