
A Toolbox of Genetically Encoded FRET-Based Biosensors for Rapid l-Lysine Analysis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction of l-Lysine Sensor Plasmids
2.2. Protein Preparation
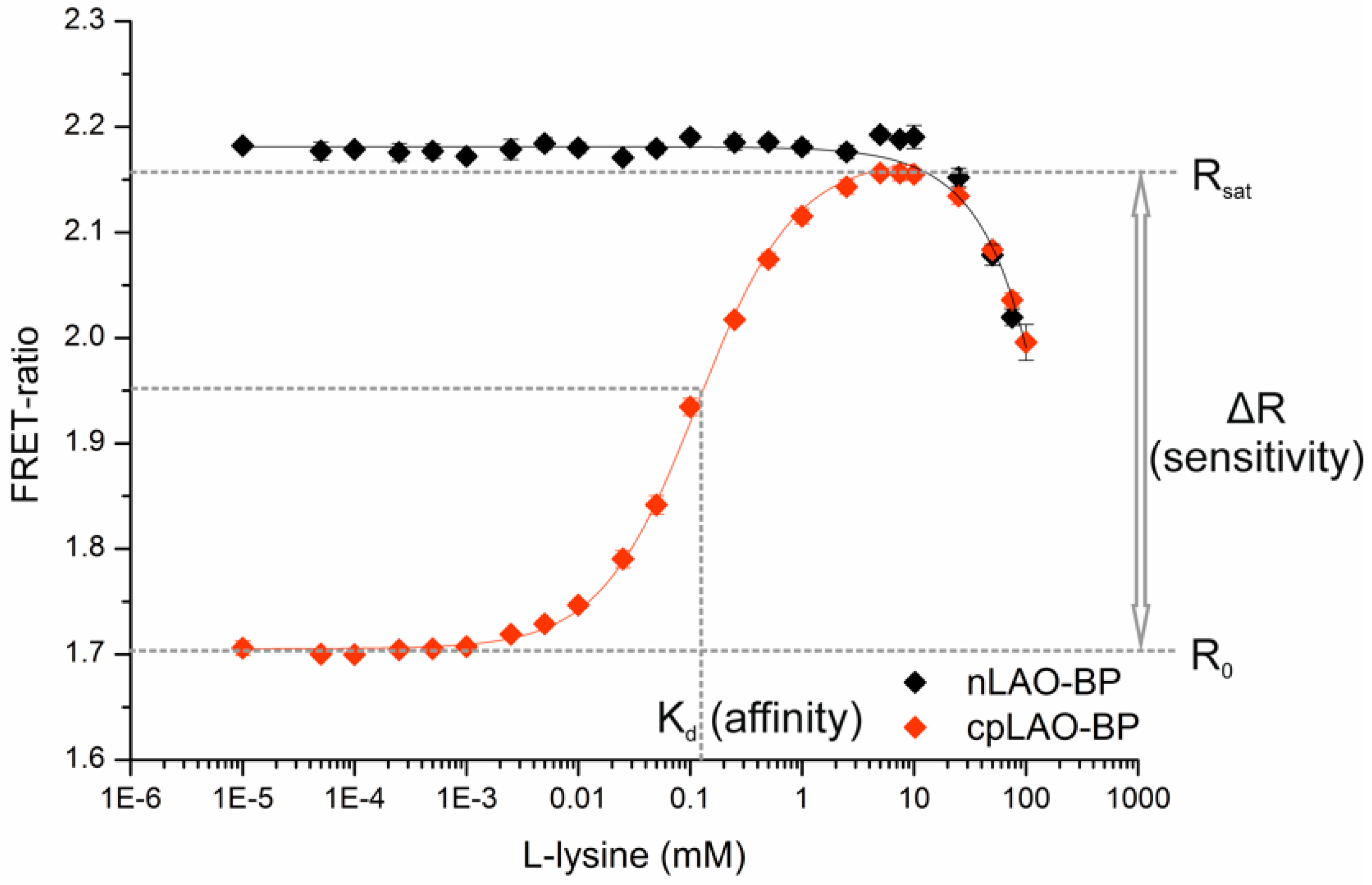
2.3. Determination of Binding Isotherms and Dissociation Constants of FRET-Based l-Lysine Sensors in Vitro
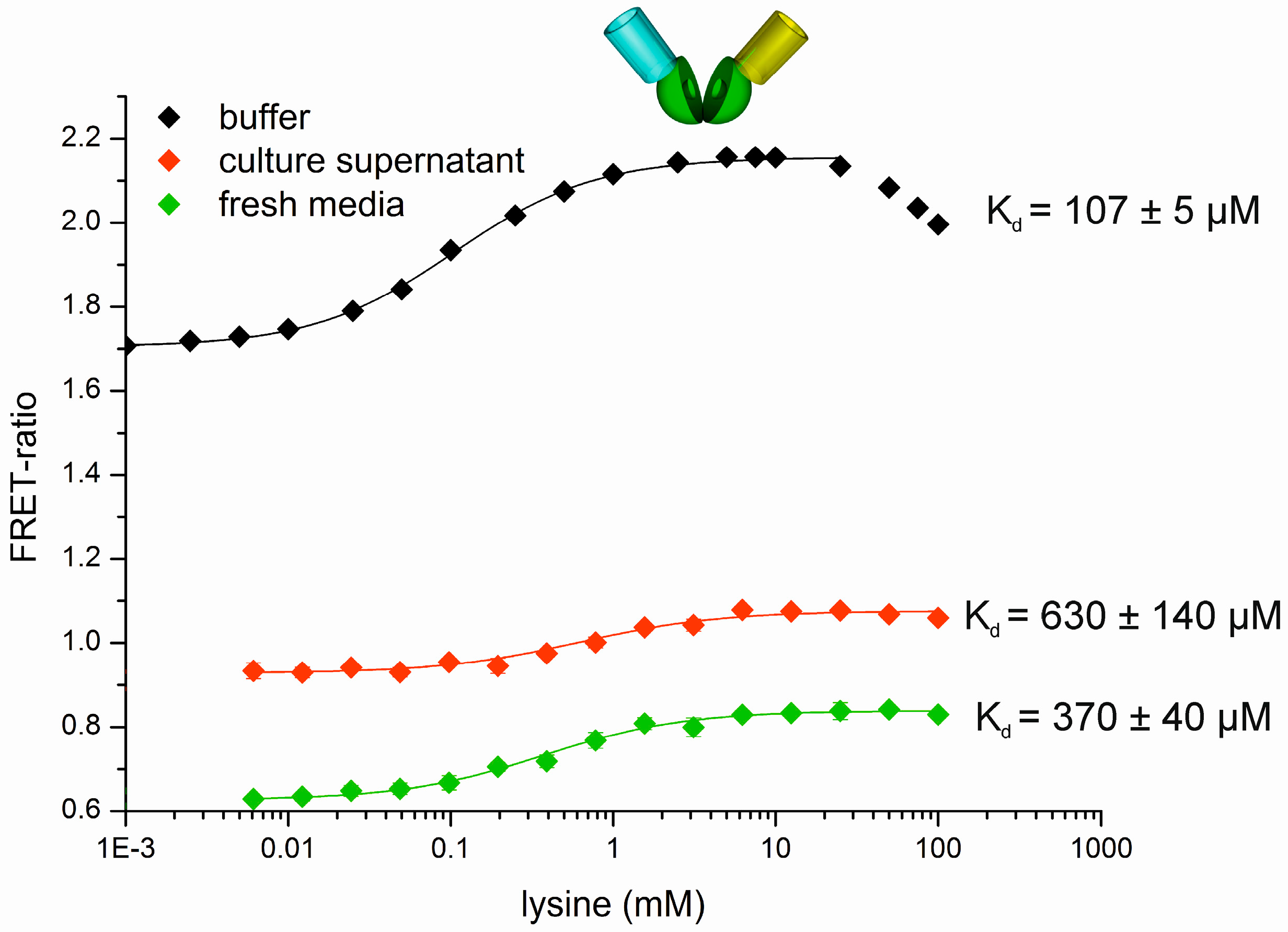
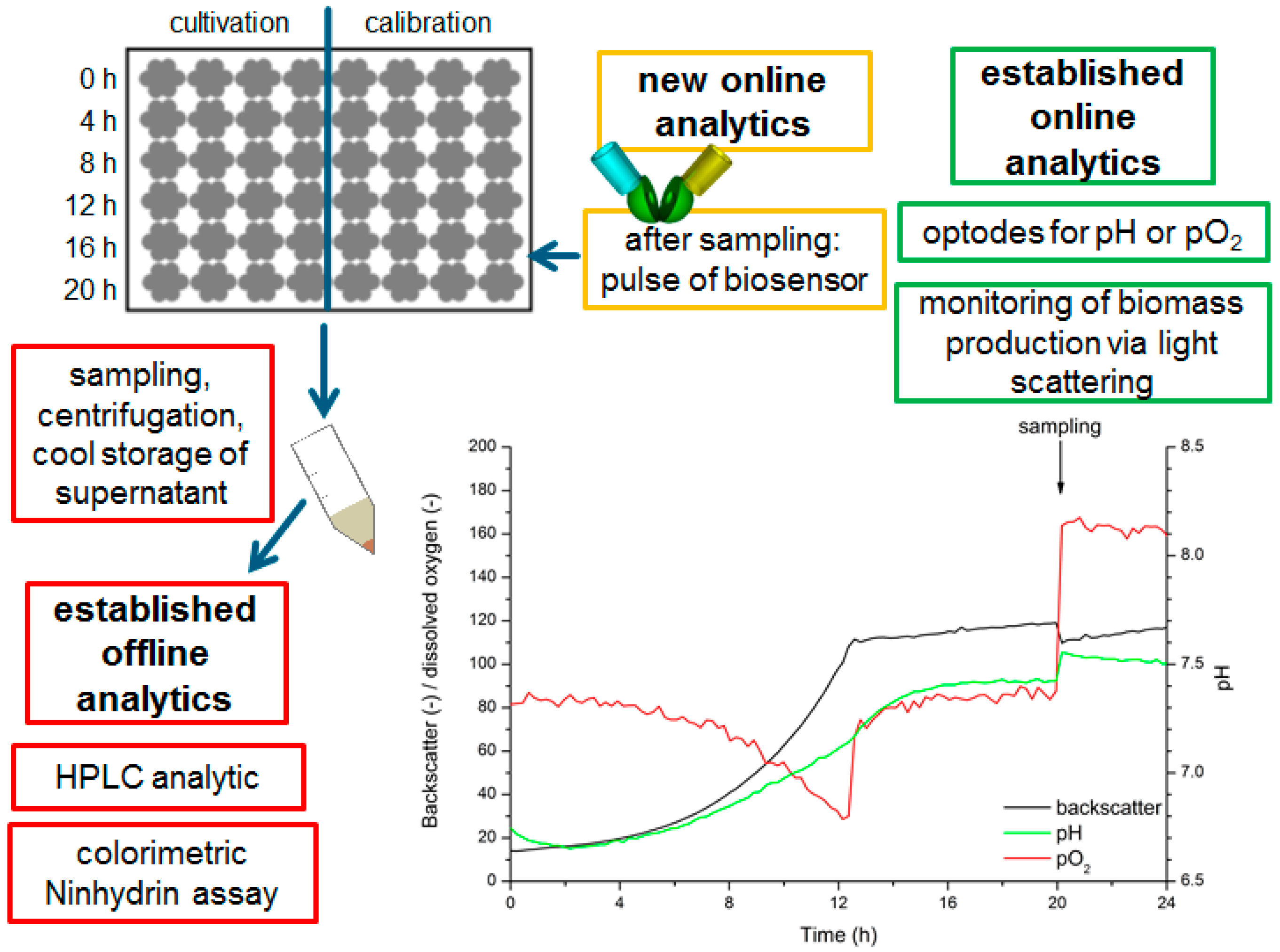
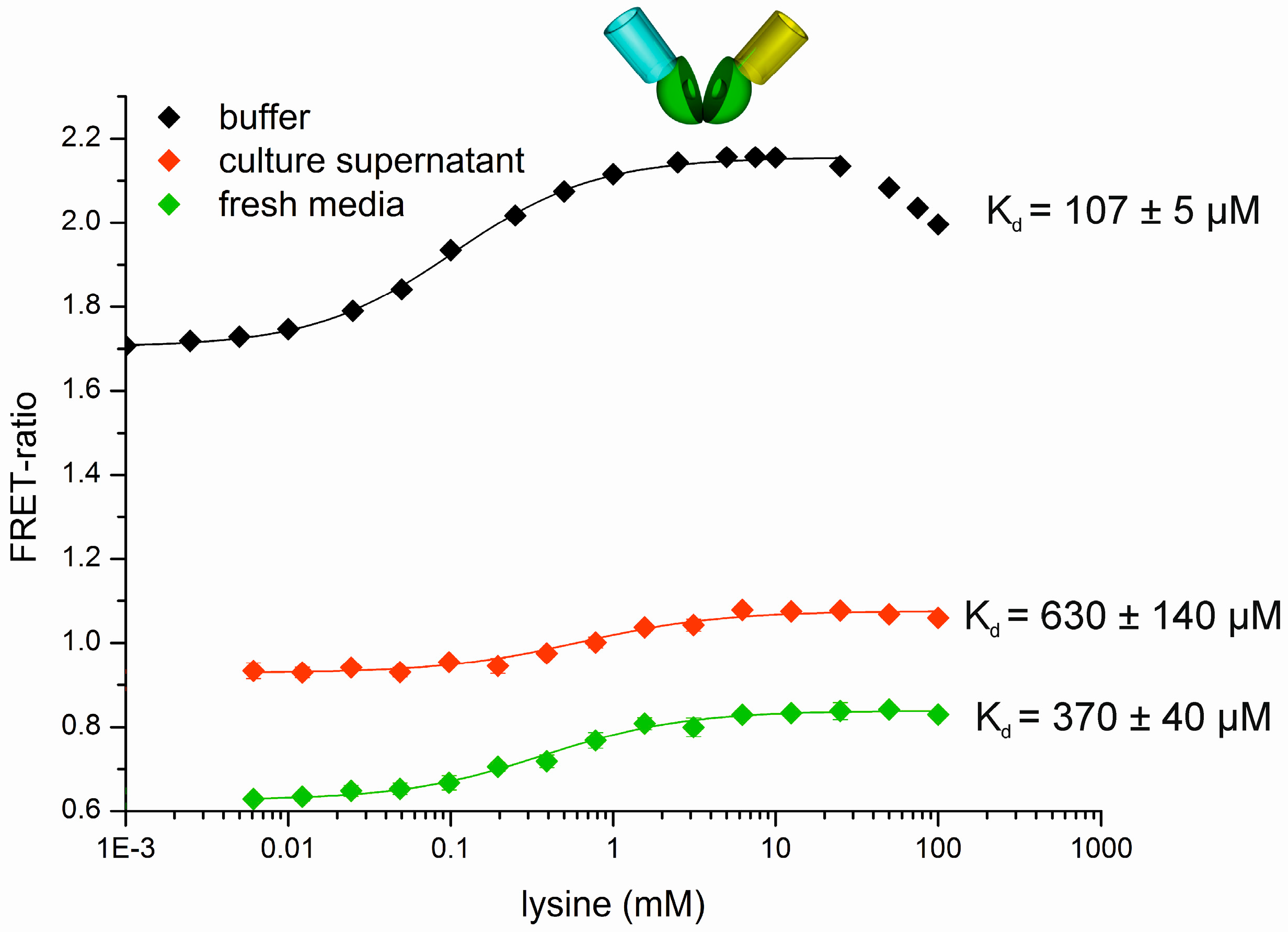
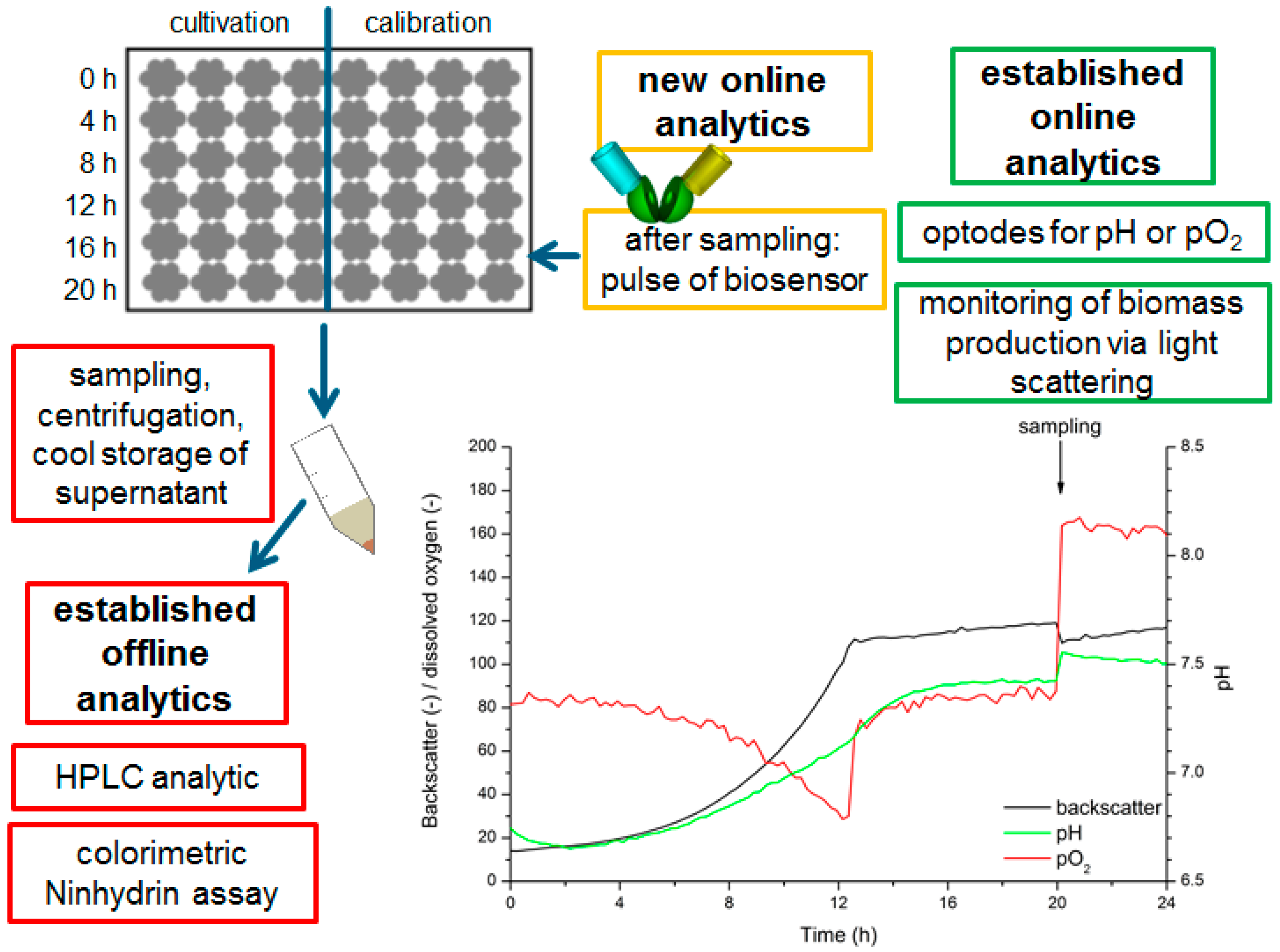
2.4. Production and Determination of l-Lysine in C. Glutamicum Culture Supernatants
3. Results and Discussion
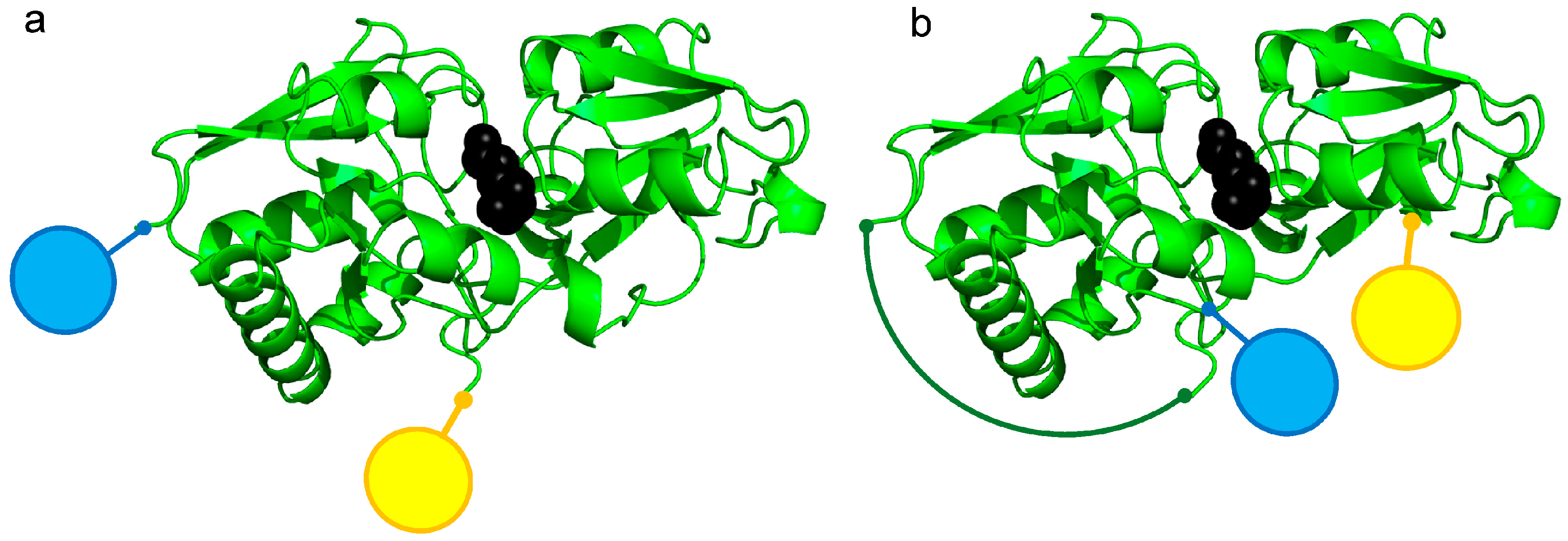
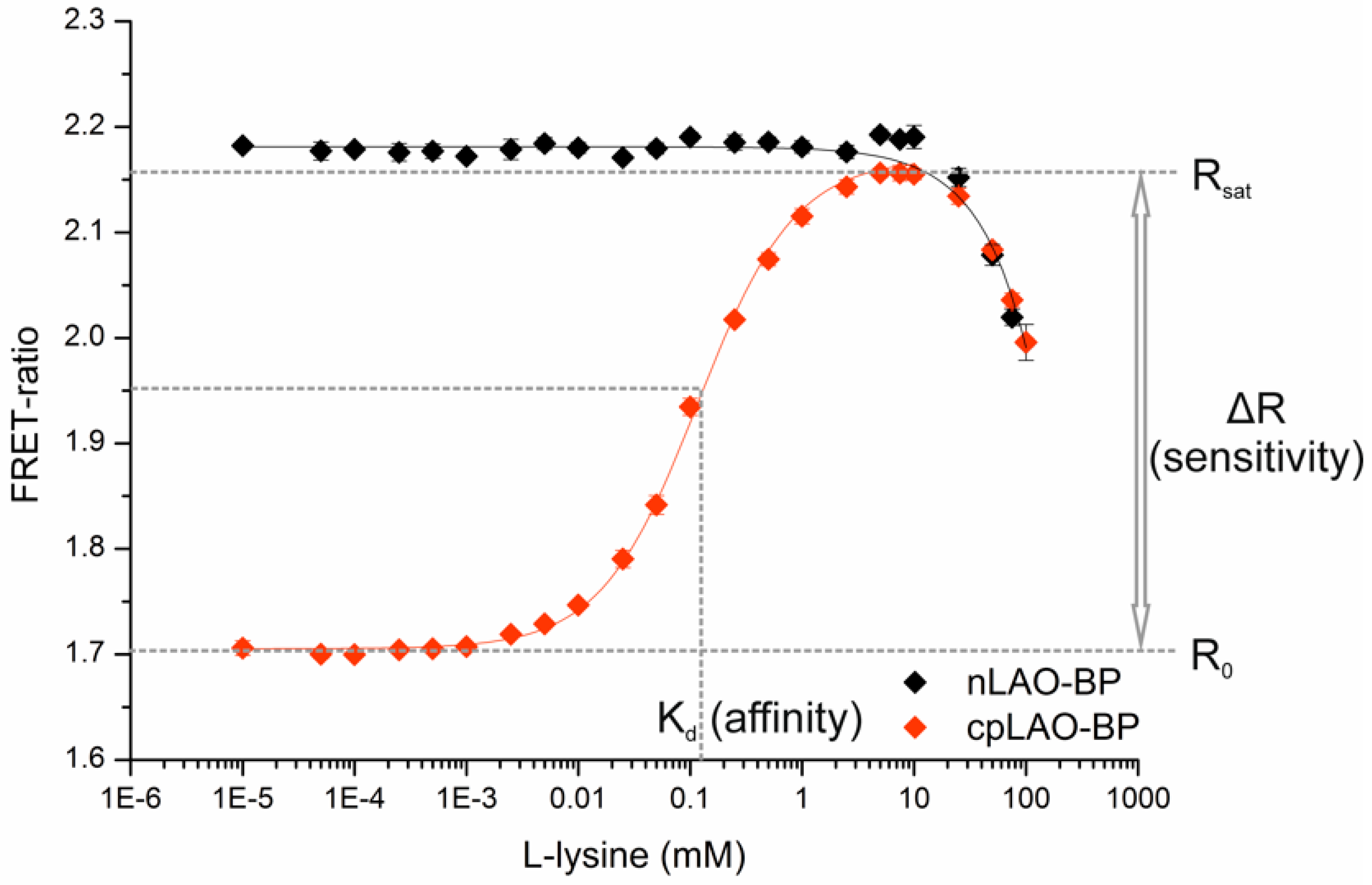
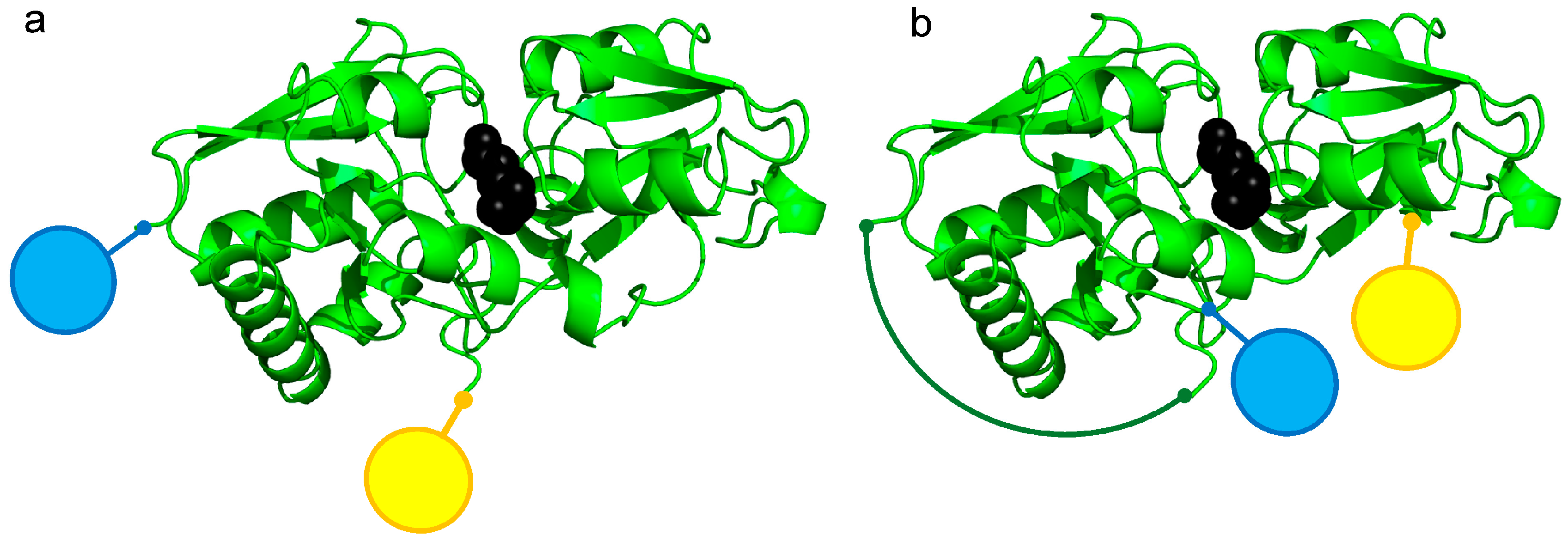
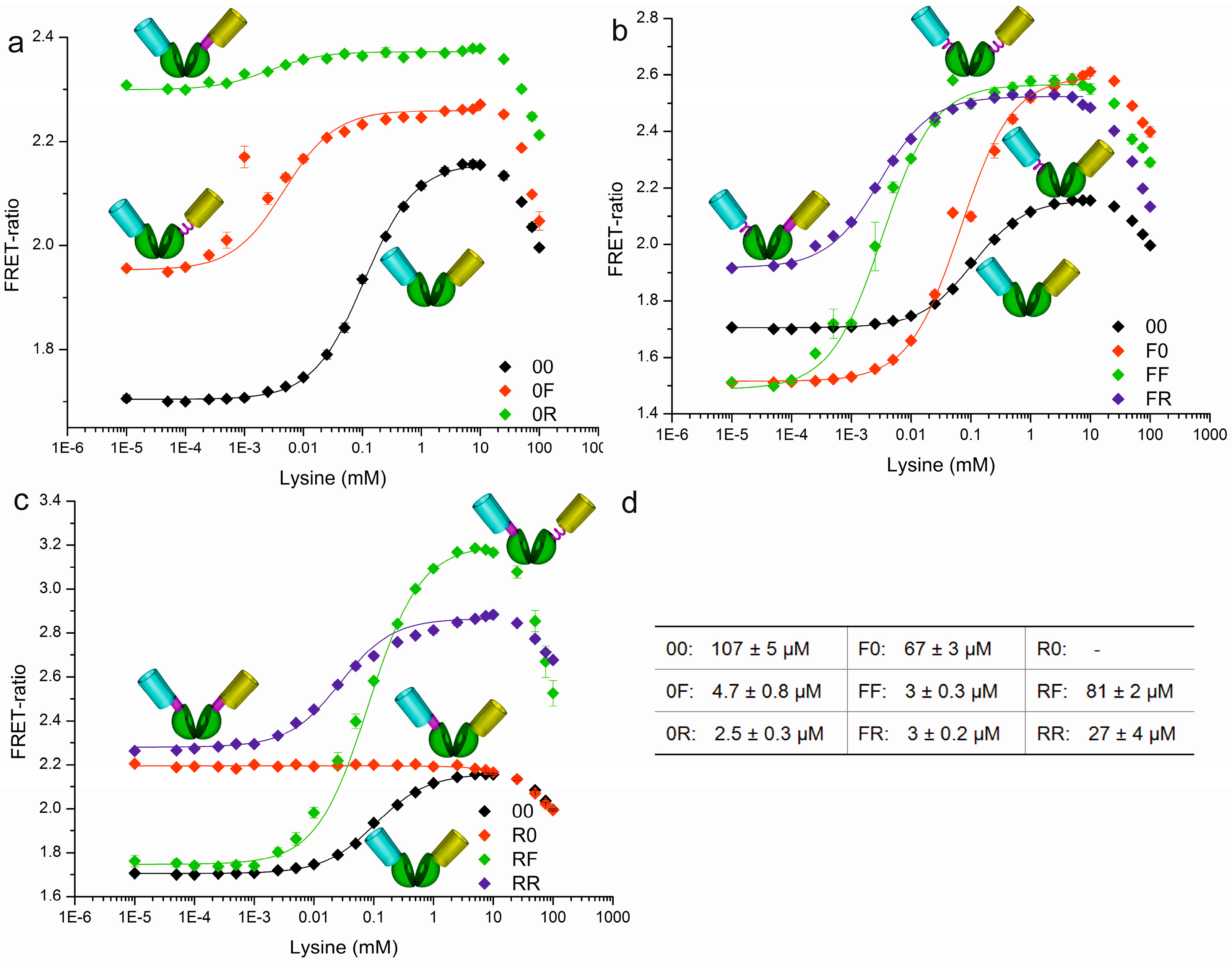
3.1. Construction of FRET-Based Biosensors without and with Circular Permutation of the LAO-Binding Protein
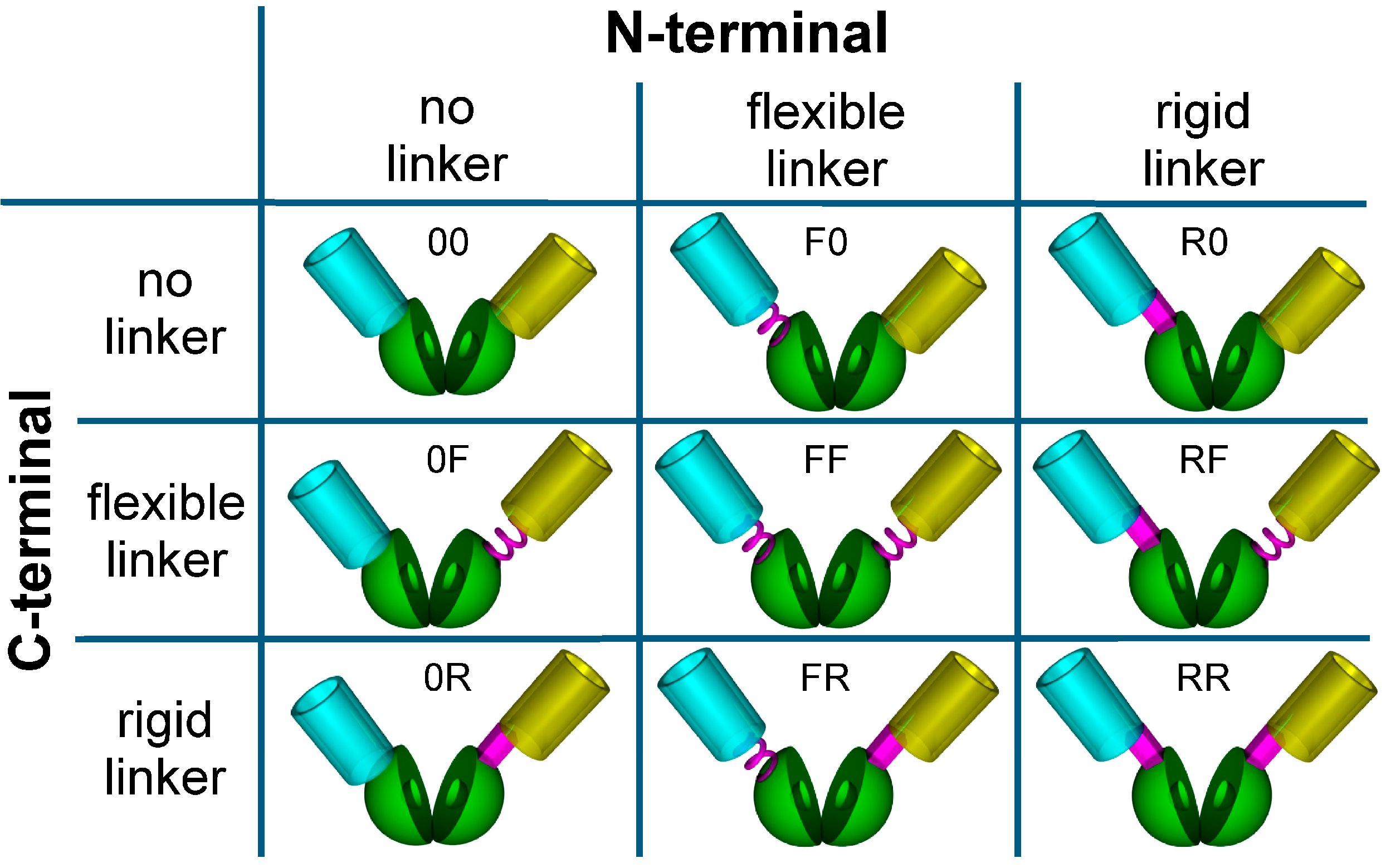
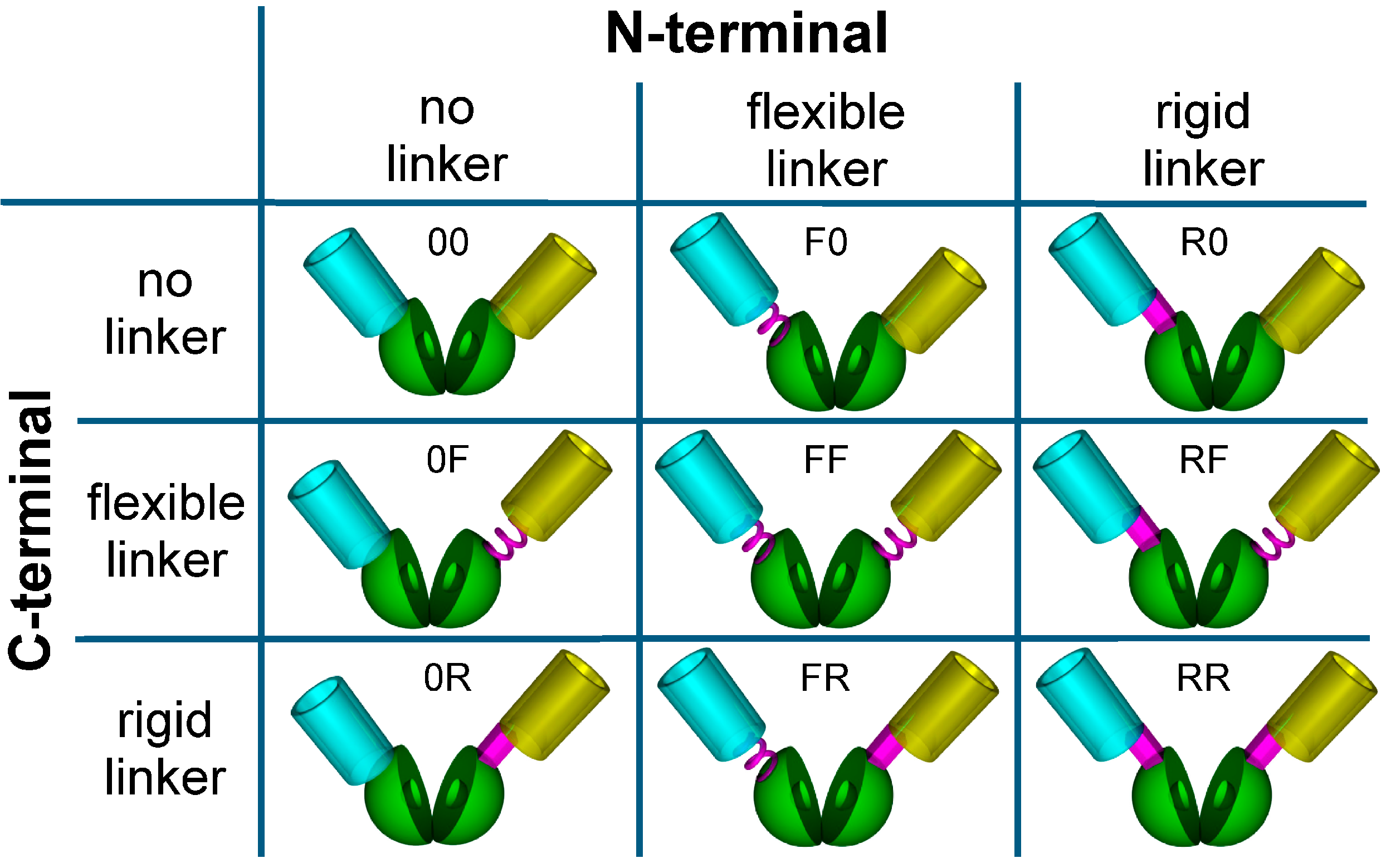
3.2. Construction of a Sensor Toolbox Using Flexible and Rigid Linkers
3.3. Development of a Measurement Protocol Applying the FRET-Based Biosensors
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| cpLAO-BP | circular permutated lysine-/arginine-/ornithine-binding protein |
| ECFP | enhanced cyan fluorescent protein |
| HPLC | high-performance liquid chromatography |
| FRET | Förster resonance energy transfer |
| ITC | isothermal titration calorimetry |
| LAO | l-lysine-/l-arginine-/l-ornithine- |
| nLAO-BP | native l-lysine-/l-arginine-/l-ornithine-binding protein |
| R0 | minimal FRET-ratio |
| Rsat | saturated FRET-ratio |
References
