
Relationships Among the Bryophytes and Vascular Plants: A Case Study in Deep-Time Reconstruction


Abstract
1. Theoretical Considerations in Deep Phylogenetic Analyses
2. Previous Studies on Early Land Plant Relationships
2.1. Morphological and Early Molecular Studies
2.2. Large-Scale Phylogenomic Studies
3. Properties of Different Types of Characters for Reconstructing Deep Relationships of Land Plants
3.1. Morphology
3.2. Nucleotide Sequences
3.3. Genome Structural Characters
3.4. Fossil Evidence
4. Criteria for Successful Phylogenetic Reconstruction
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mishler, B.D. Deep phylogenetic relationships among “plants“ and their implications for classification. Taxon 2000, 49, 661–683. [Google Scholar] [CrossRef]
- Felsenstein, J. Cases in which parsimony or compatibility methods will be positively misleading. Syst. Zool. 1978, 27, 401–410. [Google Scholar] [CrossRef]
- Mishler, B.D.; Churchill, S.P. A cladistic approach to the phylogeny of the bryophytes. Brittonia 1984, 36, 406–424. [Google Scholar] [CrossRef]
- Parenti, L.R. A phylogenetic analysis of the land plants. Biol. J. Linn. Soc. 1980, 13, 225–242. [Google Scholar] [CrossRef]
- Samigullin, T.K.; Yacentyuk, S.P.; Degtyaryeva, G.V.; Valiehoroman, K.M.; Bobrova, V.K.; Capesius, I.; Martin, W.M.; Troitsky, A.V.; Filin, V.R.; Antonov, A.S. Paraphyly of bryophytes and close relationship of hornworts and vascular plants inferred from analysis of chloroplast rDNA ITS (cpITS) sequences. Arctoa 2002, 11, 31–43. [Google Scholar] [CrossRef]
- Qiu, Y.-L.; Li, L.B.; Wang, B.; Chen, Z.D.; Knoop, V.; Groth-Malonek, M.; Dombrovska, O.; Lee, J.; Kent, L.; Rest, J.; et al. The deepest divergences in land plants inferred from phylogenomic evidence. Proc. Natl. Acad. Sci. USA 2006, 103, 15511–15516. [Google Scholar] [CrossRef]
- Wolf, P.G.; Karol, K.G.; Mandoli, D.F.; Kuehl, J.; Arumuganathan, K.; Ellis, M.W.; Mishler, B.D.; Kelch, D.G.; Olmstead, R.G.; Boore, J.L. The first complete chloroplast genome sequence of a lycophyte, Huperzia lucidula (Lycopodiaceae). Gene 2005, 350, 117–128. [Google Scholar] [CrossRef]
- Chang, Y.; Graham, S.W. Inferring the higher-order phylogeny of mosses (Bryophyta) and relatives using a large, multigene plastid data set. Am. J. Bot. 2011, 98, 839–849. [Google Scholar] [CrossRef]
- Qiu, Y.-L.; Taylor, A.B.; Fine, I.M. A molecular temporal evolutionary framework of land plants and the age of angiosperms. Ann. Mo. Bot. Gard. 2025, in press. [Google Scholar]
- Nishiyama, T.; Wolf, P.G.; Kugita, M.; Sinclair, R.B.; Sugita, M.; Sugiura, C.; Wakasugi, T.; Yamada, K.; Yoshinaga, K.; Yamaguchi, K.; et al. Chloroplast phylogeny indicates that bryophytes are monophyletic. Mol. Biol. Evol. 2004, 21, 1813–1819. [Google Scholar] [CrossRef]
- Ruhfel, B.R.; Gitzendanner, M.A.; Soltis, P.S.; Soltis, D.E.; Burleigh, J.G. From algae to angiosperms–inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evol. Biol. 2014, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Xi, Z.; Goremykin, V.V.; Fong, R.; Mclenachan, P.A.; Novis, P.M.; Davis, C.C.; Penny, D. Streptophyte algae and the origin of land plants revisited using heterogeneous models with three new algal chloroplast genomes. Mol. Biol. Evol. 2014, 31, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Sahu, S.K.; Yang, L.; Liu, Y.; Mu, W.; Liu, X.; Strube, M.L.; Liu, H.; Zhong, B. Comparative analyses of 3,654 plastid genomes unravel insights into evolutionary dynamics and phylogenetic discordance of green plants. Front. Plant Sci. 2022, 13, 808156. [Google Scholar] [CrossRef] [PubMed]
- Turmel, M.; Otis, C.; Lemieux, C. Tracing the evolution of streptophyte algae and their mitochondrial genome. Genome Biol. Evol. 2013, 5, 1817–1835. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cox, C.J.; Wang, W.; Goffinet, B. Mitochondrial phylogenomics of early land plants: Mitigating the effects of saturation, compositional heterogeneity, and codon-usage bias. Syst. Biol. 2014, 63, 862–878. [Google Scholar] [CrossRef] [PubMed]
