Sensing Lanthanide Metal Content in Biological Tissues with Magnetic Resonance Spectroscopy

Abstract
: The development and validation of MRI contrast agents consisting of a lanthanide chelate often requires a determination of the concentration of the agent in ex vivo tissue. We have developed a protocol that uses 70% nitric acid to completely digest tissue samples that contain Gd(III), Dy(III), Tm(III), Eu(III), or Yb(III) ions, or the MRI contrast agent gadodiamide. NMR spectroscopy of coaxial tubes containing a digested sample and a separate control solution of nitric acid was used to rapidly and easily measure the bulk magnetic susceptibility (BMS) shift caused by each lanthanide ion and gadodiamide. Each BMS shift was shown to be linearly correlated with the concentration of each lanthanide ion and gadodiamide in the 70% nitric acid solution and in digested rat kidney and liver tissues. These concentration measurements had outstanding precision, and also had good accuracy for concentrations ≥10 mM for Tm(III) Eu(III), and Yb(III), and ≥3 mM for Gd(III), gadodiamide, and Dy(III). Improved sample handling methods are needed to improve measurement accuracy for samples with lower concentrations.1. Introduction
MRI contrast agents that consist of Gd(III) chelates are often used to enhance the image contrast of anatomical features during clinical diagnoses. Examples include the identification cerebral lesions during neuroimaging [1], the localization of ischemia and acutely infarcted myocardium during cardiac imaging [2], and the diagnoses of solid tumor morphology during oncological imaging [3]. These MRI contrast agents are also used to assess tissue function, including the tracking of vascular flow for large arteries and veins using MRI angiography [4], and the evaluation of vascular perfusion and permeability in capillary networks using Dynamic Contrast Enhancement MRI [5]. Gd(III) and Dy(III) chelates have been used for similar diagnoses of animal models [6–8].
Lanthanide chelates have also been used for molecular imaging. Gd(III) chelates have been used to detect enzyme activities, metabolites, ions, pH, and temperature by monitoring changes in T1- or T2-relaxation [9]. Dy(III), Tm(III), Eu(III) and Yb(III) chelates have also been developed for molecular imaging, primarily through the mechanism of Chemical Exchange Saturation Transfer (CEST) [10]. These lanthanide chelates contain a labile proton that can be selectively saturated, which reduces the coherent MR signal of the proton. The labile proton of the agent can exchange with water, which transfers the saturation to water and effectively reduces the MR signal of water. The magnitude of this change in MR water signal is sensitive to the chemical exchange rate of the CEST agent, which can be modulated by enzymatic catalysis of the agent's chemical structure, a change in temperature or pH that directly affect the kinetic rates of chemical exchange processes, or binding interactions with metabolites and ions [9].
Clinical translation of MRI contrast agents that consist of lanthanide chelates requires evaluations of biodistributions to assess potential toxicities. In particular, slow clearance from the body can lead to extended retention and subsequent dissociation of a lanthanide ion from the organic chelator, which can cause Nephrogenic Systemic Fibrosis [11]. Ex vivo measurements of lanthanide chelate concentrations in various tissues of animal models are often one of the first steps of toxicological assessments. In addition, molecular imaging studies of animal models require the accurate measurement of tissue concentrations of MRI contrast agents to validate imaging results. However, T1 relaxation caused by a Gd(III) chelate, and CEST generated from other lanthanide chelates, are dependent on multiple physico-chemical characteristics, so that converting T1-weighted MR images or CEST MRI results to a measurement of contrast agent concentration is not necessarily accurate [12,13]. Therefore, ex vivo validation of contrast agent concentrations can improve in vivo studies with MRI contrast agents.
Two methods are currently used to measure concentration of lanthanide chelates in ex vivo tissues. Inductively coupled plasma mass spectrometry (ICP-MS) provides outstanding measurement accuracy of concentrations of lanthanide ions as low as 1 ng/mL [14]. However, ICP-MS is relatively expensive, has limited availability at many research institutions, requires frequent calibration with known samples, and requires careful sample preparation and handling. A colorimetric test with xylenol orange, methyl thymol blue, or arsenazo dye can also be used to measure concentration of lanthanide ions as low as 8–50 μM depending on the pH and temperature of the sample [15–17]. These spectrophotometric methods are popular because spectrophotometers are relatively inexpensive and available at mot research institutions. Yet colorimetric analyses require samples that are optically transparent except for the dye, which severely limits the ability to assess tissue samples for lanthanide chelate concentration.
The concentrations of lanthanide chelates have also been measured by employing the Evans method with a NMR spectrometer [18]. Paramagnetic lanthanide ions have an effective magnetic moment, μeff, that can cause a bulk magnetic susceptibility (BMS) shift of the solvent [19]. This shift in MR frequency can be compared to the shift of a solvent without the lanthanide ion, which is directly related to the concentration of the ion, [Ln] (Equation (1)) [20]. In this relationship, T represents temperature and s is set to 1/3, −1/6, or 0 for a sample geometry that consists of a cylinder parallel to the main magnetic field, a cylinder perpendicular to the main field, or a sphere, respectively [21,22]. This relationship assumes that the measurement temperature is above the Curie-Weiss temperature and that the diamagnetic and the hyperfine shifts are negligible relative to the BMS shift, which is a good assumption for all paramagnetic lanthanide ions. Equation (1) also assumes that direct interactions between paramagnetic lanthanide chelates have negligible influence on the BMS shift, which has been shown to be a good assumption at practical concentrations [23]:
We investigated whether a protocol could be developed that can measure the BMS shift with NMR spectroscopy to accurately measure lanthanide ion concentrations in ex vivo tissues of animal models. We evaluated the accuracies and precisions of measuring the concentrations of Gd(III), Dy(III), Tm(III), Eu(III), and Yb(III) ions and a Gd(III) chelate in solutions of 70% nitric acid. We tested conditions of the NMR spectrometer and sample that may affect the concentration measurement. After establishing correlations for these ions, we then performed similar evaluations with ex vivo rat liver and kidney tissues that were complete digested in 70% nitric acid. Our results establish a protocol and analytical criteria for measuring ex vivo concentrations of lanthanide-based MRI concentrations using the BMS shift.
2. Experimental Section
TmCl3 hydrate was acquired from Strem Chemicals Inc. (Newburyport, MA, USA). EuCl3, YbCl3, DyCl3, GdCl3 were obtained from Acros Organics (Fair Lawn, NJ, USA). Bovine serum albumin and NaCl were purchased from Thermo Fisher Scientific Inc. (Waltham, MA, USA). ACS-grade, 68%–70% nitric acid was purchased from Mallinckrodt Chemical Inc. (St. Louis, MO, USA). Gadodimide (Omniscan™, GE Healthcare Inc., Waukesha, WI, USA) was obtained from the Department of Radiology at Case Western Reserve University (Cleveland, OH, USA). Rat liver and kidney tissues were provided by the Small Animal Medical Imaging Services of the University of Arizona (Tucson, AZ, USA).
Stock solutions were prepared at 5 mM for GdCl3, 10 mM for DyCl3, and 50 mM for TmCl3, EuCl3, and YbCl3 using distilled water. The concentration of each stock solution was verified with ICP-MS (Department of Geosciences, University of Arizona). A stock solution of gadodiamide was prepared at 5 mM concentration based on the concentration of the clinical product. A series of concentrations ranging between 0.1–5.0 mM for GdCl3 and gadodiamide, 0.5–10 mM for DyCl3, and 1.0–50.0 mM for TmCl3, EuCl3, and YbCl3 were then created by serially diluting each stock with distilled water. To maintain accuracy, solutions with concentrations less that 5 mM were created with a final volume of 5 mL. Solutions with higher concentrations were created with a final volume of 1 mL. These samples were then frozen in liquid nitrogen and lyophilized for 24–48 h depending on the sample volume. Concentrated nitric acid was then added to the dried sample, using a volume of 5 mL for dilute samples and 1 mL for concentrated samples.