- Schallmey, M.; Singh, A.; Ward, O.P. Developments in the use of Bacillus species for industrial production. Can. J. Microbiol. 2004, 50, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, E.J. Production of vitamins, coenzymes and related biochemicals by biotechnological processes. J. Chem. Technol. Biotechnol. 1992, 53, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Hermann, T. Industrial production of amino acids by coryneform bacteria. J. Biotechnol. 2003, 104, 155–172. [Google Scholar] [CrossRef]
- De Graaf, A.A.; Eggeling, L.; Sahm, H. Metabolic engineering for l-lysine production by Corynebacterium glutamicum. Adv. Biochem. Eng. Biotechnol. 2001, 73, 9–29. [Google Scholar] [PubMed]
- Chen, Y.; Nielsen, J. Biobased organic acids production by metabolically engineered microorganisms. Curr. Opin. Biotechnol. 2016, 37, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Mahr, R.; Frunzke, J. Transcription factor-based biosensors in biotechnology: Current state and future prospects. Appl. Microbiol. Biotechnol. 2016, 100, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Rowland, C.E.; Brown, C.W.; Medintz, I.L.; Delehanty, J.B. Intracellular FRET-based probes: A review. Methods Appl. Fluoresc. 2015, 3, 042006. [Google Scholar] [CrossRef]
- Miyawaki, A.; Llopis, J.; Heim, R.; McCaffery, J.M.; Adams, J.A.; Ikura, M.; Tsien, R.Y. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 1997, 388, 882–887. [Google Scholar] [PubMed]
- Romoser, V.A.; Hinkle, P.M.; Persechini, A. Detection in living cells of Ca2+-dependent changes in the fluorescence emission of an indicator composed of two green fluorescent protein variants linked by a calmodulin-binding sequence. A new class of fluorescent indicators. J. Biol. Chem. 1997, 272, 13270–13274. [Google Scholar] [CrossRef] [PubMed]
