1. Introduction
The genus
Salix (family of
Salicaceae) is one of the most important taxonomic entities of the world for the great number of species and varieties, with around 300 taxa of deciduous trees and shrubs widespread both in the boreal and austral hemisphere [
1,
2,
3]. Almost half of these taxa are distributed in the Eurasian continent, one third are found in the American continent from Alaska to Patagonia, and the remaining are present in Europe and the Mediterranean basin [
4,
5]. The genus
Salix shows all forms of development from tall trees (hot-temperate climates), to shrubs (cold-temperate climates) and to creeping and dwarf shrubs (cold and arctic zones). More clearly than in other genera of phanerophytes, each single species of willow is related to very specific environmental conditions, so that the corresponding plant associations show distinctive ecophysionomic aspects due to the climatic and edaphic characteristics of their own habitats [
5]. Hybridization between
Salix species is very common. In addition to the naturally occurring hybrids in various parts of the world, a considerable number of others have been artificially produced by controlled matings [
3]. Spontaneous hybridization is determined by dioecism and is affected by variation of flowering times in different species. In the genus
Salix, both balanced and introgressed hybrids seem to dominate when considering only morphological traits, but studies based on biochemical and molecular markers revealed that genetic differentiation within populations is low as well as genetic flow between populations [
6,
7]. This finding is consistent with historical data available for forest trees with a dioecious-type breeding system and wind-dispersed seeds [
8]. Nevertheless, a striking absence of geographical effects in the haplotype distributions of
Salix species was recently discovered by [
9] and likely owed to recent and continual plastid capture events, aided by wide-spread hybridization and long-range seed dispersal, but primarily propelled by one or more trans-species selective sweeps. In addition, [
10] reported that
S. alba,
S. fragilis, and their hybrid
S. ×
rubens are genetically recognizable and that only few hybrids and any backcrosses to the parental species are present in mixed stands. Overall findings supported a clear separation of the pure species,
S. alba and
S. fragilis, and an intermediate position of
S. ×
rubens individuals.
Morphology indicates that pure species of the
Salix alba L. (white willow)
—Salix fragilis L. (crack willow) complex and their natural hybrids form a polyploid complex of closely related willows [
11,
12,
13,
14]. The problem of the separation of the hybrid entities between
S. alba and
S. fragilis is one of the critical matters of systematics of the genus
Salix [
3]. This complex includes mostly tetraploid trees, which are dioecious and thus obligate outcrossers. As a consequence, natural populations of these willows can be represented by a mixture of highly heterozygous genotypes, sharing a common gene pool as a result of recurring hybridization [
15]. Species like
S. babylonica L.,
S. pentandra L. and
S. triandra L. are the only ones that successfully intercross with both
S. alba and
S. fragilis [
5,
16]. Although they have similar ecological characteristics,
S. fragilis and
S. alba are distinguishable in terms of temperature requirements and areas of spontaneous occurrence, being the former native to Western Asia and naturalized in central and northern Europe, and the latter more spread in temperate regions and the Mediterranean basin [
11,
12]. On the basis of plant morphology, European populations seem to include mainly balanced and introgressed
S. alba ×
S. fragilis hybrids [
13,
17]. The analysis of leaves, buds and twigs has indicated that many potential hybrids are present within natural populations [
18]. This finding agrees with the observations of several botanists [
11,
19,
20] that the vast majority of willows in the
S. alba—S. fragilis complex could be hybrids or introgressants. Owing to their continuous variation, most of the phenotypic features have however a low diagnostic value for identifying interspecific hybrid constitutions, assessing introgression patterns, and defining genetic variation structure and relatedness at the population level. Consequently, the taxonomic classification of pure species as well as the identification of hybrid individuals is still today a matter of debate.
Molecular studies carried out using controlled crosses and field clones contradict traditional hypothesis on the extensive occurrence of hybrids in the
S. alba—S. fragilis complex. In particular, molecular markers revealed that both species have kept their gene pools well separated and that interspecific hybridization actually does not seem to be a dominating process [
13]. New biotechnological developments have expanded the range of plant DNA polymorphism assays for characterizing and investigating germplasm resources and genetic relatedness, as well as for linkage mapping, gene targeting and assisted breeding. The area of willow research showing the greatest development with respect to the use of molecular marker technology is that of population genetics [
7,
13,
14,
18,
21,
22,
23,
24], whereas information on structural genomics is scanty. In fact, nothing is known about willow genomic constitution and origin, and whether species are autopolyploid or allopolyploid remains an open question. Because of the many basic chromosomes (
x = 19), willow species with 2
n = 38 may represent ancient polyploid derivations [
25,
26,
27], that however behave like functionally diploidized genomes [
28,
29]. In fact, diploidization affects the chromosome behavior during meiosis along with gene dosages and inheritance models in polyploids. Cytological observations of the pairing modes of tetraploid plants with 2
n = 76, useful to reveal bivalent or multivalent formation, are difficult owing to the high chromosome number and small chromosome size of willow species. Moreover, genetic analysis in willow has been restricted not only because of genome architecture complexity, but also due to the lack of discriminant molecular markers and the need of experimental populations amenable to molecular analysis. As a result, a few genetic linkage maps have been constructed and data on segregation patterns or recombination estimates are scanty for tetraploid willow [
14].