To test the effect of salt, samples at 15 mM TmCl3 or 5 mM GdCl3 were prepared with NaCl concentrations ranging from 40 mOsm/L to 300 mOsm/L, using a 1 Osm/L stock solution of NaCl and the TmCl3 and GdCl3 stock solutions listed above. In addition, samples ranging between 0.1–5.0 mM for GdCl3 and 1.0–50.0 mM for TmCl3 were prepared with NaCl at 300 mOsm/L. To test the effect of protein content, the same series of samples of TmCl3 and GdCl3 were prepared with 90 μM of albumin, using a 300 μM stock solution of albumin and the TmCl3 and GdCl3 stock solutions listed above.
Rat liver and kidney tissues were excised and placed into 10% neutral buffered formalin and stored at 4°C before they were used for each tissue sample preparation. Rat kidney and liver tissue samples weighting 100 mg were tested, and the density of each tissue was assumed to be 1 g/mL to determine the volume of the tissue. Tissue samples that weighed 50, 100, 150, 200 and 250 gm were also tested. A stock solution of each lanthanide ion or gadodiamide was then added to a tissue sample, and diluted with distilled water, to create a range of tissue samples that ranged between 0.1–5.0 mM for GdCl3 and gadodiamide, 0.5–10 mM for DyCl3, and 1.0–50.0 mM for TmCl3, EuCl3, and YbCl3. A final volume of 5 mL was used for tissue samples with a concentration less that 5 mM, and tissue samples with a greater concentration were prepared with a final volume of 1 mL. The tissue samples were allowed to stand at room temperature for 30 min before lyophilization. Upon complete removal of water, the samples were suspended in 1 mL or 5 mL of conc. nitric acid for 3–5 h to allow for complete decomposition of the biological material.
A volume of 200 μL of each sample was added to a clean 3 mm NMR tube, and this narrow NMR tube was then placed in a 5 mm NMR tube that contained 300 μL of concentrated nitric acid as a reference solution. The heights of the sample volumes in the co-axial tubes exceeded the height of the NMR transceiver coil. Thus the sample approximated an infinite cylinder parallel to the static magnetic field of the NMR spectrometer, which provided the greatest BMS shift (i.e., s = 1/3 in Equation (1)). Samples were equilibrated at each temperature before acquiring spectra. A one-dimensional NMR spectrum was acquired for each sample, with a 2.19 s repetition time; 7 μsec excitation pulse; 12.5 ppm spectral width; 16,384 acquired data points; 0.114 