- Sousa, F.; Civan, P.; Brazao, J.; Foster, P.G.; Cox, C.J. The mitochondrial phylogeny of land plants shows support for Setaphyta under composition-heterogeneous substitution models. PeerJ 2020, 8, e8995. [Google Scholar] [CrossRef] [PubMed]
- Mishler, B.D.; Kelch, D.G. Phylogenomics and early land plant evolution. In Bryophyte Biology, 2nd ed.; Goffinet, B., Shaw, A.J., Eds.; Cambridge University Press: Cambridge, MA, USA, 2009; pp. 173–197. [Google Scholar]
- Harris, B.J.; Clark, J.W.; Schrempf, D.; Szollosi, G.J.; Donoghue, P.C.J.; Hetherington, A.M.; Williams, T.A. Divergent evolutionary trajectories of bryophytes and tracheophytes from a complex common ancestor of land plants. Nat. Ecol. Evol. 2022, 6, 1634–1643. [Google Scholar] [CrossRef]
- Mishler, B.D.; Lewis, L.A.; Buchheim, M.A.; Renzaglia, K.S.; Garbary, D.J.; Delwiche, C.F.; Zechman, F.W.; Kantz, T.S.; Chapman, R.L. Phylogenetic relationships of the “green-algae“ and “bryophytes“. Ann. Mo. Bot. Gard. 1994, 81, 451–483. [Google Scholar] [CrossRef]
- Sousa, F.; Civan, P.; Foster, P.G.; Cox, C.J. The chloroplast land plant phylogeny: Analyses employing better-fitting tree- and site-heterogeneous composition models. Front. Plant Sci. 2020, 11, 1062. [Google Scholar] [CrossRef] [PubMed]
- Renzaglia, K.S.; Duff, R.J.; Nickrent, D.L.; Garbary, D.J. Vegetative and reproductive innovations of early land plants: Implications for a unified phylogeny. Philos. Trans. R. Soc. Lond. Ser. B 2000, 355, 769–793. [Google Scholar] [CrossRef]
- Hedderson, T.A.; Chapman, R.L.; Rootes, W.L. Phylogenetic relationships of bryophytes inferred from nuclear-encoded rRNA gene sequences. Plant Syst. Evol. 1996, 200, 213–224. [Google Scholar] [CrossRef]
- Hedderson, T.A.; Chapman, R.; Cox, C.J. Bryophytes and the origins and diversification of land plants: New evidence from molecules. In Bryology for the Twenty-First Century; Bates, J.W., Ashton, N.W., Duckett, J.G., Eds.; W.S. Maney & Son Ltd.: Leeds, UK, 1998; pp. 65–77. [Google Scholar]
- Nickrent, D.L.; Parkinson, C.L.; Palmer, J.D.; Duff, R.J. Multigene phylogeny of land plants with special reference to bryophytes and the earliest land plants. Mol. Biol. Evol. 2000, 17, 1885–1895. [Google Scholar] [CrossRef]
- Wickett, N.J.; Mirarab, S.; Nguyen, N.; Warnow, T.; Carpenter, E.; Matasci, N.; Ayyampalayam, S.; Barker, M.S.; Burleigh, J.G.; Gitzendanner, M.A.; et al. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Acad. Sci. USA 2014, 111, E4859–E4868. [Google Scholar] [CrossRef]
- Sousa, F.d.; Foster, P.G.; Donoghue, P.C.J.; Schneider, H.; Cox, C.J. Nuclear protein phylogenies support the monophyly of the three bryophyte groups (Bryophyta Schimp.). New Phytol. 2019, 222, 565–575. [Google Scholar] [CrossRef]
- One Thousand Plant Transcriptomes Initiative. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 2019, 574, 679–685. [Google Scholar] [CrossRef]
- Goremykin, V.V.; Hellwig, F.H. Evidence for the most basal split in land plants dividing bryophyte and tracheophyte lineages. Plant Syst. Evol. 2005, 254, 93–103. [Google Scholar] [CrossRef]
- Gitzendanner, M.A.; Soltis, P.S.; Wong, G.K.-S.; Ruhfel, B.R.; Soltis, D.E. Plastid phylogenomic analysis of green plants: A billion years of evolutionary history. Am. J. Bot. 2018, 105, 291–301. [Google Scholar] [CrossRef]
- Puttick, M.N.; Morris, J.L.; Williams, T.A.; Cox, C.J.; Edwards, D.; Kenrick, P.; Pressel, S.; Wellman, C.H.; Schneider, H.; Pisani, D.; et al. The Interrelationships of land plants and the nature of the ancestral embryophyte. Curr. Biol. 2018, 28, 733–745. [Google Scholar] [CrossRef]
- Harris, B.J.; Harrison, C.J.; Hetherington, A.M.; Williams, T.A. Phylogenomic evidence for the monophyly of bryophytes and the reductive evolution of stomata. Curr. Biol. 2020, 30, 2001–2012. [Google Scholar] [CrossRef]
- Su, D.; Yang, L.; Shi, X.; Ma, X.; Zhou, X.; Hedges, S.B.; Zhong, B. Large-scale phylogenomic analyses reveal the monophyly of bryophytes and Neoproterozoic origin of land plants. Mol. Biol. Evol. 2021, 38, 3332–3344. [Google Scholar] [CrossRef]