Molecular investigations of tetraploid species belonging to the S. alba—S. fragilis complex could provide valuable data about the genetic inheritance and genomic structure of willows. The genetic segregation assay of single-dose markers was used to assess the inheritance pattern of tetraploid types (disomy vs. tetrasomy), whereas genomic in situ hybridization technology was exploited to shed light on the genome constitution of tetraploid willows (allotetraploidy vs. autotetraploidy). New findings supporting the genome organization and gene transmission patterns in tetraploid Salix spp. are presented and a working hypothesis that explains why the gene pools of S. alba and S. fragilis species have remained well separated is formulated and discussed.
3. Results
3.1. Genomic DNA Fingerprint Analysis
A total of 305 amplicons were scored across all 36 genomic DNA fingerprints, including reproducible polymorphisms among the assayed willow plants. In fact, the adoption of replicated samples for each clonal genotype allowed us to verify a full reliability of the obtained molecular results. This characterization of Salix germplasm was based on three EcoRI/MseI and 3 PstI/MseI primer combinations, selected during preliminary experiments for their polymorphism information content, which enabled a successful evaluation of the genetic relationships within as well as between species.
UPGMA grouping methods were useful to assess genetic variation within single species and genetic relationships among pure species (
Figure 1).
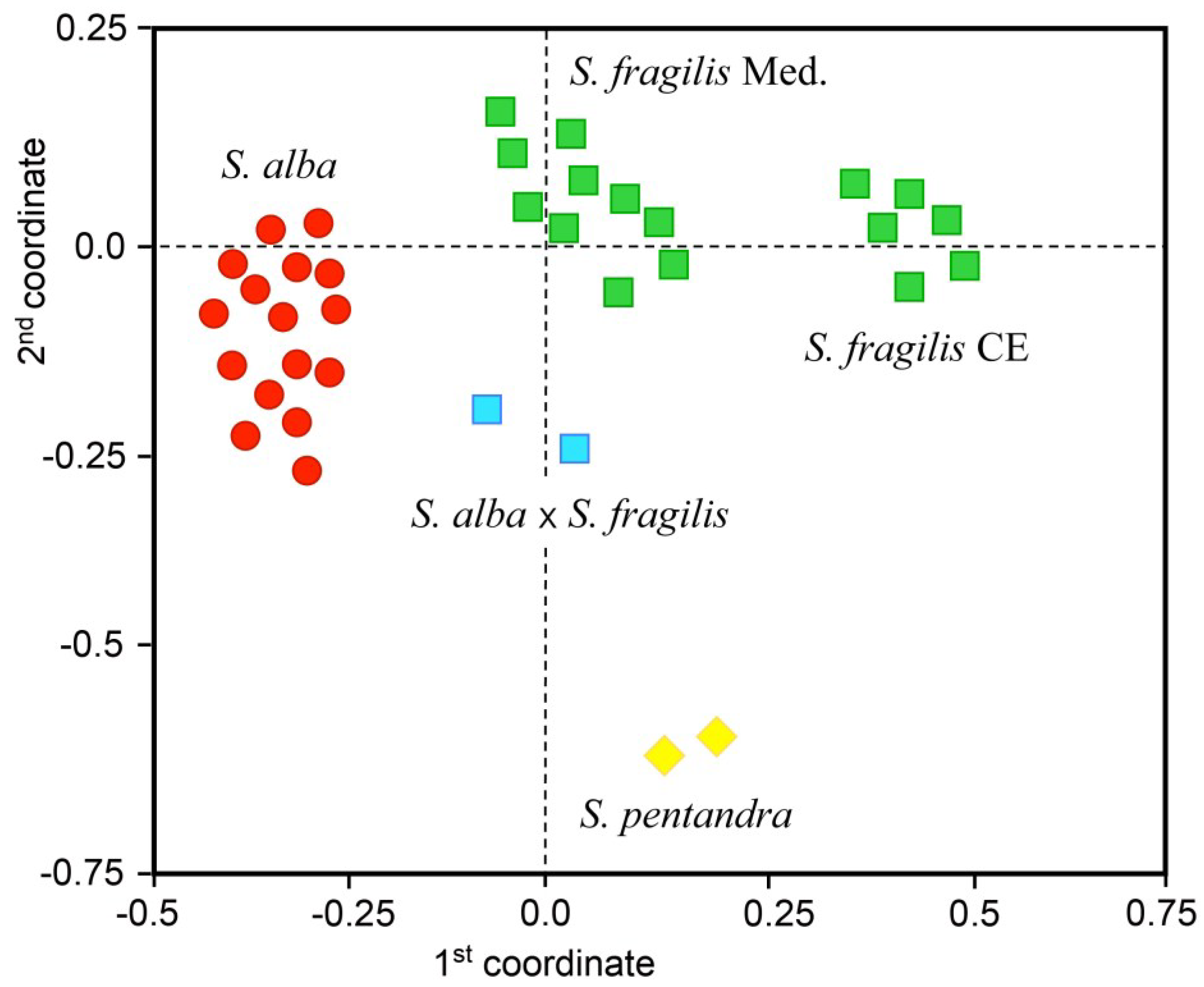
Figure 1.