- Okumoto, S.; Jones, A.; Frommer, W.B. Quantitative imaging with fluorescent biosensors. Annu. Rev. Plant Biol. 2012, 63, 663–706. [Google Scholar] [CrossRef] [PubMed]
- Hamers, D.; van Voorst Vader, L.; Borst, J.W.; Goedhart, J. Development of FRET biosensors for mammalian and plant systems. Protoplasma 2014, 251, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Mohsin, M.; Ahmad, A.; Iqbal, M. FRET-based genetically-encoded sensors for quantitative monitoring of metabolites. Biotechnol. Lett. 2015, 37, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter, B.; Garcia, A.; Schmid, J. Fluorescent proteins as genetically encoded FRET biosensors in life sciences. Sensors 2015, 15, 26281–26314. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Kamioka, Y.; Matsuda, M. Fluorescence resonance energy transfer imaging of cell signaling from in vitro to in vivo: basis of biosensor construction, live imaging, and image processing. Dev. Growth Differ. 2013, 55, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Moussa, R.; Baierl, A.; Steffen, V.; Kubitzki, T.; Wiechert, W.; Pohl, M. An evaluation of genetically encoded FRET-based biosensors for quantitative metabolite analyses in vivo. J. Biotechnol. 2014, 191, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Eggeling, L.; Bott, M. Handbook of Corynebacterium Glutamicum; Taylor & Francis: Boca Raton, FL, USA, 2005; pp. 9–36. [Google Scholar]
- Bott, M.; Brocker, M. Two-component signal transduction in Corynebacterium glutamicum and other corynebacteria: On the way towards stimuli and targets. Appl. Microbiol. Biotechnol. 2012, 94, 1131–1150. [Google Scholar] [CrossRef] [PubMed]