- Karol, K.G.; Arumuganathan, K.; Boore, J.L.; Duffy, A.M.; Everett, K.D.E.; Hall, J.D.; Hansen, S.K.; Kuehl, J.V.; Mandoli, D.F.; Mishler, B.D.; et al. Complete plastome sequences of Equisetum arvense and Isoetes flaccida: Implications for phylogeny and plastid genome evolution of early land plant lineages. BMC Evol. Biol. 2010, 10, 321. [Google Scholar] [CrossRef]
- Lemieux, C.; Otis, C.; Turmel, M. Comparative chloroplast genome analyses of streptophyte green algae uncover major structural alterations in the Klebsormidiophyceae, Coleochaetophyceae and Zygnematophyceae. Front. Plant Sci. 2016, 7, 679. [Google Scholar] [CrossRef]
- Kenrick, P.; Crane, P.R. The Origin and Early Diversification of Land Plants: A Cladistic Study; Smithsonian Institution Press: Washington, DC, USA, 1997; p. 441. [Google Scholar]
- Duff, R.J.; Nickrent, D.L. Phylogenetic relationships of land plants using mitochondrial small-subunit rDNA sequences. Am. J. Bot. 1999, 86, 372–386. [Google Scholar] [CrossRef]
- Lewis, L.A.; Mishler, B.D.; Vilgalys, R. Phylogenetic relationships of the liverworts (Hepaticae), a basal embryophyte lineage, inferred from nucleotide sequence data of the chloroplast gene rbcL. Mol. Phylogen. Evol. 1997, 7, 377–393. [Google Scholar] [CrossRef]
- Mishler, B.D. The logic of the data matrix in phylogenetic analysis. In Parsimony, Phylogeny, and Genomics; Albert, V.A., Ed.; Oxford University Press: Oxford, UK, 2005; pp. 57–70. [Google Scholar]
- Garbary, D.J.; Renzaglia, K.S.; Duckett, J.G. The phylogeny of land plants—A cladistic analysis based on male gametogenesis. Plant Syst. Evol. 1993, 188, 237–269. [Google Scholar] [CrossRef]
- Chapman, R.L.; Buchheim, M.A. Green algae and the evolution of land plants: Inferences from nuclear-encoded rRNA gene sequences. Biosystems 1992, 28, 127–137. [Google Scholar] [CrossRef]
- Soltis, P.S.; Soltis, D.E.; Wolf, P.G.; Nickrent, D.L.; Chaw, S.; Chapman, R.L. The phylogeny of land plants inferred from 18S rDNA sequences: Pushing the limits of rDNA signal? Mol. Biol. Evol. 1999, 16, 1774–1784. [Google Scholar] [CrossRef]
- Manhart, J.R. Phylogenetic analysis of green plant rbcL sequences. Mol. Phylogen. Evol. 1994, 3, 114–127. [Google Scholar] [CrossRef]
- Kallersjo, M.; Farris, J.S.; Chase, M.W.; Bremer, B.; Fay, M.F.; Humphries, C.J.; Petersen, G.; Seberg, O.; Bremer, K. Simultaneous parsimony jackknife analysis of 2538 rbcL DNA sequences reveals support for major clades of green plants, land plants, seed plants and flowering plants. Plant Syst. Evol. 1998, 213, 259–287. [Google Scholar] [CrossRef]
- Malek, O.; Lattig, K.; Hiesel, R.; Brennicke, A.; Knoop, V. RNA editing in bryophytes and a molecular phylogeny of land plants. EMBO J. 1996, 15, 1403–1411. [Google Scholar] [CrossRef]
- Manhart, J.R.; Palmer, J.D. The gain of two chloroplast transfer-RNA introns marks the green algal ancestors of land plants. Nature 1990, 345, 268–270. [Google Scholar] [CrossRef]
- Raubeson, L.A.; Jansen, R.K. Chloroplast DNA evidence on the ancient evolutionary split in vascular land plants. Science 1992, 255, 1697–1699. [Google Scholar] [CrossRef]
- Qiu, Y.-L.; Cho, Y.R.; Cox, J.C.; Palmer, J.D. The gain of three mitochondrial introns identifies liverworts as the earliest land plants. Nature 1998, 394, 671–674. [Google Scholar] [CrossRef]
- Kelch, D.G.; Driskell, A.; Mishler, B.D. Inferring phylogeny using genomic characters: A case study using land plant plastomes. In Molecular Systematics of Bryophytes; Goffinet, B., Hollowell, V., Magill, R., Eds.; Missouri Botanical Garden Press: St. Louis, MO, USA, 2004; pp. 3–11. [Google Scholar]
- Soltis, D.E.; Soltis, P.S.; Mort, M.E.; Chase, M.W.; Savolainen, V.; Hoot, S.B.; Morton, C.M. Inferring complex phylogenies using parsimony: An empirical approach using three large DNA data sets for angiosperms. Syst. Biol. 1998, 47, 32–42. [Google Scholar] [CrossRef]