Outcome from UPGMA (unweighted pair-group method with arithmetic mean) grouping analysis of Salix spp. samples on the basis of molecular amplified fragment length polymorphism (AFLP) fingerprints, depicting genetic relationships among S. alba and S. fragilis pure species, S. alba × S. fragilis hybrids, using S. pentandra as outgroup entities. Centroids of the 36 willow trees were obtained from the mean genetic similarity matrix and bi-dimensionally plotted according to the first two components that explain about 60% of the total genetic variation detected across all marker loci.
Figure 1.
Outcome from UPGMA (unweighted pair-group method with arithmetic mean) grouping analysis of Salix spp. samples on the basis of molecular amplified fragment length polymorphism (AFLP) fingerprints, depicting genetic relationships among S. alba and S. fragilis pure species, S. alba × S. fragilis hybrids, using S. pentandra as outgroup entities. Centroids of the 36 willow trees were obtained from the mean genetic similarity matrix and bi-dimensionally plotted according to the first two components that explain about 60% of the total genetic variation detected across all marker loci.
In particular, cluster analysis was useful to plot the centroids of willow accessions into four main subgroups according to the first two coordinates, which explained about 41% and 19% of the total genetic variation: pure
S. fragilis individuals from the Mediterranean area formed a first subgroup, while a second subgroup included pure
S. fragilis individuals from Central-Europe and a third subgroup was composed by pure
S. alba individuals. The
S. alba ×
S. fragilis hybrids were plotted separately from the pure species subgroups. In addition, the reference
S. pentandra individuals were clustered apart forming a fourth well-separated subgroup. The first coordinate grouped separately the Central-European accessions of
S. fragilis from the other accessions of the same species deriving from the Mediterranean area, and also from all the accessions of
S. alba. The second coordinate enabled to distinguish clearly the accessions of
S. pentandra from the
S. alba accessions and from all
S. fragilis accessions, irrespectively of their Mediterranean or Central-European origin, along with the
S. alba ×
S. fragilis hybrids (see
Figure 1).
The finding of genetically differentiated subgroups and the lack of genotypes with intermediary characteristics confirm that both species of the S. alba—S. fragilis complex have kept their gene pools well separated and that interspecific hybridization S. alba × S. fragilis does not seem to be a dominating process in natural populations. It is worth mentioning that the subgroup of Central-European accessions of S. fragilis resulted more similar to the Mediterranean accessions of S. fragilis than to the S. alba accessions (0.748 against 0.644, respectively). The greatest genetic differentiation was observed between Central-European accessions of S. fragilis and S. pentandra (0.558). However, the fact that accessions of Mediterranean S. fragilis were genetically more similar to accessions of S. alba (0.830) rather than to Central-European accessions of S. fragilis (0.748) underlines the importance in evolutionary terms of the pedo-climatic environment.
On the basis of molecular data, it is likely that the spatial isolation, together with the habitat characteristics, could play an important role in the process of differentiation of separate subgroups within the same species.
3.2. Segregation Pattern Analysis of Marker Alleles
The experimental F1 populations stemmed from S. alba × S. fragilis crosses and reciprocals were analyzed with 10 EcoRI/MseI and 10 PstI/MseI primer combinations, yielding an average of 56 reliable markers per single AFLP fingerprint (ranging from a minimum of 48 up to a maximum of 72 markers).