- Unthan, S.; Radek, A.; Wiechert, W.; Oldiges, M.; Noack, S. Bioprocess automation on a mini pilot plant enables fast quantitative microbial phenotyping. Microb. Cell Fact. 2015, 14, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M. Applications of the ninhydrin reaction for analysis of amino acids, peptides, and proteins to agricultural and biomedical sciences. J. Agric. Food Chem. 2004, 52, 385–406. [Google Scholar] [CrossRef] [PubMed]
- Rohe, P.; Venkanna, D.; Kleine, B.; Freudl, R.; Oldiges, M. An automated workflow for enhancing microbial bioprocess optimization on a novel microbioreactor platform. Microb. Cell Fact. 2012, 11, 144. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Ota, K.; Ito, T. Circular permutation of ligand-binding module improves dynamic range of genetically encoded FRET-based nanosensor. Protein Sci. 2009, 18, 2518–2527. [Google Scholar] [CrossRef] [PubMed]
- Rosen, B.P. Basic amino acid transport in Escherichia coli. J. Biol. Chem. 1971, 246, 3653–3662. [Google Scholar] [PubMed]
- Heim, R.; Tsien, R.Y. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr. Biol. 1996, 6, 178–182. [Google Scholar] [CrossRef]
- Kremers, G.J.; Goedhart, J.; van Munster, E.B.; Gadella, T.W., Jr. Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET Forster radius. Biochemistry 2006, 45, 6570–6580. [Google Scholar] [CrossRef] [PubMed]
- Griesbeck, O.; Baird, G.S.; Campbell, R.E.; Zacharias, D.A.; Tsien, R.Y. Reducing the environmental sensitivity of yellow fluorescent protein. J. Biol. Chem. 2001, 276, 29188–29194. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, K.; Ames, G.F.-L. Purification and characterization of the periplasmic lysine-binding, arginine-binding, ornithine-binding protein (LAO) from Salmonella typhimurium. J. Biol. Chem. 1992, 267, 20706–20712. [Google Scholar] [PubMed]