Hz/point spectra resolution after zero-filling; and four repetitions with phase cycling. A deuterium lock was not used, because adding deuterium to the sample may have changed the BMS effect. Sample spinning was not used and shimming was not required, because the resulting lineshapes were sufficiently narrow and symmetric for accurately measuring the BMS shifts in each spectrum. The separation between the two spectral peaks was recorded in Hz to ensure measurement precision, and the Larmor frequency was used to convert this measurement to units of ppm. NMR spectra were recorded using a 300 MHz (7 T) Varian Unity+ spectrometer with an inverse tranceiver probe, and a 600 MHz (14 T) Varian Inova spectrometer with an inverse tranceiver cryoprobe. Measurements were acquired at 22.0 °C, except for some measurements performed at temperatures as high as 67.0 °C, using the calibrated temperature unit of the spectrometer.
For each tissue study, three samples at each concentration of lanthanide ion and gadodiamide were prepared and measured with BMS NMR spectroscopy to ensure reproducibility. The other studies were performed with only one sample per concentration of lanthanide ion and gadodiamide. The BMS shift measured for each tissue sample was converted to a concentration of lanthanide ion and gadodiamide using each calibration determined with lanthanide ion and gadodiamide solutions. The calculated concentrations were compared with the concentrations used during preparation of the sample for BMS NMR spectroscopic analyses.
3. Results and Discussion
A variety of methods for preparing samples were initially tested, including combinations of burning tissue samples to ash in a 600 °C furnace, homogenizing tissue samples to a paste, and high-speed centrifugation that have been previously used to extract metals from tissues [24–26]. These methods provided disappointing 8%–10% yields of recovered lanthanide ion in tissues. Therefore, these other methods were abandoned when a simplistic treatment with nitric acid was found to provide excellent yields [27]. The nitric acid completely digested tissues within 20 min, resulting in clear solution with a yellow color.
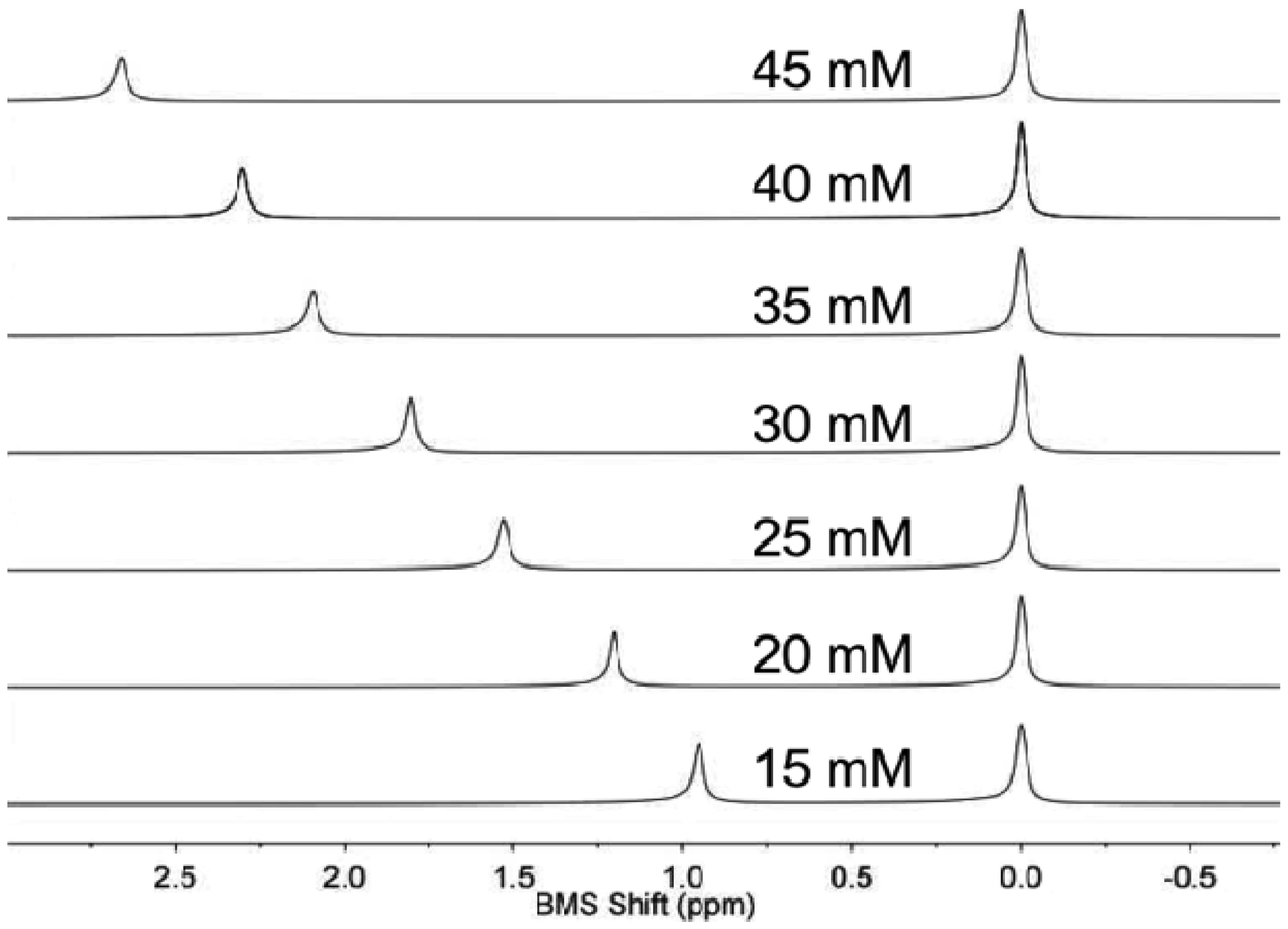
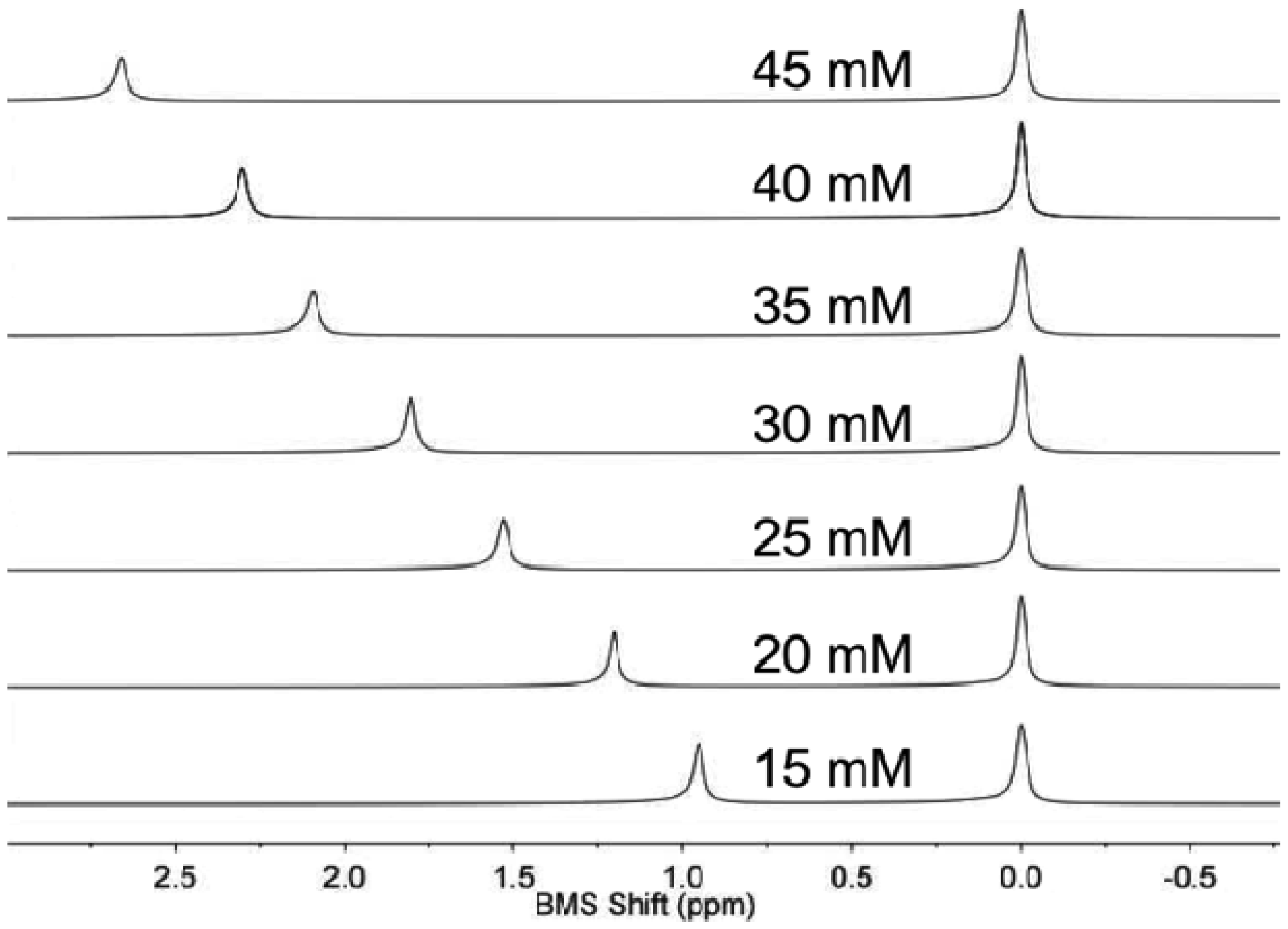