Main statistics for marker alleles that were scored as polymorphic or shared between parents, and that were analyzed for segregation patterns are summarized in
Table 1. A total of 1,122 AFLP marker loci were detected in the F
1 progenies using the selected primer combinations: 870 marker alleles (77.5%) were polymorphic between parental species, whereas 252 marker alleles (22.5%) were shared. Of the polymorphisms, 568 were
S. alba-specific (51% of total marker alleles) and 320 were
S. fragilis-specific (28% of total marker alleles). The difference between the amount of
S. alba-specific and
S. fragilis-specific markers, as assessed by a contingency test, was highly significant (χ
2 = 30.4***, 1 d.f.). Within each species, the gender contribution in terms of maternal and paternal polymorphisms was comparable in both
S. alba (282
vs. 268) and
S. fragilis (132
vs. 142).
Chi-square analyses for various segregation ratios (from 1:1 up to 11:1, presence
vs. absence) were performed to determine segregation patterns of
S. alba and
S. fragilis marker alleles according to allo- and/or auto-tetraploidy (see
Table 1). The most represented segregation ratios ranged from 0.75 to 1.25 as expected for SDMs, with the mean ratios of 0.99 for
S. alba and 1.08 for
S. fragilis markers. Low proportions of marker loci with skewed segregation patterns were observed in both species (6.0% in
S. alba and 4.6% in
S. fragilis).
Table 1.
Information on the inheritance patterns observed for single-dose and multiple-dose marker alleles. Marker types refer to the parental individuals whereas segregating, distorted and monomorphic markers refer to the F1 progenies.
Table 1.
Information on the inheritance patterns observed for single-dose and multiple-dose marker alleles. Marker types refer to the parental individuals whereas segregating, distorted and monomorphic markers refer to the F1 progenies.
| Marker Types | | Segregating Markers (p > 0.05)* | Distorted Markers | Monomorphic Markers |
|---|
| 1:1 ratio | 3:1 ratio | 5:1 ratio | 11:1 ratio |
|---|
| Polymorphic | Total | 292 (33.6) | - | 24 (2.8) | - | 48 (5.5) | 506 (58.1) |
| Species-specific | S. alba | 194 (34.2) | - | 11 (1.9) | - | 34 (6.0) | 329 (57.9) |
| S. fragilis | 98 (32.5) | - | 13 (4.3) | - | 14 (4.6) | 177 (58.6) |
| Gender-specific | Female | 149 (34.0) | - | 11 (2.5) | - | 19 (4.3) | 259 (59.1) |
| Male | 143 (33.1) | - | 13 (3.0) | - | 29 (6.7) | 247 (57.2) |
| Shared | Total | | 22 (8.7) | | 6 (2.7) | 11 (4.4) | 213 (84.5) |
A total of 316 (35.5%) marker alleles of those polymorphic between parents were shown to segregate in the F
1 progenies, with 292 (89.6%) that were inherited as SDMs. In particular, 194
S. alba (81 paternal and 113 maternal) and 98
S. fragilis (62 paternal and 36 maternal) SDMs polymorphic between parents segregated in a 1:1 Mendelian fashion (
p ≤ 0.05), whereas 22 SDMs shared between parents segregated in a 3:1 Mendelian fashion (
p ≤ 0.05). Of the remaining marker loci analyzed, only 29 paternal and 19 maternal were markedly distorted (
p < 0.01). In particular, segregation distortion of marker alleles inherited from
S. alba and
S. fragilis did not differ significantly (χ
2 = 0.419, d.f. = 1). While 1:1 and 3:1 are the only segregation ratios expected for allotetraploid genomes and detectable uniquely with SDMs, additional segregation patterns in autotetraploids may also be identified using DDMs. Of the 870 and 252 marker loci, respectively, polymorphic and shared between parents, only 24 (2.8%) plus 6 (2.7%) showed a segregation attributable to DDMs on the basis of chi-square values. Inheritance patterns observed for marker alleles of these loci correspond to segregation ratios, respectively, of 5:1 and 11:1 expected in duplex by nulliplex and duplex by simplex crosses (see
Table 1).
In conclusion, segregation patterns of most markers support allotetraploidy for willows and only a few markers scored segregation patterns concordant with autotetraploidy.
3.3.Chromosome Pairing Behavior
The chromosome pairing was successfully assessed discriminating between preferential or random models. S. alba-specific and S. fragilis-specific SDMs were ordered in maternal and paternal co-segregation groups and then tested for the linkage phase (i.e., coupling vs. repulsion) in all possible pairwise comparisons among marker loci.