- Oh, B.H.; Pandit, J.; Kang, C.H.; Nikaido, K.; Gokcen, S.; Ames, G.F.-L.; Kim, S.H. Three-dimensional structures of the periplasmic lysine/arginine/ornithine-binding protein with and without a ligand. J. Biol. Chem. 1993, 268, 11348–11355. [Google Scholar] [PubMed]
- Rosen, B. Basic amino acid transport in Escherichia coli. II. Purification and properties of an arginine-specific binding protein. J. Biol. Chem. 1972, 248, 1211–1218. [Google Scholar]
- Deuschle, K.; Okumoto, S.; Fehr, M.; Looger, L.L.; Kozhukh, L.; Frommer, W.B. Construction and optimization of a family of genetically encoded metabolite sensors by semirational protein engineering. Protein Sci. 2005, 14, 2304–2314. [Google Scholar] [CrossRef] [PubMed]
- Tillett, D.; Neilan, B.A. Enzyme-free cloning: a rapid method to clone PCR products independent of vector restriction enzyme sites. Nucleic Acids Res. 1999, 27, e26–e28. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Itoh, T.Q. Self-assembly cloning: A rapid construction method for recombinant molecules from multiple fragments. Biotechniques 2011, 51, 55–56. [Google Scholar] [CrossRef] [PubMed]
- Chester, N.; Marshak, D.R. Dimethyl sulfoxide-mediated primer Tm reduction: A method for analyzing the role of renaturation temperature in the polymerase chain reaction. Anal. Biochem. 1993, 209, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 1983, 166, 557–580. [Google Scholar] [CrossRef]