NMR spectroscopy of the sample and control solution in co-axial tubes generated outstanding spectral results (Figure 1). An initial study demonstrated that the BMS shift was invariant within one minute of inserting the sample in the magnet at 22.0 °C, indicating that the sample equilibrated from room temperature to 22.0 °C within one minute. The acquisition of the NMR spectrum required 8.76 s. Data processing and analysis required less than one minute. Therefore, the total time to analyze one sample was approximately two minutes. Thirty samples were routinely analyzed within one hour.
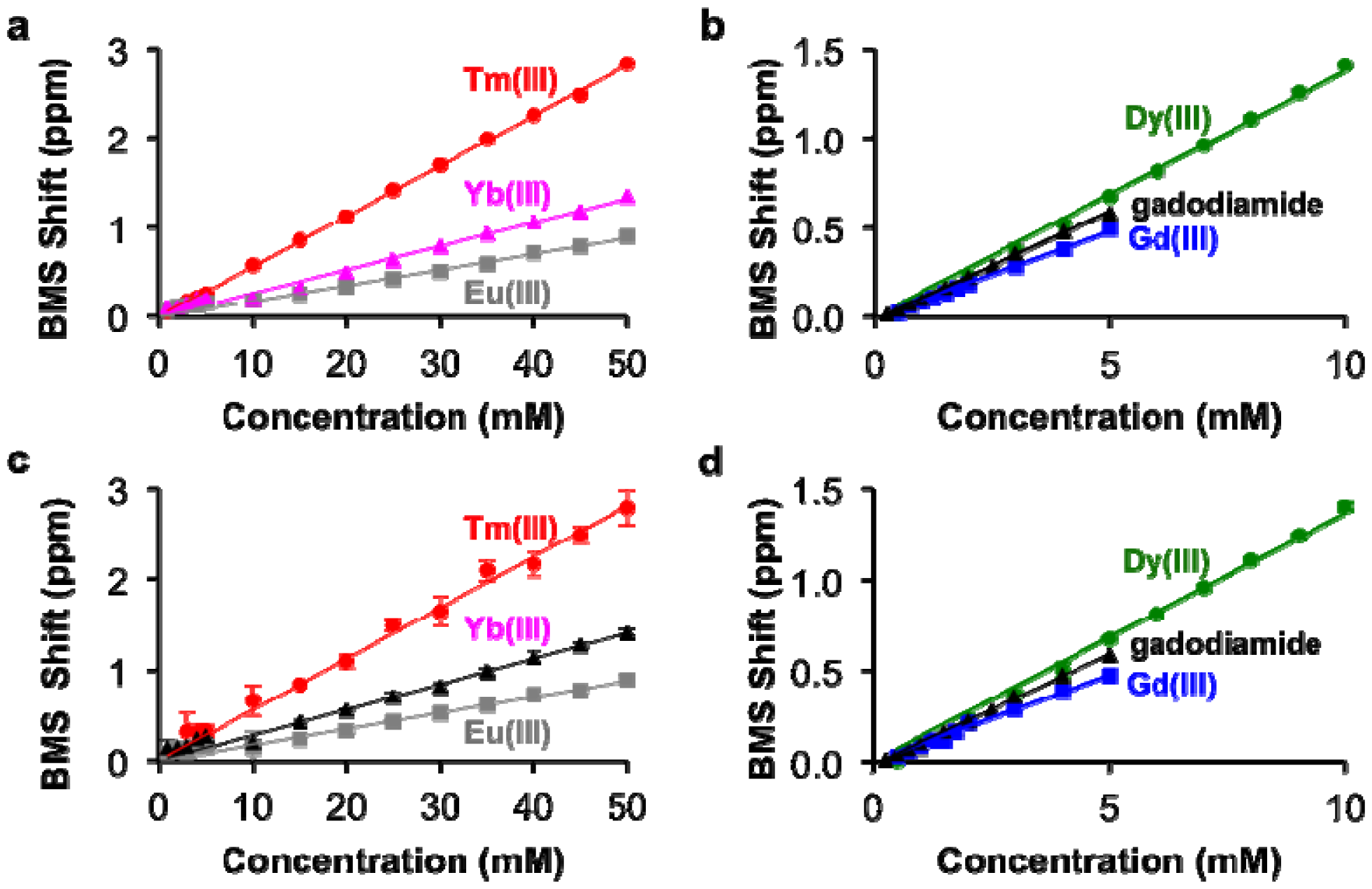
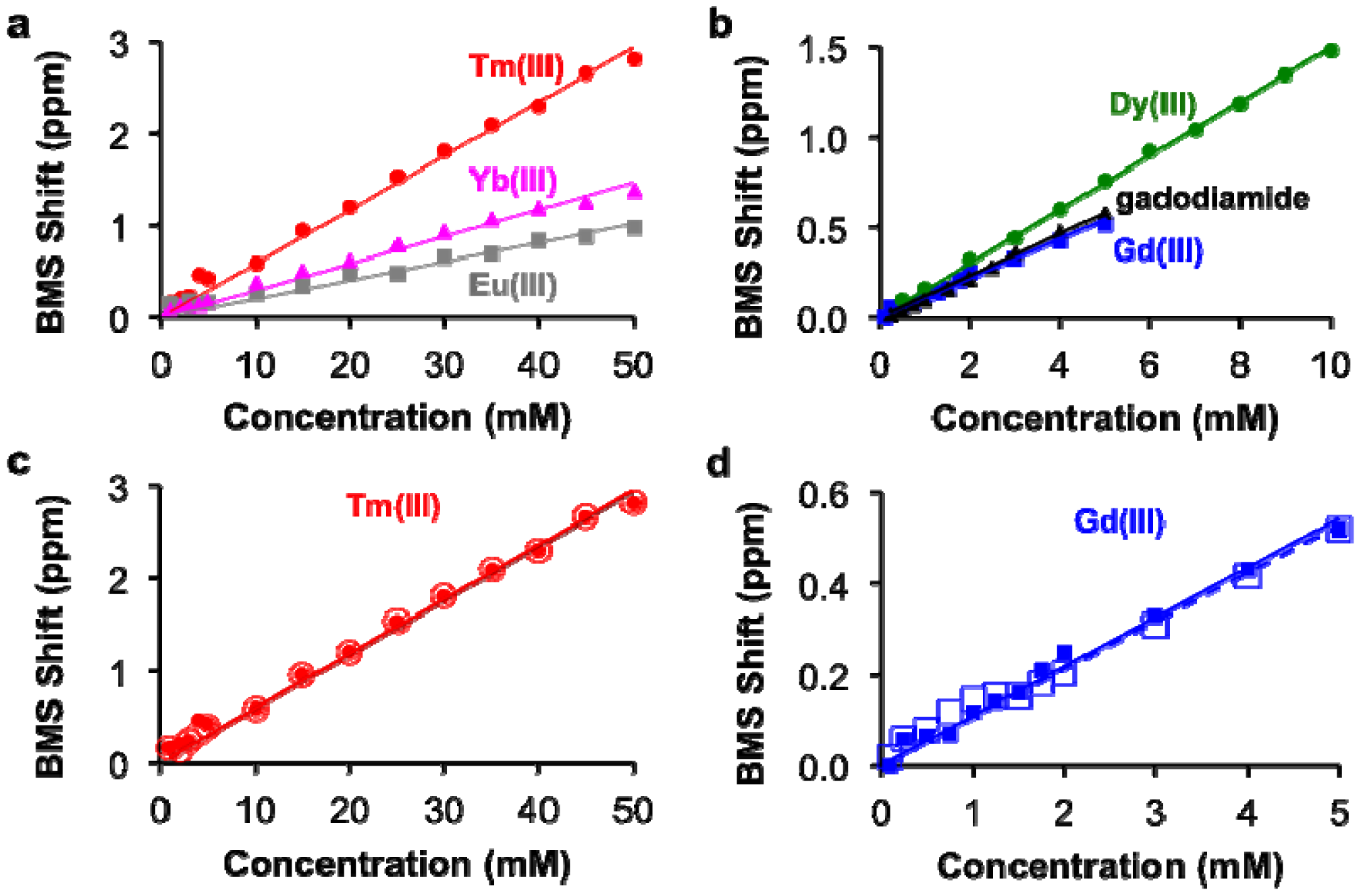
The concentrations of each lanthanide ion and gadodiamide in nitric acid were linearly correlated with BMS shifts acquired at 7T magnetic field strength. (Figure 2a,b). The calibrations with Gd(III) and gadodiamide were identical, indicating that the nitric acid digested the chelator, or the chelator had negligible effect on the BMS shift. Gd(III), gadodiamide, and Dy(III) were analyzed using concentration ranges that were lower than the ranges used for the other lanthanide ions, because very high concentrations for Gd(III) and Dy(III) caused rapid T2 relaxation that led to substantial line broadening in the NMR spectrum. These correlations showed outstanding linearity with R2 correlation coefficients greater than 0.98. The slope of each calibration was used to determine μeff for each lanthanide ion, which agreed with previously published results (Table 1; Equation (2)). This agreement validated that the concentration dependence of the measured chemical shifts arose from a BMS shift:
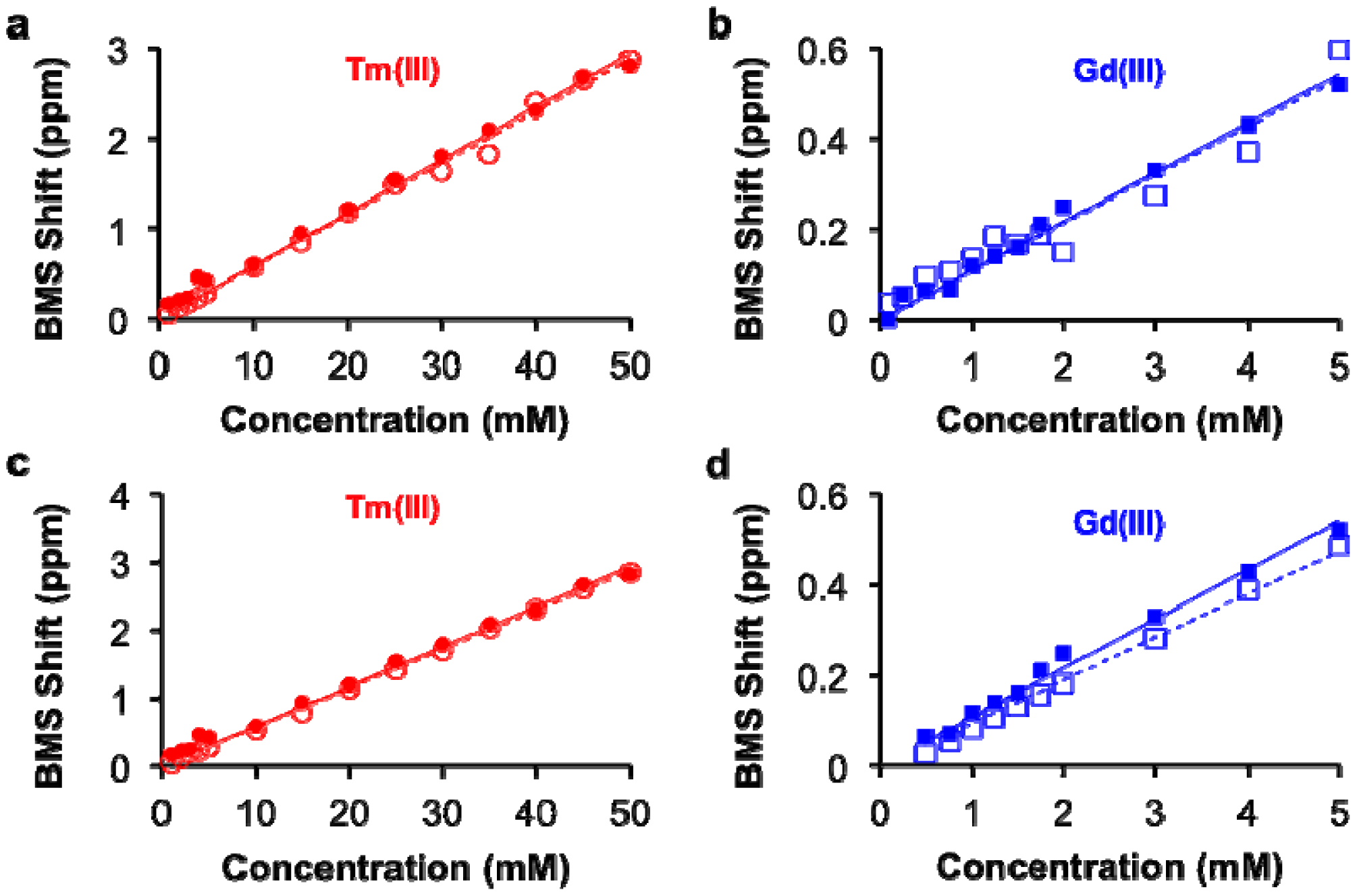
This study with Tm(III) and Gd(III) samples at different concentrations was repeated at 14 T magnetic field strength, which generated the same calibration (Figure 2c,d). This result indicated that NMR spectrometers at any field strength may be used to measure lanthanide ion concentration via BMS shift measurements. The calibration of BMS shift with the concentration of Tm(III) or Gd(III) was not affected by the addition of 300 mOsm/L salt or 90 μM protein (Figure 3). In addition, samples of 15 mM Tm(III) and 5 mM Gd(III) that had varying concentrations of salt generated identical BMS shifts (data not shown), which further verified that salt had no effect on the BMS measurement. These results indicated that estimates of lanthanide ion concentrations via BMS shift measurements with sample preparation using nitric acid could be translated to tissue samples that have high salt and protein contents.