The maternal and paternal SDMs were ordered in 55 co-segregation groups according to the Mather test. SDMs of each pair of co-segregation groups were then tested for the linkage phase on the basis of parental or recombinant patterns. For pairwise combinations of associated genomic loci, the number of marker alleles linked in coupling and the number of marker alleles linked in repulsion were assayed to fit the 1 to 1 expected ratio. Of the
S. alba co-segregation groups, 14 of the 19 maternal linkage groups and 13 of the 16 paternal linkage groups showed non-significant deviations (
p ≤ 0.05), suggesting preferential chromosome pairing. In
S. fragilis, of the co-segregation groups detected, only 3 out of 20 maternal and paternal linkage groups scored significant deviations (
p ≤ 0.01), further supporting preferential chromosome pairing. The linkage phase of marker loci belonging to
S. alba and
S. fragilis co-segregational female and male marker loci groups is reported in
Table 2.
Table 2.
Main statistics about linkage phase (coupling vs. repulsion) of marker loci belonging to S. alba and S. fragilis co-segregational female and male linkage groups. A comparable number of markers linked in coupling and repulsion were found for most linkage groups, suggesting that basic chromosomes pair preferentially as occurs for allotetraploid species with disomic inheritance.
Table 2.
Main statistics about linkage phase (coupling vs. repulsion) of marker loci belonging to S. alba and S. fragilis co-segregational female and male linkage groups. A comparable number of markers linked in coupling and repulsion were found for most linkage groups, suggesting that basic chromosomes pair preferentially as occurs for allotetraploid species with disomic inheritance.
| Species (Gender) | Linkage Groups * | Linkage Phase | Size (cM) | No. loci | Chromosome Pairing (%) |
|---|
| Coupling | Repulsion |
|---|
| S. alba ♀ | 19 | 929 | 728 | 709 | 73 | Preferential (74) |
| S. alba ♂ | 16 | 424 | 374 | 340 | 48 | Preferential (81) |
| S. fragilis ♀ | 6 | 36 | 43 | 98 | 13 | Preferential (100) |
| S. fragilis ♂ | 14 | 156 | 186 | 321 | 33 | Preferential (79) |
In conclusion, a comparable number of markers linked in coupling and repulsion for most co-segregation groups suggests that most basic chromosomes pair preferentially as it happens for allotetraploid species with disomic inheritance.
3.4. Cytological Observations of Chromosome Morphology
Cytological analysis carried out on chromosomes isolated from somatic cells belonging to root tip meristems proved that about half of the chromosome complements at pro-metaphase are actually much more extended and much less colorable than the remaining chromosomes, which appear almost completely contracted and fully colorable (for details, see
Figure 2). The finding of two sets of chromosome complements of 38 elements each characterized by antagonist contraction ratios support the existence of two distinct diploid ancestral genomes in the tetraploid species of
S. alba and
S. fragilis.
A variable morphology for all basic chromosomes is in agreement with allotetraploidy, involving hybridization between divergent diploid species.
3.5. Genomic in situ Hybridization Analysis
Chromosome preparations of pure species and hybrids were hybridized using
S. alba (or
S. fragilis) total genomic DNA as probe labeled with FITC. The GISH experiments showed signals of both
S. fragilis and
S. alba labeled genomes prevalently on contracted chromosomes of the hybrids (
Figure 2, panels A–H). When a pure species (
S. alba or
S. fragilis) was hybridized with the other, signals were still evident on the same set of contracted chromosomes (
Figure 2, panels I–P).
Figure 2.
Main results from genomic in situ hybridization (GISH) experiments. Hybridization signals and patterns obtained using S. alba as probe and S. fragilis as template (panels A-D) and S. fragilis as probe and S. alba as template (panels E-H). Hybridization signals and patterns obtained using S. alba as probe and interspecific hybrid as template (panels I-L) and S. fragilis as probe and interspecific hybrid as template (panels M-P). Both male and female S. alba and S. fragilis accessions are the parental genotypes used for S. alba × S. fragilis and S. fragilis × S. alba crosses, whereas interspecific hybrids refer to two intermediate F1 progeny genotypes. Note the great variation of genomic in situ hybridization signals and patterns in contracted (i.e. large spots) and extended (i.e. small dots) chromosomes across all experiments. Magnifications of specific contracted (C) and extended (E) chromosomes are reported for each GISH-derived karyotype.
Figure 2.