- Studier, F.W.; Moffatt, B.A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 1986, 189, 113–130. [Google Scholar] [CrossRef]
- Bertani, G. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 1951, 62, 293–300. [Google Scholar] [PubMed]
- Studier, F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Okumoto, S. Imaging approach for monitoring cellular metabolites and ions using genetically encoded biosensors. Curr. Opin. Biotechnol. 2010, 21, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, S.; Udaka, S.; Shimono, M. Studies on the amino acid fermentation. Part 1. Production of l-glutamic acid by various microorganisms. J. Gen. Appl. Microbiol. 1957, 50, 331–343. [Google Scholar]
- Blombach, B.; Hans, S.; Bathe, B.; Eikmanns, B.J. Acetohydroxyacid synthase, a novel target for improvement of l-lysine production by Corynebacterium glutamicum. Appl. Environ. Microbiol. 2009, 75, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Limberg, M.H.; Pooth, V.; Wiechert, W.; Oldiges, M. Plug flow vs. stirred tank reactor flow characteristics in two compartment scale down bioreactor: Setup specific influence on the metabolic phenotype and bioprocess performance of Corynebacterium glutamicum. Eng. Life Sci. 2016, 35, 1–10. [Google Scholar] [CrossRef]
- Fukami-Kobayashi, K.; Tateno, Y.; Nishikawa, K. Domain dislocation: A change of core structure in periplasmic binding proteins in their evolutionary history. J. Mol. Biol. 1999, 286, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Berntsson, R.P.A.; Smits, S.H.J.; Schmitt, L.; Slotboom, D.J.; Poolman, B. A structural classification of substrate-binding proteins. FEBS Lett. 2010, 584, 2606–2617. [Google Scholar] [CrossRef] [PubMed]