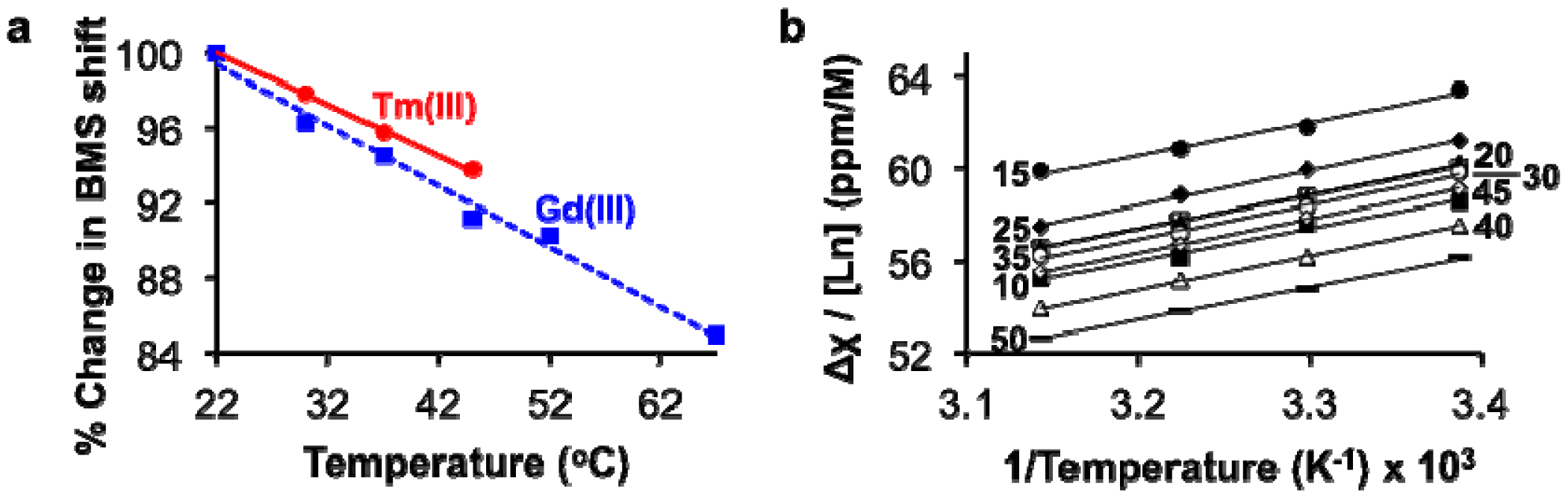
The BMS shifts of Tm(III) and Gd(III) decreased with increasing temperature, as predicted by Equation (1) (Figure 4a). This result indicated that analyses of lanthanide ion concentrations should be conducted at lower temperatures to improve measurement precision. We elected to perform measurements at 22 °C, the lowest temperature tested in our studies, to avoid delays required to equilibrate the sample at lower temperatures. The values of μeff for Tm(III) and Gd(III) were estimated based on the dependence of the BMS shift on concentration and temperature (Table 1; Equation (3); Figure 4b). Once again, these estimates of μeff agreed with published results, which further validated that the BMS shift was the primary mechanism responsible for the differences in chemical shifts observed using NMR spectroscopy:
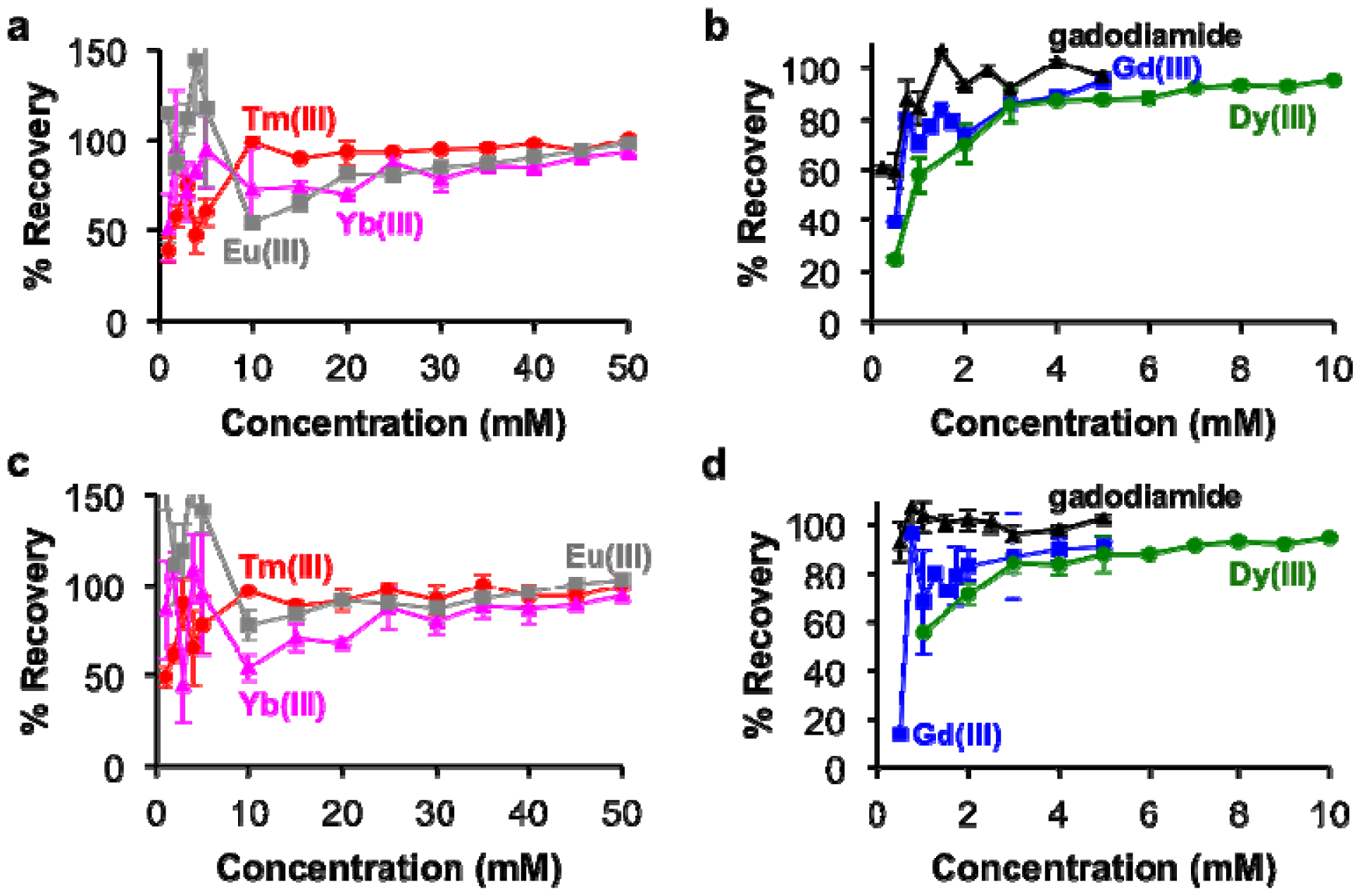
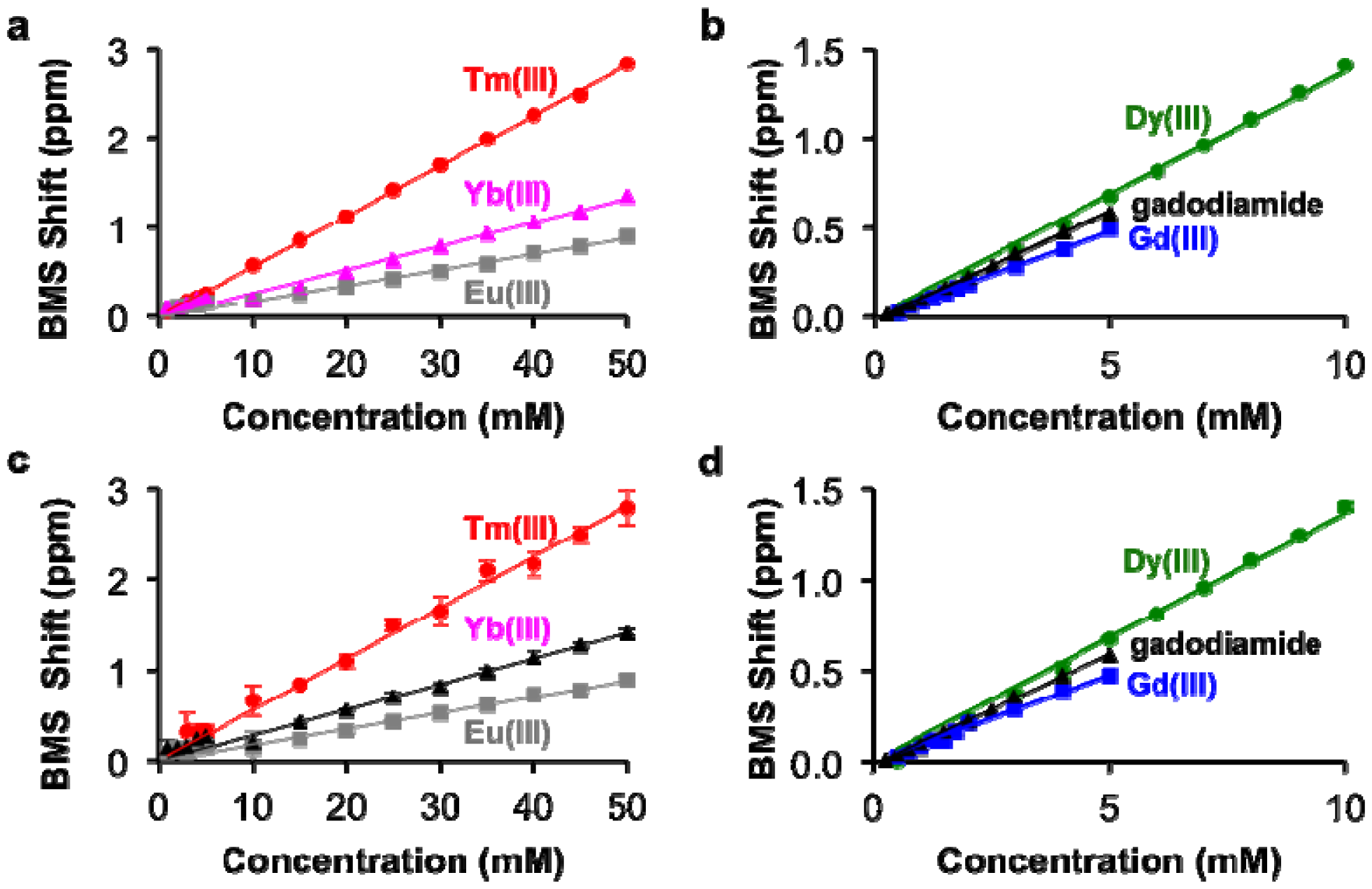
The concentrations of each lanthanide ion and gadodiamide in kidney and liver tissues treated with nitric acid were linearly correlated with BMS shifts acquired at 7T magnetic field strength. (Figure 5). These relationships had R2 correlation coefficients greater than 0.96. These outstanding linear correlations with a 0 y-intercept indicated that endogenous metals such as iron had negligible effects on the BMS measurements. Furthermore, the average standard deviations from triplicate evaluations of each sample were less than 10%, and the great majority of standard deviations were much smaller than 10%, as evidenced by many error bars that are smaller than the size of the data symbol in