Main results from genomic in situ hybridization (GISH) experiments. Hybridization signals and patterns obtained using S. alba as probe and S. fragilis as template (panels A-D) and S. fragilis as probe and S. alba as template (panels E-H). Hybridization signals and patterns obtained using S. alba as probe and interspecific hybrid as template (panels I-L) and S. fragilis as probe and interspecific hybrid as template (panels M-P). Both male and female S. alba and S. fragilis accessions are the parental genotypes used for S. alba × S. fragilis and S. fragilis × S. alba crosses, whereas interspecific hybrids refer to two intermediate F1 progeny genotypes. Note the great variation of genomic in situ hybridization signals and patterns in contracted (i.e. large spots) and extended (i.e. small dots) chromosomes across all experiments. Magnifications of specific contracted (C) and extended (E) chromosomes are reported for each GISH-derived karyotype.
It is worth emphasizing that, at the tetraploid level (2n = 76 chromosomes), each of the species-specific genomic probes was found to hybridize the other target-species genomic templates across all technical and biological replicates, always involving the 38 contracted chromosomes. This finding strongly suggests that S. alba and S. fragilis share a common diploid ancestor species.
The absence of hybridization signals on extended chromosomes over all GISH experiments suggests that these chromosomes are not homeologous to the
S. alba—
S. fragilis contracted ones and that the genomes of pure species should have a quite different origin. In particular, hybridization signals on contracted chromosomes were usually strong and uniform along one or both arms, whereas the extended chromosomes totally lacked fluorescent signals or revealed only small fluorescent dots (see
Figure 2). The presence of hybridization spots on the extended chromosomes may indicate the occurrence of inter-genomic translocations and re-arrangements that could have taken place through genetic recombination among homeologous chromosomes.
4. Discussion
Polyploidy has long been recognized to be associated with novel morphologies and adaptations, but how genome duplication ultimately translates into novel evolutionary opportunity has remained obscure in many species. Understanding of evolution forces, mechanisms and outcomes at genomic and genetic levels is a prerequisite to developing a fuller appreciation of the role of polyploidy to morphological evolution and ecological adaption of willows.
In general, allotetraploids are those polyploids that have arisen through the processes of interspecific hybridization and chromosome doubling, whereas autotetraploids are those polyploids that have arisen from conspecific parents [
40]. The former are characterized by fixed (
i.e., non-segregating) heterozygosity, resulting from the combination of divergent parental genomes, such that bivalent formation occurs at meiosis and disomic inheritance operates at each locus. In autopolyploids all homologous chromosomes can pair with each other, so they usually exhibit multivalent formation at meiosis and are characterized by polysomic inheritance, because more than two different alleles occur at each locus. Verification of segregation patterns and construction of linkage maps for polyploids can have great implications not only for marker-assisted selection, but also for plant genomics and population genetics in evolution studies. During the last decades, a few theoretical, simulation and explorative studies have been performed to estimate linkage in polyploids [
37,
39,
41,
42,
43,
44,
45].
Here we report genetic segregation and genomic hybridization data, which help shedding light on the genomic structure and genic inheritance in tetraploid willows of the S. alba—S. fragilis complex.
Of the total molecular markers found to be polymorphic between parents, more than half did not segregate as a consequence of multiple-dose alleles at the loci being tested, whereas about one-third (33.6%) were inherited as SDMs. A total of 292 amplified fragment length polymorphism (AFLP) markers, segregating properly in the mapping populations were used in genetic linkage analysis. The fact that most loci scored only 1:1 and 3:1 segregation ratios, as expected for single-dose marker alleles, and very few showed 5:1 or 11:1 segregation ratios, as expected for double-dose marker alleles, is a first strong evidence supporting an allotetraploid nature and disomic inheritance in
Salix spp. Moreover, in
S. alba, 73 maternal and 48 paternal SDMs were mapped to 19 and 16 linkage groups, in
S. fragilis, 13 maternal and 33 paternal SDMs were mapped in 6 and 14 linkage groups (see
Table 2). A comparable number of markers linked in either coupling or repulsion was identified for most co-segregation groups. In polyploid species, chromosome pairing may be preferential, random, or of either type suggesting an allopolyploid, autopolyploid, or mixed genomic origin, respectively. Our findings clearly suggest that most of the chromosomes pair preferentially as occurs in species exhibiting an allotetraploid genome with disomic inheritance. In fact, the proportion of single-dose marker alleles linked in each group according to a coupling or repulsion phase was in agreement with the expected 1:1 ratio.
An important, still open issue concerns the genomic affinity and relationship between
S. alba and
S. fragilis. The fact that of the 1122 marker loci totally identified in the two male and female parents, the vast majority (77.5%) were proven to be polymorphic and only 22.5% were shared between parental species, highlights that
S. alba and
S. fragilis individuals have genotypes deeply differentiated. If it is true that derived species are more rare and contain less alleles, the highly significant difference between
S. alba-specific and
S. fragilis-specific markers found in both parental combinations (on average, 65.3%
vs. 34.7%, respectively) supports the phylogenetic hypothesis primarily formulated by [
24], that
S. fragilis is a derivative of
S. alba-like progenitors. Moreover, the pairwise analysis of co-segregation groups linked in repulsion indicated that most of the willow chromosomes pair preferentially in
S. alba—
S. fragilis interspecific hybrids. This finding strongly supports a low genomic affinity between these two species and consequently suggests that the recombination potential between the two species is limited.