- Hillis, D.M. Inferring complex phylogenies. Nature 1996, 383, 130–131. [Google Scholar] [CrossRef]
- Lake, J.A. Reconstructing evolutionary trees from DNA and protein sequences: Paralinear distances. Proc. Natl. Acad. Sci. USA 1994, 91, 1455–1459. [Google Scholar] [CrossRef]
- Lockhart, P.J.; Steel, M.A.; Hendy, M.D.; Penny, D. Recovering evolutionary trees under a more realistic model of sequence evolution. Mol. Biol. Evol. 1994, 11, 605–612. [Google Scholar]
- Mirarab, S.; Reaz, R.; Bayzid, M.S.; Zimmermann, T.; Swenson, M.S.; Warnow, T. ASTRAL: Genome-scale coalescent-based species tree estimation. Bioinformatics 2014, 30, i541–i548. [Google Scholar] [CrossRef]
- Steel, M.; Rodrigo, A. Maximum likelihood supertrees. Syst. Biol. 2008, 57, 243–250. [Google Scholar] [CrossRef]
- Szöllosi, G.J.; Tannier, E.; Lartillot, N.; Daubin, V. Lateral gene transfer from the dead. Syst. Biol. 2013, 62, 386–397. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. STRIDE: Species tree root inference from gene duplication events. Mol. Biol. Evol. 2017, 34, 3267–3278. [Google Scholar] [CrossRef] [PubMed]
- Sack, F.D. The development and structure of stomata. In Stomatal Function; Zieger, E., Farquhar, G.D., Cowan, I.R., Eds.; Stanford University Press: Stanford, CA, USA, 1987; pp. 59–89. [Google Scholar]
- Mishler, B.D. Cladistic analysis of molecular and morphological data. Am. J. Phys. Anthropol. 1994, 94, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. Maximum-likelihood estimation of phylogeny from DNA sequences when substitution rates differ over sites. Mol. Biol. Evol. 1993, 10, 1396–1401. [Google Scholar] [PubMed]
- Pagel, M.; Meade, A. A phylogenetic mixture model for detecting pattern-heterogeneity in gene sequence or character-state data. Syst. Biol. 2004, 53, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Blanquart, S.; Lartillot, N. A site- and time-heterogeneous model of amino acid replacement. Mol. Biol. Evol. 2008, 25, 842–858. [Google Scholar] [CrossRef] [PubMed]
- Philippe, H.; de Vienne, D.M.; Ranwez, V.; Roure, B.; Baurain, D.; Delsuc, F. Pitfalls in supermatrix phylogenomics. Eur. J. Taxon. 2017, 283, 1–25. [Google Scholar] [CrossRef]
- Thorne, J.L.; Goldman, N.; Jones, D.T. Combining protein evolution and secondary structure. Mol. Biol. Evol. 1996, 13, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Philippe, H. A bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 2004, 21, 1095–1109. [Google Scholar] [CrossRef]
- Goldman, N.; Thorne, J.L.; Jones, D.T. Assessing the impact of secondary structure and solvent accessibility on protein evolution. Genetics 1998, 149, 445–458. [Google Scholar] [CrossRef]
- Redmond, A.K.; McLysaght, A. Evidence for sponges as sister to all other animals from partitioned phylogenomics with mixture models and recoding. Nat. Commun. 2021, 12, 1783. [Google Scholar] [CrossRef]
- Dunn, C.W.; Hejnol, A.; Matus, D.Q.; Pang, K.; Browne, W.E.; Smith, S.A.; Seaver, E.; Rouse, G.W.; Obst, M.; Edgecombe, G.D.; et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 2008, 452, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Laumer, C.E.; Fernández, R.; Lemer, S.; Combosch, D.; Kocot, K.M.; Riesgo, A.; Andrade, S.C.S.; Sterrer, W.; Sørensen, M.V.; Giribet, G. Revisiting metazoan phylogeny with genomic sampling of all phyla. Proc. R. Soc. B 2019, 286, 20190831. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-C.; Minh, B.Q.; Susko, E.; Roger, A.J. Modeling site heterogeneity with posterior mean site frequency profiles accelerates accurate phylogenomic estimation. Syst. Biol. 2018, 67, 216–235. [Google Scholar] [CrossRef]
- Leebens-Mack, J.; Raubeson, L.A.; Cui, L.Y.; Kuehl, J.V.; Fourcade, M.H.; Chumley, T.W.; Boore, J.L.; Jansen, R.K.; de Pamphilis, C.W. Identifying the basal angiosperm node in chloroplast genome phylogenies: Sampling one’s way out of the felsenstein zone. Mol. Biol. Evol. 2005, 22, 1948–1963. [Google Scholar] [CrossRef]