Figure 5. These results demonstrated that the concentration of lanthanide ion or gadodiamide in tissues was measured with outstanding precision. In addition, the correlations for Gd(III) and gadodiamide were the same, which once again indicated that nitric acid digested the chelator during tissue digestion, or the chelator had negligible effect on the BMS shift. Identical results were obtained using samples that ranged from 50 to 250 mg of tissue, indicating that the tissue volume did not influence the measurement precision (data not shown), which further verified that protein content within the processed solution did not influence the measurement of lanthanide ion concentration with BMS NMR spectroscopy.
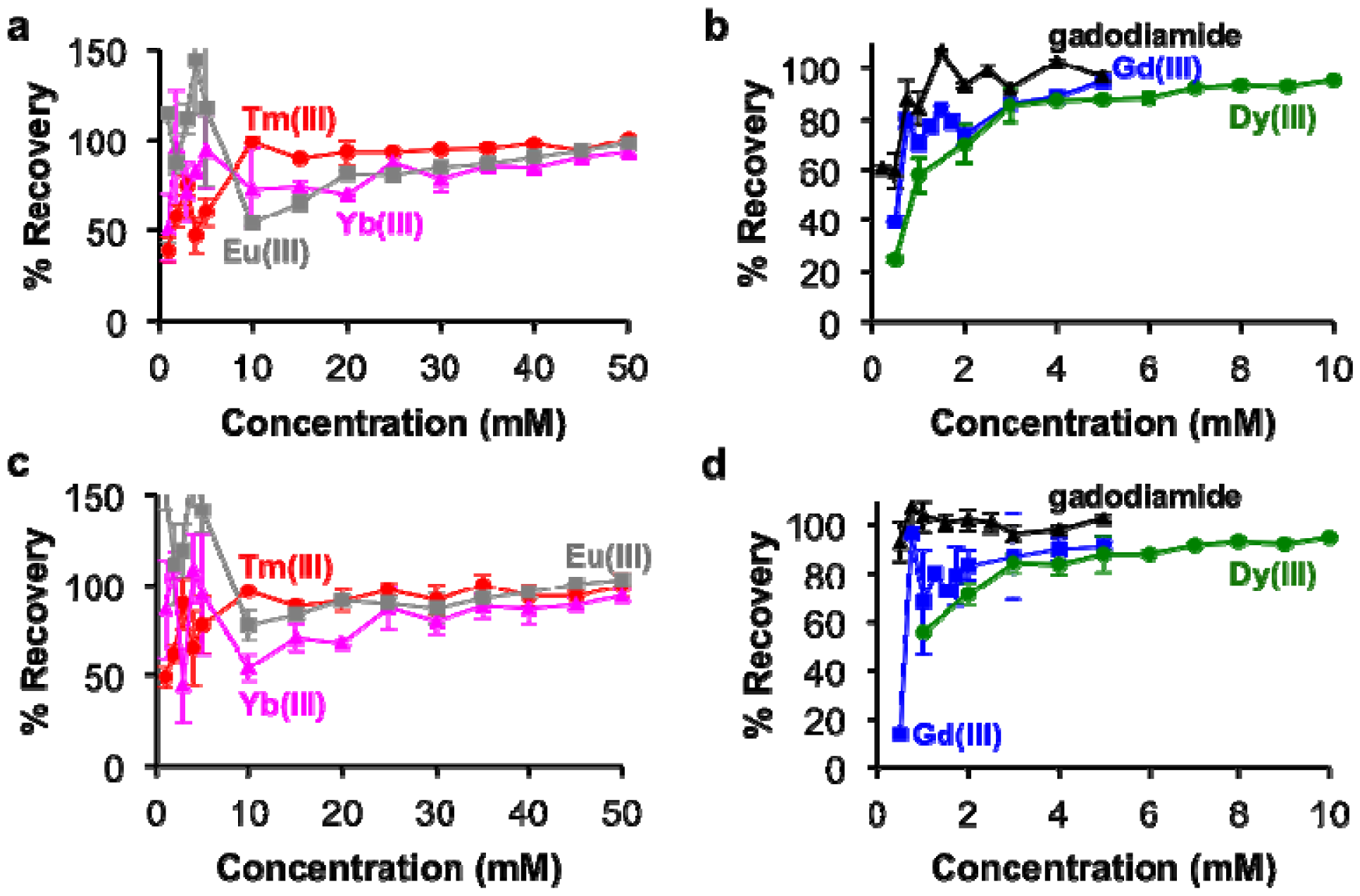
The percent recovery of lanthanide ions from the tissue samples was determined by using the calibrations in Figure 2 to estimate the ion concentrations from the BMS shifts of the tissue samples (Figure 6). These results showed greater than 85% recovery with samples containing at least 3 mM with Gd(III), gadodiamide, or Dy(III), and greater than 80% recovery with samples containing at least 25 mM of Tm(III), Yb(III), and Eu(III). However, the recovery was less accurate for more dilute samples. This result reflects the difficulty in sample handling, which affects dilute samples to a greater extend than concentrated samples. In particular, the development of our methodology showed that care needs to be taken to handle nitric acid without loss due to evaporation, which can be substantial over time and especially during outdoor transport in a southwest desert environment.