The analysis of DNA fingerprints in
S. alba and
S.
fragilis natural populations showed the presence of diversified groups of willows and absence of genotypes with intermediary characteristics: this result confirms that both species have kept their gene pools well separated and that interspecific hybridization
S. alba ×
S. fragilis does not seem to be a dominating process. A high genetic differentiation and a low gene flow among populations were found by [
14], suggesting a low presence of spontaneous hybridization between these two willow species in natural populations. Recently, [
10] reported that
S. alba,
S. fragilis, and their hybrid
S. ×
rubens are distinctly separated and that only few hybrids and any backcrosses to the parental species are present in mixed stands. Furthermore, as a preliminary investigation, it is worth mentioning that all capsules collected from a subsample of plants belonging to either
S. alba ×
S. fragilis or
S. fragilis ×
S. alba hybrid progenies set seeds unable to germinate (Meneghetti, pers. comm.). If this finding will be confirmed by additional tests, it may be asserted not only that inter-specific plants deriving from both
S. alba ×
S. fragilis crosses and reciprocals form unviable seeds, but also hypothesized that back-crosses of inter-specific hybrids with pure
S. alba or
S.
fragilis species are impossible. As a consequence, the
S. alba—
S. fragilis complex would have a predominance of balanced hybrids, but it could not include fertile introgressed types within natural populations. It is worth mentioning that a massive non-random coalescence failure (
i.
e., incomplete lineage sorting) along with a striking absence of geographical effects (
i.
e., isolation by distance) in the haplotype distributions of many
Salix species was recently discovered and it is likely owed to recent and continual plastid capture events, aided by wide-spread hybridization and long-range seed dispersal, but primarily propelled by one or more trans-species selective sweeps [
9].
Although the analysis of pairing behavior of tetraploids in Salix spp. could help explaining the genomic organization of willow trees, cytological observations in this species complex are difficult to perform owing to the high number and small size of chromosomes. Nevertheless, our cytological observations of chromosomes isolated from root tip meristematic cells proved to be informative and revealed that about half of the chromosome complements at pro-metaphase are more extended than the remaining chromosomes, which are already almost completely contracted. This finding, which is supported by well-documented karyotypes, is very important because it further agrees with allotetraploidy in Salix spp.
In addition, the GISH experiments showed hybridization signals of both
S. fragilis and
S. alba labeled genomes prevalently on contracted chromosomes of the
S. alba ×
S. fragilis hybrids used as template genomes. Surprisingly, when the two pure species were hybridized one with the other on both ways (
i.
e., using
S. alba genome as template and
S. fragilis chromosomes as probe and vice versa), signals were still evident on the same set of contracted chromosomes showing reproducible patterns. The absence of large hybridization spots on non-contracted chromosomes over all experiments, exception made for a few small hybridization dots, suggests that the extended chromosomes are not homeologous to the
S. alba—S. fragilis contracted ones. If this is true, the genomes belonging to the diploid ancestors of the two pure species should have different origin and could share half of the chromosome complements at the tetraploid levels (
Figure 3).