- Qiu, Y.-L.; Li, L.; Wang, B.; Xue, J.-Y.; Hendry, T.A.; Li, R.; Brown, J.W.; Liu, Y.; Hudson, G.T.; Chen, Z. Angiosperm phylogeny inferred from sequences of four mitochondrial genes. J. Syst. Evol. 2010, 48, 391–425. [Google Scholar] [CrossRef]
- Xi, Z.; Liu, L.; Rest, J.S.; Davis, C.C. Coalescent versus concatenation methods and the placement of Amborella as sister to water lilies. Syst. Biol. 2014, 63, 919–932. [Google Scholar] [CrossRef]
- Ohno, S. Evolution by Gene Duplication; Springer: New York, NY, USA, 1970. [Google Scholar]
- Wendel, J.F. Genome evolution in polyploids. Plant Mol. Biol. 2000, 42, 225–249. [Google Scholar] [CrossRef]
- Kingman, J.F.C. On the genealogy of large populations. J. Appl. Probab. 1982, 19, 27–43. [Google Scholar] [CrossRef]
- Hudson, R.R. Gene genealogies and the coalescent process. In Oxford Surveys in Evolutionary Biology; Futuyma, D.J., Antonovics, J.D., Eds.; Oxford University Press: New York, NY, USA, 1990; pp. 1–44. [Google Scholar]
- Maddison, W.P. Gene trees in species trees. Syst. Biol. 1997, 46, 523–536. [Google Scholar] [CrossRef]
- Rannala, B.; Yang, Z. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics 2003, 164, 1645–1656. [Google Scholar] [CrossRef]
- Adams, R.H.; Castoe, T.A. Statistical binning leads to profound model violation due to gene tree error incurred by trying to avoid gene tree error. Mol. Phylogen. Evol. 2019, 134, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Naser-Khdour, S.; Minh, B.Q.; Zhang, W.; Stone, E.A.; Lanfear, R. The prevalence and impact of model violations in phylogenetic analysis. Genome Biol. Evol. 2019, 11, 3341–3352. [Google Scholar] [CrossRef]
- Zitnik, M.; Sosic, R.; Feldman, M.W.; Leskovec, J. Evolution of resilience in protein interactomes across the tree of life. Proc. Natl. Acad. Sci. USA 2019, 116, 4426–4433. [Google Scholar] [CrossRef]
- Arabidopsis Interactome Mapping Consortium. Evidence for network evolution in an Arabidopsis interactome map. Science 2011, 333, 601–607. [Google Scholar] [CrossRef]
- Fitch, W.M. Distinguishing homologous from analogous proteins. Syst. Zool. 1970, 19, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Al Jewari, C.; Baldauf, S.L. An excavate root for the eukaryote tree of life. Sci. Adv. 2023, 9, eade4973. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Zhang, S.; Paterson, A.H. Pervasive genome duplications across the plant tree of life and their links to major evolutionary innovations and transitions. Comput. Struct. Biotechnol. J. 2023, 20, 3248–3256. [Google Scholar] [CrossRef]
- Popper, K. The Logic of Scientific Discovery; Routledge Classics: London, UK; New York, NY, USA, 2002. [Google Scholar]
- Boore, J.L.; Lavrov, D.V.; Brown, W.M. Gene translocation links insects and crustaceans. Nature 1998, 392, 667–668. [Google Scholar] [CrossRef]
- Macey, J.R.; Schulte, J.A.I.; Larson, A. Evolution and phylogenetic information content of mitochondrial genomic structural features illustrated with acrodont lizards. Syst. Biol. 2000, 49, 257–277. [Google Scholar] [CrossRef]
- Zhao, T.; Zwaenepoel, A.; Xue, J.-Y.; Kao, S.-M.; Li, Z.; Schranz, M.E.; Van de Peer, Y. Whole-genome microsynteny-based phylogeny of angiosperms. Nat. Commun. 2021, 12, 3498. [Google Scholar] [CrossRef]
- Spang, A.; Saw, J.H.; Jørgensen, S.L.; Zaremba-Niedzwiedzka, K.; Martijn, J.; Lind, A.E.; van Eijk, R.; Schleper, C.; Guy, L.; Ettema, T.J.G. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 2015, 521, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J. Gene trees and species trees: Molecular systematics as one-character taxonomy. Syst. Bot. 1992, 17, 144–163. [Google Scholar] [CrossRef]
- Miyamoto, M.M.; Fitch, W.M. Testing species phylogenies and phylogenetic methods with congruence. Syst. Biol. 1995, 44, 64–76. [Google Scholar] [CrossRef]
- Bell, D.; Lin, Q.; Gerelle, W.K.; Joya, S.; Chang, Y.; Taylor, Z.N.; Rothfels, C.J.; Larsson, A.; Villarreal, J.C.; Li, F.-W.; et al. Organellomic data sets confirm a cryptic consensus on (unrooted) land-plant relationships and provide new insights into bryophyte molecular evolution. Am. J. Bot. 2019, 107, 91–115. [Google Scholar] [CrossRef] [PubMed]