These results established a protocol for measuring the lanthanide ion concentration in ex vivo tissues. Nitric acid can be used to digest any tissue or lanthanide chelate, which allows this method to be applied to many biomedical studies. NMR spectrometers at any magnetic field strength are commonly available at many research institutions for a nominal usage fee. Many samples can be quickly analyzed with this method. As an additional benefit, the completely digested tissues are not biohazardous, which facilitates waste handling (however, care should be taken to properly dispose of samples with lanthanide ions).
Our results show that lanthanide ion content can be measured in ex vivo tissues with outstanding precision. In addition, accurate measurements can be made with concentrations ≥ 25 mM for Tm(III), Eu(III) or Yb(III), and ≥ 3 mM for Gd(III), gadodiamide, or Dy(III). Unfortunately, T1 and T2 MRI contrast agents typically accumulate in tissues at 10 μM to 1 mM concentrations, and paramagnetic CEST agents likely accumulate in tissues at concentrations less than 10 mM. Therefore, the current concentration thresholds for accurate measurements are too high to apply this method for most biodistribution studies of MRI contrast agents. The lower yields at low concentrations are attributed to the loss of lanthanide ions or chelates during tissue processing. Therefore, improved tissue handling methods are warranted to facilitate this methodology. For example, extreme care is needed for handling and processing lanthanide chelates for optical imaging studies, and these careful handling methods may be adopted for measuring BMS shifts with NMR spectroscopy [28]. As an alternative, lyophilization of a large volume of tissue may be digested with a small volume of nitric acid, which would effectively concentrate the lanthanide ion in the acidic solution used for BMS NMR measurements. The volume of tissue that can be digested by 1 or 5 mL of nitric oxide (the volumes of acid used in this study) would need to be tested for each tissue type to assess the improvement in detection sensitivity by using this approach. Also, the influence of concentrating endogenous metals such as iron would also need to be assessed.
Other biological and non-biological samples besides ex vivo tissues also require testing for lanthanide concentrations. For example, biosorption of lanthanide metals has been monitoried in microbial species [29], plants [30], and biomass [31] to assess environmental quality and bioremediation. The concentrations of lanthanide metals are also assessed in soils [32,33] and stream waters [34,35], especially near mining sites and industrial complexes. These environmental assessments are likely to continue, considering the ∼10%/year increase in lanthanide ore production to meet rising demands for glass polishing, catalysts, phosphors, magnets and electronic products [36]. As with the analyses of tissues in this report, the application of acid treatment and BMS shifts for measuring lanthanide concentrations in other samples will require consideration for the minimum detection level and/or consideration for concentrating the sample to meet the minimum detection level for accurate and precise measurements of lanthanide metal concentrations.
4. Conclusions/Outlook
The BMS shift was successfully used to measure the concentration of lanthanide ions and a MRI contrast agent, gadodiamide, in solution and within ex vivo tissue samples. Key steps in this process included the use of nitric acid to completely digest tissues, and the use of coaxial tubes for simultaneous NMR spectroscopy of experimental and control samples. The concentration dependence of the measured chemical shifts was shown to be due to the BMS shift. Although the methodology had outstanding precision, concentration measurements were accurate only for samples with higher concentrations. Improvements to sample handling during tissue processing are warranted to improve the accuracy of measuring lanthanide ion concentrations of more dilute samples.
Acknowledgments
This work was supported by the Phoenix Friends of the Arizona Cancer Center, and NIH grants R01 CA169774-01 and P50 CA95060. DVH was sponsored by the TRIF Fellowship Program of the University of Arizona. SIG was sponsored by the MARC Program of the University of Arizona through NIH grant T34 GM008718.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Runge, V.M.; Muroff, L.R.; Wells, J.W. Principles of contrast enhancement in the evaluation of brain diseases: An overview. J. Mag. Reson. Imag. 2005, 7, 5–13. [Google Scholar]
- Moriarty, J.M.; Finn, J.P.; Fonesca, C.G. Contrast agents used in cardiovascular magnetic resonance imaging: Current issues and future directions. Am. J. Cardiovasc. Drugs 2010, 10, 227–237. [Google Scholar]
- Zhou, Z.; Lu, Z.R. Gadolinium-based contrast agents for magnetic resonance cancer imaging. Nanomed. Nanobiotechnol. 2013, 5, 1–18. [Google Scholar]
- Stafford Johnson, D.B.; Prince, M.R.; Chenevert, T.L. Magnetic resonance angiography: A review. Acad. Radiol. 1998, 5, 289–305. [Google Scholar]
- O'Connor, J.P.B.; Jackson, A.; Parker, G.J.M.; Jayson, G.C. DCE-MRI biomarkers in the clinical evaluation of antiangiogenic and vascular disrupting agents. Brit. J. Cancer 2007, 96, 189–195. [Google Scholar]
- Dijkhuizen, R.M. Advances in MRI-Based detection of cerebrovascular changes after experimental traumatic brain injury. Translat. Stroke Res. 2011, 2, 524–532. [Google Scholar]
- Geelen, T.; Paulis, L.E.M.; Coolen, B.F.; Nicolay, K.; Strijkers, G.J. Contrast-enhanced MRI of murine myocardial infarction–part I. NMR Biomed. 2012, 25, 953–968. [Google Scholar]
- Gore, J.C.; Manning, H.C.; Quarles, C.C.; Waddell, K.W.; Yankeelov, T.E. Magnetic resonance in the era of molecular imaging of cancer. Mag. Reson. Imag. 2011, 29, 587–600. [Google Scholar]
- Yoo, B.; Pagel, M.D. An overview of responsive MRI contrast agents for molecular imaging. Front. Biosci. 2008, 13, 1733–1752. [Google Scholar]
- Sheth, V.R.; Pagel, M.D. CEST & PARACEST MRI Contrast Agents for Imaging Cancer Biomarkers. In Molecular Imaging Probes for Cancer Research; Chen, X., Ed.; World Scientific Publishing: Hackensack, NJ, USA, 2012; pp. 689–713. [Google Scholar]
- Kuo, P.H.; Kanal, E.; Abu-Alfa, A.K.; Cowper, S.E. Gadolinium-based MR contrast agents and nephrogenic systemic fibrosis. Radiology 2007, 242, 847–849. [Google Scholar]