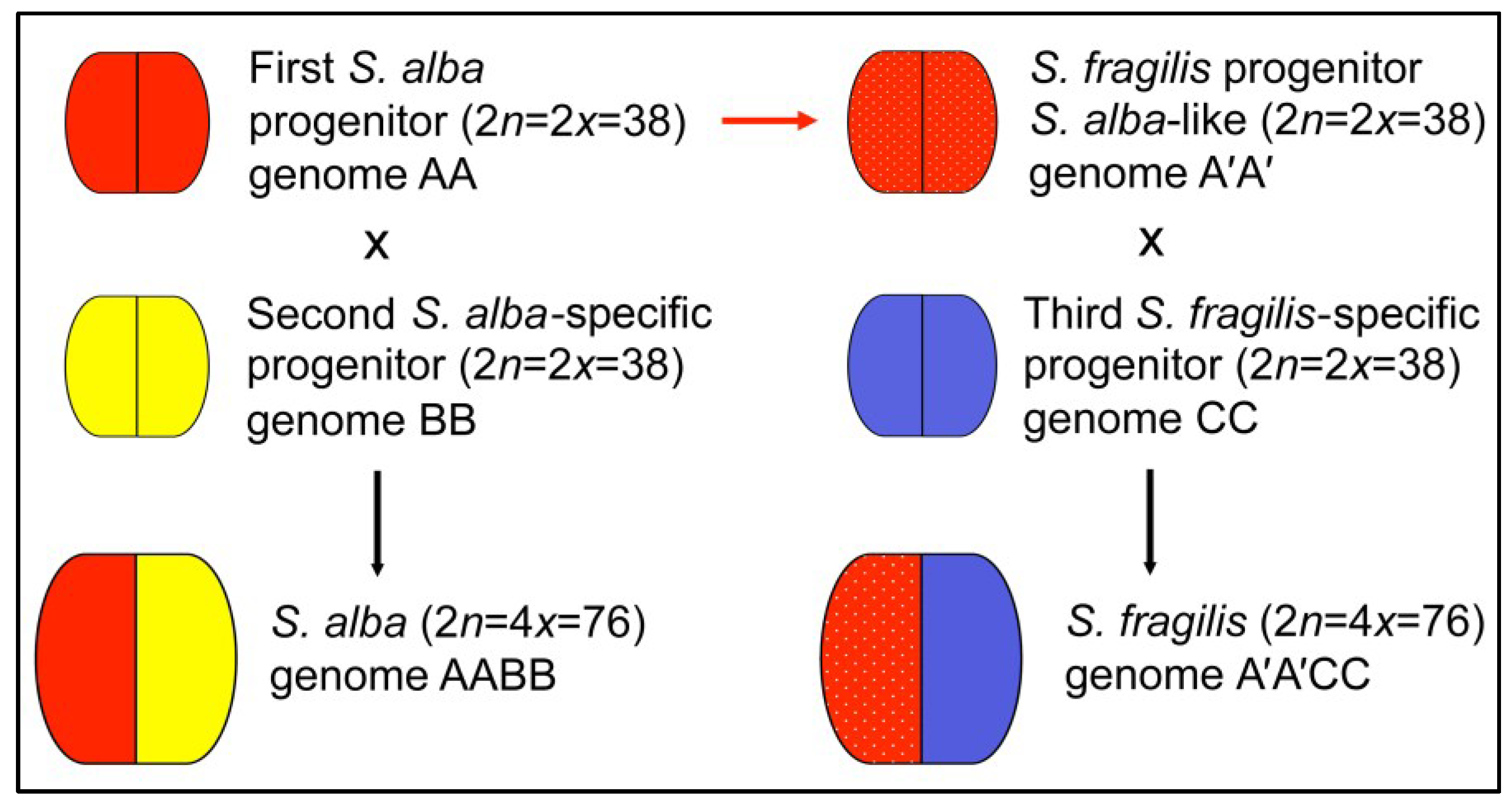
Figure 3.
Working hypothesis relating to the genomic organization of S. alba and S. fragilis genomes, along with their origin and relationship with putative diploid ancestor species. Letters A and A′ refer to the shared or closely related diploid progenitor species of S. alba and S. fragilis whose genome(s) contributed with contracted chromosomes (i.e., first common ancestor), whereas letters B and C indicate the two additional and different progenitor’ genomes which transmitted the extended chromosomes to both S. alba and S. fragilis species (i.e., S. alba-specific and S. fragilis-specific second and third progenitor, respectively).
Figure 3.
Working hypothesis relating to the genomic organization of S. alba and S. fragilis genomes, along with their origin and relationship with putative diploid ancestor species. Letters A and A′ refer to the shared or closely related diploid progenitor species of S. alba and S. fragilis whose genome(s) contributed with contracted chromosomes (i.e., first common ancestor), whereas letters B and C indicate the two additional and different progenitor’ genomes which transmitted the extended chromosomes to both S. alba and S. fragilis species (i.e., S. alba-specific and S. fragilis-specific second and third progenitor, respectively).
On the whole, genomic analyses based on DNA fingerprints and GISH patterns revealed that
S. alba and
S. fragilis are much differentiated genotypically and that
S. fragilis species may have derived from
S. alba-like progenitors. The fact that both
S. alba and
S. fragilis show halved chromosome complements with different contraction ratios in tetraploid genomes and that hybridization signals are evident on the same set of contracted chromosomes and absent in the rest of extended chromosomes, allow us to hypothesize that two sets of non-contracted chromosomes found in both
S. alba and
S. fragilis belong to different diploid ancestors (see
Figure 3). Such explanation of the genomic structure could also explain why interspecific
S. alba ×
S. fragilis hybrids do not set viable seeds.




{kind=link}
{kind=link}
{kind=link}