- Gray, J. Major Paleozoic land plant evolutionary bio-events. Palaeogeogr. Palaeoclimatol. Palaeoecol. 1993, 104, 153–169. [Google Scholar] [CrossRef]
- Strother, P.K.; Taylor, W.A. A fossil record of spores before sporophytes. Diversity 2024, in press. [Google Scholar]
- Edwards, D.; Morris, J.L.; Richardson, J.B.; Kenrick, P. Cryptospores and cryptophytes reveal hidden diversity in early land floras. New Phytol. 2014, 202, 50–78. [Google Scholar] [CrossRef] [PubMed]
- Wellman, C.H.; Osterloff, P.L.; Mohiuddin, U. Fragments of the earliest land plants. Nature 2003, 425, 282–285. [Google Scholar] [CrossRef]
- Edwards, D.; Wellman, C. Embryophytes on land: The Ordovician to Lochkovian (Lower Devonian) record. In Plants Invade the Land: Evolutionary & Environmental Perspectives; Gensel, P.G., Edwards, D., Eds.; Columbia University Press: New York, NY, USA, 2001; pp. 3–28. [Google Scholar]
- Labandeira, C.C.; Tremblay, S.L.; Bartowski, K.E.; Hernick, L.V. Middle Devonian liverwort herbivory and antiherbivor defence. New Phytol. 2014, 202, 247–258. [Google Scholar] [CrossRef]
- Hernick, L.V.; Landing, E.; Bartowski, K.E. Earth’s oldest liverworts—Metzgeriothallus sharonae sp. nov. from the Middle Devonian (Giventian) of eastern New York, USA. Rev. Palaeobot. Palynol. 2008, 148, 154–162. [Google Scholar] [CrossRef]
- Hueber, F.M. Hepaticites devonicus: A new fossil liverwort from the Devonian of New York. Ann. Mo. Bot. Gard. 1961, 48, 125–131. [Google Scholar] [CrossRef]
- Gray, J.; Massa, D.; Boucot, A.J. Caradocian land plant microfossils from Libya. Geology 1982, 10, 197–201. [Google Scholar] [CrossRef]
- Strother, P.K.; Al-Hajri, S.; Traverse, A. New evidence for land plants from the lower Middle Ordovician of Saudia Arabia. Geology 1996, 24, 55–58. [Google Scholar] [CrossRef]
- Rubinstein, C.V.; Gerrienne, P.; de la Puente, G.S.; Astini, R.A.; Steemans, P. Early Middle Ordovician evidence for land plants in Argentina (eastern Gondwana). New Phytol. 2010, 188, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Strother, P.K.; Foster, C. A fossil record of land plant origins from charophyte algae. Science 2021, 373, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Strother, P.K.; Traverse, A.; Vecoli, M. Cryptospores from the Hanadir Shale Member of the Qasim Formation, Ordovician (Darriwilian) of Saudi Arabia: Taxonomy and systematics. Rev. Palaeobot. Palynol. 2015, 212, 97–110. [Google Scholar] [CrossRef]
- Wang, K.; Xu, H.-H.; Yin, L.-M. A palynological assemblage from the Cambrian (Series 2, Stage 4) of Shandong Province, China, and its implications to the transition from algae to land plants. Rev. Palaeobot. Palynol. 2022, 301, 104645. [Google Scholar] [CrossRef]
- Haeckel, E. Generelle Morphologie der Organismen. Zweiter Band: Allgemeine Entwickelungsgeschichte; Georg Reimer: Berlin, Germany, 1866. [Google Scholar]
- Mayr, E. The Growth of Biological Thought: Diversity, Evolution, and Inheritance; The Belknap Press of Harvard University Press: Cambridge, MA, USA, 1982; p. 974. [Google Scholar]
- Hennig, W. Phylogenetic Systematics; University of Illinois Press: Urbana, IL, USA, 1966. [Google Scholar]
- Pamilo, P.; Nei, M. Relationships between gene trees and species trees. Mol. Biol. Evol. 1988, 5, 568–583. [Google Scholar] [PubMed]
- Atchley, W.R.; Fitch, W.M. Gene trees and the origins of inbreed mice. Science 1991, 254, 554–558. [Google Scholar] [CrossRef]
- Leigh, J.W.; Lapointe, F.-J.; Lopez, P.; Bapteste, E. Evaluating phylogenetic congruence in the post-genomic era. Genome Biol. Evol. 2011, 3, 571–587. [Google Scholar] [CrossRef]
- Keating, J.N.; Garwood, R.J.; Sansom, R.S. Phylogenetic congruence, conflict and consilience between molecular and morphological data. BMC Ecol. Evol. 2023, 23, 30. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L.; Olsen, O.J.; Waddell, P.J.; Hillis, D.M. Phylogenetic inference. In Molecular Systematics, 2nd ed.; Hillis, D.M., Moritz, C., Mable, B.K., Eds.; Sinauer: Sunderland, MA, USA, 1996; pp. 407–514. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Tree Topology | Data Source | Characters | OTUs ** Outgroup Liverworts Mosses Hornworts Vascular Plants | Method(s) of Analysis | Reference |