- Buckley, D.L.; Parker, G.J.M. Measuring Contrast Agent Concentration in T1-Weighted Dynamic Contrast-Enhanced MRI. In Medical Radiology—Diagnostic Imaging and Radiation Oncology; Jackson, A., Buckley, D., Parker, G.J.M., Eds.; Springer: New York, NY, USA, 2005; pp. 69–79. [Google Scholar]
- Ali, M.M.; Liu, G.; Shah, T.; Flask, C.A.; Pagel, M.D. Using two chemical exchange saturation transfer magnetic resonance imaging contrast agents for molecular imaging studies. Acc. Chem. Res. 2009, 42, 915–924. [Google Scholar]
- Crayton, S.H.; Elias, D.R.; Al Zaki, A.; Cheng, Z.; Tsourkas, A. ICP-MS analysis of lanthanide-doped nanoparticles as a non-radiative, multiplex approach to quantify biodistribution and blood clearance. Biomat 2012, 33, 1509–1519. [Google Scholar]
- Barge, A.; Cravotto, G.; Gianolio, E.; Fedeli, F. How to determine free Gd and free ligand in solution of Gd chelates. A technical note. Contrast Media Mol. Imag. 2006, 1, 184–188. [Google Scholar]
- Brown, R.; Clarke, D.W.; Daffner, R.H. Is a mixture of gadolinium and iodinated contrast material safe during MR arthrography? Am. J. Roentgenol. 2000, 175, 1087–1090. [Google Scholar]
- Rohwer, H.; Collier, N.; Hosten, E. Spectrophotometric study of arsenazo III and its interactions with lanthanides. Anal. Chim. Acta. 1995, 314, 219–223. [Google Scholar]
- Evans, D.F. The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J. Chem. Soc. 1959. [Google Scholar] [CrossRef]
- Peters, J.A.; Huskens, J.; Raber, D.J. Lanthanide induced shifts and relaxation rate enhancements. Prog. Nucl. Mag. Reson. Spect. 1996, 28, 283–350. [Google Scholar]
- Corsi, D.M.; Platas-Iglesias, C.; von Bekkum, H.; Peters, J.A. Determination of paramagnetic lanthanide(III) concentrations from bulk magnetic susceptibility shifts in NMR spectra. Mag. Reson. Chem. 2001, 39, 723–726. [Google Scholar]
- Gysling, H.; Tsutsui, M. Organolanthanides and Organoactinides. Adv. Organomet. Chem. 1971, 9, 361–395. [Google Scholar]
- Gerothanassis, I.P.; Lauterwein, J. Oxygen-17 NMR spectroscopy: Referencing in diamagnetic and paramagnetic solutions. Mag. Reson. Chem. 1986, 24, 1034–1038. [Google Scholar]
- Fossheim, S.; Johansson, C.; Fahlvik, A.K.; Grace, D.; Klaveness, J. Lanthanide-based susceptibility contrast agents: Assessment of the magnetic properties. Mag. Reson. Med. 1996, 35, 201–206. [Google Scholar]
- Parker, M.M.; Humoller, F.L.; Mahler, D.J. Determination of copper and zinc in biological material. Clin. Chem. 1967, 13, 40–48. [Google Scholar]
- Schulten, H.R.; Ziskoven, R. Determination of thallium in brain tissue of the mouse by stable isotope dilution and field desorption mass spectrometry. J. Physiol. 1978, 284, 170P–171P. [Google Scholar]
- Dos Santos, W.P.C.; Hatje, V.; da Santil, D.; Fernandes, A.P.; Korn, M.G.A.; de Souza, M.M. Optimization of a centrifugation and ultrasound-assisted procedure for the determination of trace and major elements in marine invertebrates by ICP OES. Microchemical. J. 2010, 95, 169–173. [Google Scholar]
- O'Brien, R.D. Nitric acid digestion of tissues for liquid scintillation counting. Anal. Biochem. 1964, 7, 251–254. [Google Scholar]
- Josan, J.S.; de Silva, C.R.; Yoo, B.; Lynch, R.M.; Pagel, M.D.; Vagner, J.; Hruby, V.J. Fluorescent and lanthanide labeling for ligand screens, assays, and imaging. Meth. Mol. Biol. 2011, 716, 89–126. [Google Scholar]
- Andres, Y.; Thouand, G.; Boulalam, M.; Mergeay, M. Factors influencing the biosorption of gadolinium by microorganisms and its mobilisation from sand. Appl. Microbiol. Biotech. 2000, 54, 262–267. [Google Scholar]
- Anan, Y.; Awaya, Y.; Ogihara, Y.; Yosida, M.; Yawata, A.; Ogra, Y. Comparison in accumulation of lanthanide elements among three brassicaceae plant sprouts. Bull. Environ. Contam. Toxicol. 2012, 89, 133–137. [Google Scholar]
- Parsons, J.G.; Peralta-Videa, J.R.; Tiemann, K.J.; Saupe, G.B.; Gardea-Torresdey, J.L. Use of chemical modification and spectroscopic techniques to determine the binding and coordination of gadolinium(III) and neodymium(III) ions by alfalfa biomass. Talanta 2005, 67, 34–45. [Google Scholar]
- Vodyanitskii, Y.N.; Savichev, A.T. Lanthanides in Soils: X-ray Determination, Spread in Background and Contaminated Soils in Russia. In Geochemistry: Earth's System Processes; InTech: New York, NY, USA, 2012; pp. 389–412. [Google Scholar]
- Shumilin, E.; Rodriguez-Figueroa, G.; Sapozhnikov, D. Lanthanide contamination and lanthanide concentration and strong positive Europium Anomalies in the Surface Sediments of the Santa Rosalia Copper Mining Region, Baja California Peninsula, Mexico. Bull. Environ. Contamin. Toxicol. 2005, 75, 308–315. [Google Scholar]
- Neal, C. Rare earth element concentrations in dissolved and acid available particulate forms for eastern UK rivers. Hydrol. Earth Syst. Sci. 2007, 11, 313–327. [Google Scholar]
- Protano, G.; Riccobono, F. High contents of rare earth elements (REEs) in stream waters of a Cu-Pb-Zn mining area. Environ. Pollut. 2002, 117, 499–514. [Google Scholar]
- Naumov, A.V. Review of the world market of rare-earth metals. Russ. J. Non-Ferrous Metals 2008, 49, 14–22. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lanthanide Ion | μeff (Figure 2a,b) | μeff (Figure 4a) | μeff ([22]) |
|---|---|---|---|
| Dy(III) | 10.38 | --- | 10.6 |
| Gd(III) | 7.86 | 8.10 | 7.94 |
| Tm(III) | 6.56 | 7.54 | 7.6 |
| Yb(III) | 3.81 | --- | 4.5 |
| Eu(III) | 3.18 | --- | 3.40–3.51 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hingorani, D.V.; Gonzalez, S.I.; Li, J.F.; Pagel, M.D. Sensing Lanthanide Metal Content in Biological Tissues with Magnetic Resonance Spectroscopy. Sensors 2013, 13, 13732-13743. https://doi.org/10.3390/s131013732
Hingorani DV, Gonzalez SI, Li JF, Pagel MD. Sensing Lanthanide Metal Content in Biological Tissues with Magnetic Resonance Spectroscopy. Sensors. 2013; 13(10):13732-13743. https://doi.org/10.3390/s131013732
Chicago/Turabian StyleHingorani, Dina V., Sandra I. Gonzalez, Jessica F. Li, and Mark D. Pagel. 2013. "Sensing Lanthanide Metal Content in Biological Tissues with Magnetic Resonance Spectroscopy" Sensors 13, no. 10: 13732-13743. https://doi.org/10.3390/s131013732
APA StyleHingorani, D. V., Gonzalez, S. I., Li, J. F., & Pagel, M. D. (2013). Sensing Lanthanide Metal Content in Biological Tissues with Magnetic Resonance Spectroscopy. Sensors, 13(10), 13732-13743. https://doi.org/10.3390/s131013732