- Ameen, S.; Ahmad, M.; Mohsin, M.; Qureshi, M.I.; Ibrahim, M.M.; Abdin, M.Z.; Ahmad, A. Designing, construction and characterization of genetically encoded FRET-based nanosensor for real time monitoring of lysine flux in living cells. J. Nanobiotechnol. 2016, 14, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, E.M.; Evers, T.H.; Dekkers, L.M.; Meijer, E.W.; Klomp, L.W.; Merkx, M. Variation of linker length in ratiometric fluorescent sensor proteins allows rational tuning of Zn(II) affinity in the picomolar to femtomolar range. J. Am. Chem. Soc. 2007, 129, 3494–3495. [Google Scholar] [CrossRef] [PubMed]
- Evers, T.H.; van Dongen, E.M.; Faesen, A.C.; Meijer, E.W.; Merkx, M. Quantitative understanding of the energy transfer between fluorescent proteins connected via flexible peptide linkers. Biochemistry 2006, 45, 13183–13192. [Google Scholar] [CrossRef] [PubMed]
- Lissandron, V.; Terrin, A.; Collini, M.; D’alfonso, L.; Chirico, G.; Pantano, S.; Zaccolo, M. Improvement of a FRET-based indicator for cAMP by linker design and stabilization of donor-acceptor interaction. J. Mol. Biol. 2005, 354, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Unthan, S.; Grünberger, A.; van Ooyen, J.; Gätgens, J.; Heinrich, J.; Paczia, N.; Wiechert, W.; Kohlheyer, D.; Noack, S. Beyond growth rate 0.6: What drives Corynebacterium glutamicum to higher growth rates in defined medium. Biotechnol. Bioeng. 2014, 111, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Paczia, N.; Nilgen, A.; Lehmann, T.; Gätgens, J.; Wiechert, W.; Noack, S. Extensive exometabolome analysis reveals extended overflow metabolism in various microorganisms. Microb. Cell Fact. 2012, 11, 122. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time | FRET-Ratio | l-Lysine FRET | l-Lysine Ninhydrin | l-Lysine HPLC |