|---|---|---|---|---|---|
| Figure 2A | morphology | 17 characters | O: 2 L: 1 M: 1 H: 1 V: 14 | MP | [4] |
| chloroplast, one DNA region: rDNA ITS | nucleotide: all positions | O: 2 L: 14 M: 20 H: 4 V: 7 | ML, MP, and NJ | [5] | |
| chloroplast, 67 genes | nucleotide: all positions | O: 6 L: 1 M: 1 H: 1 V: 27 | ML and MP | [6] | |
| nucleotide: 1st–3rd positions | |||||
| nucleotide: 2nd–3rd positions | |||||
| nucleotide: 3rd positions | MP | ||||
| multigene (cp-atpB, cp-rbcL, cp-SSU cp-LSU, mt-LSU, and nu-18S) | nucleotide: all positions | O: 9 L: 47 M: 43 H: 6 V: 88 | ML and MP | [6] | |
| chloroplast, 73 genes | nucleotide: all positions | O: 1 L: 1 M: 1 H: 1 V: 16 | ML and MP | [7] | |
| nucleotide: 1st–2nd positions | |||||
| chloroplast, 17 genes and associated non-coding regions | nucleotide: all positions | O: 5 L: 6 M: 14 H: 3 V: 15 | ML and MP | [8] | |
| multigene (cp-atpB, cp-rbcL, mt-atp1, mt-nad5, and nu18S) | nucleotide: all positions | O: 23 L: 57 M: 56 H: 5 V: 492 | ML | [9] | |
| chloroplast, 51 genes | nucleotide: 1st–2nd positions | O: 4 L: 1 M: 1 H: 1 V: 13 | MP | [10] | |
| chloroplast, 78 genes | nucleotide: all positions | O: 32 L: 3 M: 2 H: 1 V: 322 | ML | [11] | |
| nucleotide: RY-coded sequences | |||||
| chloroplast, 72 genes | nucleotide: all positions, the 45,879 matrix | O: 10 L: 2 M: 2 H: 1 V: 15 | ML (RAxML) | [12] | |
| chloroplast, 72 genes | nucleotide: all positions | O: 82 L: 6 M: 8 H: 2 V: 3556 | ML | [13] | |
| mitochondrion, 40 genes | amino acid (the 9013 and 7210 matrices) | O: 13 L: 3 M: 2 H: 2 V: 5 | BI | [14] | |
| mitochondrion, 41 genes | nucleotide: all positions under a composition heterogeneous model | O: 5 L: 6 M: 19 H: 2 V: 28 | BI | [15] | |
| amino acid: under a composition homogeneous model | ML | ||||
| mitochondrion, 36 genes | nucleotide: codon-degenerate data under a homogeneous composition model | O: 5 L: 4 M: 5 H: 2 V: 10 | BI | [16] | |
| amino acid under a homogeneous composition model | |||||
| chloroplast, 47 intron positions, presence/absence, inversion, translocation, or duplication of gene(s) | genome structural characters | O: 2 L: 1 M: 2 H: 1 V: 22 | MP | [17] | |
| nucleus, 18,560 orthogroups | gene duplication data | O: 0 L: 1 M: 4 H: 2 V: 17 | STRIDE | [18] | |
| Figure 2B | morphology | 37 characters | O: 2 L: 1 M: 1 H: 1 V: 1 | MP | [3] |
| morphology | 113 characters | O: 4 L: 7 M: 5 H: 2 V: 9 | MP | [19] | |
| Figure 2F | chloroplast, 72 genes | nucleotide: all positions, the 45,879 matrix | O: 10 L: 2 M: 2 H: 1 V: 15 | ML (nhPhyML) | [12] |
| chloroplast, 83 genes | nucleotide: all positions | O: 13 L: 2 M: 2 H: 2 V: 11 | ML | [20] | |
| mitochondrion, 41 genes | nucleotide: all positions under a homogeneous composition model | O: 5 L: 6 M: 19 H: 2 V: 28 | ML | [15] | |
| mitochondrion, 36 genes | nucleotide: all positions under a homogeneous composition model | O: 5 L: 4 M: 5 H: 2 V: 10 | BI | [16] | |
| nucleus, 18,560 orthogroups | gene duplication data | O: 0 L: 1 M: 4 H: 2 V: 17 | ALE | [18] | |
| Figure 2G | mitochondrion, 36 genes | nucleotide: all positions under a heterogeneous composition model | O: 5 L: 4 M: 5 H: 2 V: 10 | BI | [16] |
| Figure 2M | morphology | 125 characters | O: 1 L: 7 M: 5 H: 3 V: 7 | MP | [21] |
| nucleus, 18S | nucleotide: all positions | O: 3 L: 7 M: 9 H: 2 V: 6 | MP | [22] | |
| nucleus, 18S | nucleotide: all positions | O: 7 L: 8 M: 20 H: 2 V: 31 | MP | [23] | |
| multigene (cp-SSU, mt-SSU, nu-SSU, cp-rbcL) | nucleotide: all positions | O: 2 L: 2 M: 3 H: 2 V: 21 | ML | [24] | |
| nucleotide: all positions with rate variation accounted for | ML | ||||
| nucleotide: RY-coded sequences for cp-rbcL 3rd positions | MP | ||||