|---|---|---|---|---|
| 0 h | 0.69 ± 0.01 | 0-1 ± 0.12 mM | 0 ± 0 mM | 0.04 ± 0.01 mM |
| 4 h | 0.68 ± 0.01 | 0-1 ± 0.01 mM | 1.6 ± 0.1 mM | 0.7 ± 0.01 mM |
| 8 h | 0.73 ± 0.01 | 3 ± 0.01 mM | 2.8 ± 0.2 mM | 3.0 ± 0.1 mM |
| 12 h | 0.78 ± 0.01 | 15 ± 0.02 mM | 8.5 ± 0.3 mM | 9.6 ± 0.5 mM |
| 16 h | 0.78 ± 0.01 | 15 ± 0.3 mM | 12.2 ± 0.8 mM | 13.2 ± 0.9 mM |
| 20 h | 0.76 ± 0.01 | 12 ± 0.04 mM | 14.6 ± 2.6 mM | 12.3 ± 0.4 mM |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steffen, V.; Otten, J.; Engelmann, S.; Radek, A.; Limberg, M.; Koenig, B.W.; Noack, S.; Wiechert, W.; Pohl, M. A Toolbox of Genetically Encoded FRET-Based Biosensors for Rapid l-Lysine Analysis. Sensors 2016, 16, 1604. https://doi.org/10.3390/s16101604
Steffen V, Otten J, Engelmann S, Radek A, Limberg M, Koenig BW, Noack S, Wiechert W, Pohl M. A Toolbox of Genetically Encoded FRET-Based Biosensors for Rapid l-Lysine Analysis. Sensors. 2016; 16(10):1604. https://doi.org/10.3390/s16101604
Chicago/Turabian StyleSteffen, Victoria, Julia Otten, Susann Engelmann, Andreas Radek, Michael Limberg, Bernd W. Koenig, Stephan Noack, Wolfgang Wiechert, and Martina Pohl. 2016. "A Toolbox of Genetically Encoded FRET-Based Biosensors for Rapid l-Lysine Analysis" Sensors 16, no. 10: 1604. https://doi.org/10.3390/s16101604
APA StyleSteffen, V., Otten, J., Engelmann, S., Radek, A., Limberg, M., Koenig, B. W., Noack, S., Wiechert, W., & Pohl, M. (2016). A Toolbox of Genetically Encoded FRET-Based Biosensors for Rapid l-Lysine Analysis. Sensors, 16(10), 1604. https://doi.org/10.3390/s16101604