| mitochondrion, 36 genes | nucleotide, codon-degenerate data under a heterogeneous composition model | O: 5 L: 4 M: 5 H: 2 V: 10 | BI | [16] | |
| amino acid under heterogeneous composition model | |||||
| nucleus, 674 genes | nucleotide, 1st–2nd positions | O: 22 L: 6 M: 11 H: 2 V: 62 | ML | [25] | |
| nucleus, 100 genes | nucleotide: all positions | O: 6 L: 4 M: 4 H: 2 V: 10 | ML | [26] | |
| nucleus, 410 genes | nucleotide: all positions | O: 235 L: 22 M: 42 H: 10 V: 869 | ML | [27] | |
| nucleus, 18,560 orthogroups | gene duplication data | O: 0 L: 1 M: 4 H: 2 V: 17 | ALE | [18] | |
| Figure 2N | morphology | 72 characters (spermatogenesis) | O: 1 L: 4 M: 3 H: 2 V: 12 | MP | [21] |
| chloroplast, 51 genes | amino acid | O: 4 L: 1 M: 1 H: 1 V: 13 | ML | [10] | |
| chloroplast, 83 genes | amino acid | O: 13 L: 2 M: 2 H: 2 V: 11 | BI | [20] | |
| chloroplast, 57 genes | nucleotide: 1st–2nd positions (LogDet correction for compositional bias) | O: 1 L: 1 M: 1 H: 1 V: 14 | NJ | [28] | |
| amino acid (paralinear correction for compositional bias) | |||||
| chloroplast, 72 genes | nucleotide: 1st–2nd positions | O: 82 L: 6 M: 8 H: 2 V: 3556 | ML | [13] | |
| nucleus, 424 genes | gene trees | O: 22 L: 6 M: 11 H: 2 V: 62 | AA | [25] | |
| chloroplast, 78 genes | amino acid | O: 275 L: 28 M: 46 H: 11 V: 1519 | ML | [29] | |
| nucleus, 852 genes | gene trees | O: 22 L: 6 M: 11 H: 2 V: 62 | BSI | [30] | |
| nucleus, 100 genes | nucleotide: codon-degenerate data | O: 6 L: 4 M: 4 H: 2 V: 10 | ML | [26] | |
| amino acid | ML | ||||
| BI | |||||
| nucleus, 410 gene families | gene trees | O: 235 L: 22 M: 42 H: 10 V: 869 | AA | [27] | |
| nucleus, 151 orthologs | amino acid | O: 19 L: 23 M: 20 H: 7 V: 93 | ML | [31] | |
| nucleus, 1440 genes | nucleotide: codon degenerate data | O: 20 L: 18 M: 44 H: 9 V: 32 | BI and ML | [32] | |
| amino acid | |||||
| gene trees | AA | ||||
| nucleus, 160 genes | amino acid | O: 23 L: 24 M: 20 H: 9 V: 101 | BI and ML | [18] | |
| gene trees | AA | ||||
| nucleus, 18,560 orthogroups | gene duplication data | O: 0 L: 1 M: 4 H: 2 V: 17 | STRIDE | ||
| ALE | |||||
| Figure 2O | chloroplast, 67 genes | nucleotide: 1st positions | O: 6 L: 1 M: 1 H: 1 V: 27 | ML | [6] |
| nucleotide: 1st–2nd positions | ML and MP | ||||
| chloroplast, 51 genes | nucleotide: fourfold degenerate sites (LogDet correction for compositional bias) | O: 4 L: 1 M: 1 H: 1 V: 13 | NJ | [10] | |
| chloroplast, 49 genes | nucleotide: all positions | O: 6 L: 1 M: 2 H: 1 V: 33 | BI and ML | [33] | |
| chloroplast, 78 genes | nucleotide: 1st–2nd positions | O: 32 L: 3 M: 2 H: 1 V: 322 | ML | [11] | |
| amino acid | |||||
| chloroplast, 72 genes | nucleotide: the 45,879 matrix | O: 10 L: 2 M: 2 H: 1 V: 15 | BI (PhyloBayes) | [12] | |
| chloroplast, 88 genes | nucleotide: all positions | O: 18 L: 1 M: 2 H: 1 V: 6 | BI and ML | [34] | |
| amino acid | |||||
| nucleus, 142 genes | gene trees | O: 22 L: 6 M: 11 H: 2 V: 62 | BSI | [30] | |
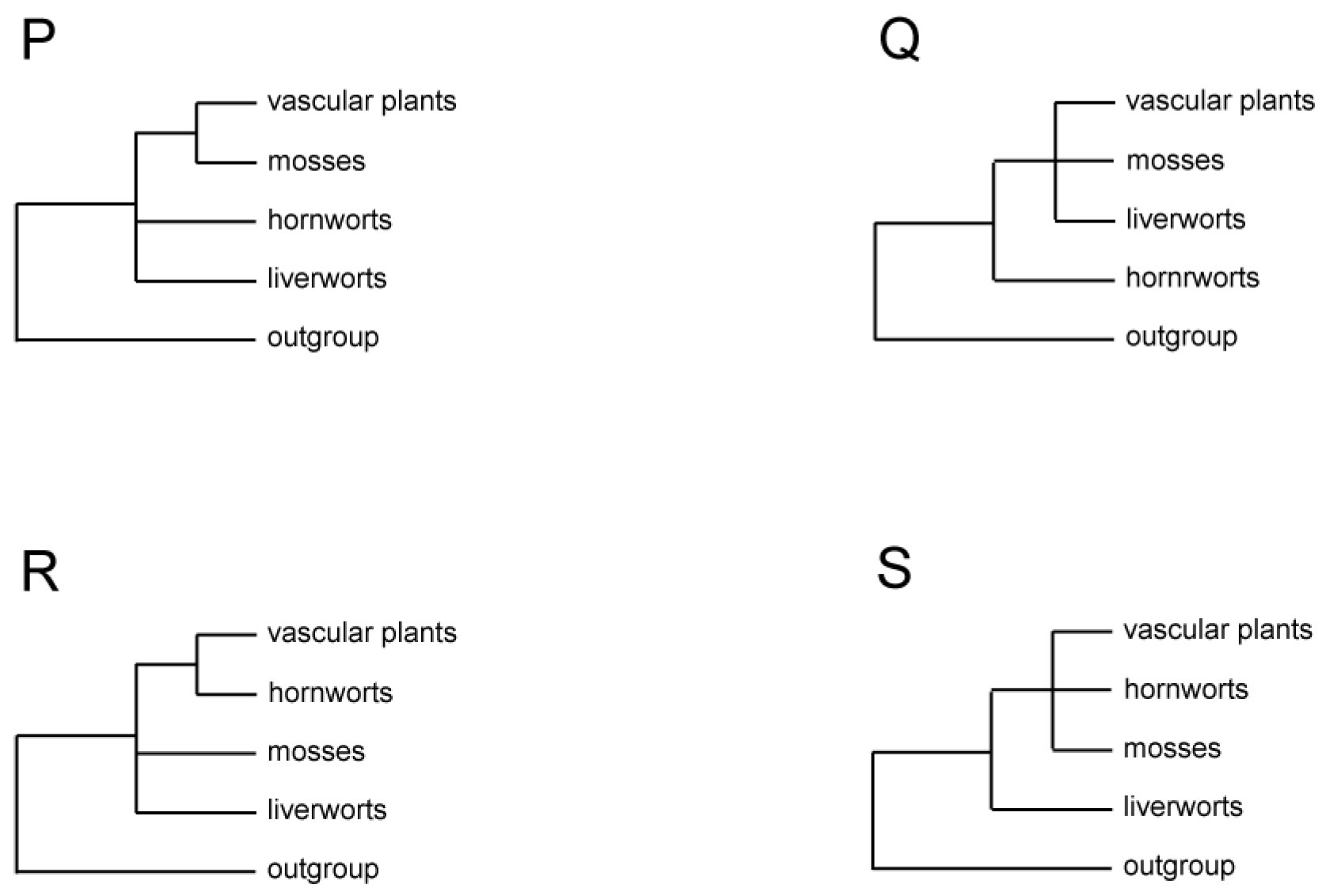
| Figure 3P | morphology | 34 characters | O: 2 L: 3 M: 5 H: 2 V: 3 | MP | [35] |
| Figure 3Q | multigene (nu-18S and mt-SSU) | nucleotide: all positions | O: 4 L: 2 M: 5 H: 4 V: 11 | MP | [21] |
| mitochondrion, SSU | nucleotide: all positions | O: 1 L: 2 M: 4 H: 3 V: 10 | MP | [36] | |
| Figure 3R | chloroplast, rbcL | nucleotide: all positions | O: 2 L: 25 M: 4 H: 3 V: 6 | ML and MP | [37] |
| chloroplast, 67 genes | nucleotide: 1st positions | O: 6 L: 1 M: 1 H: 1 V: 27 | MP | [6] | |
| nucleotide: 3rd positions | ML | ||||
| chloroplast, 73 genes | amino acid | O: 1 L: 1 M: 1 H: 1 V: 16 | ML and MP | [7] | |
| chloroplast, 83 genes | nucleotide: codon-degenerate data | O: 13 L: 2 M: 2 H: 2 V: 11 | ML | [20] | |
| mitochondrion, 72 genes | amino acid (the 9013 and 7210 matrices) | O: 13 L: 3 M: 2 H: 2 V: 5 | ML | [14] | |
| Figure 3S | mitochondrion, 28 intron positions | genome structural characters | O: 3 L: 2 M: 2 H: 3 V: 6 | MP | [6] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, Y.-L.; Mishler, B.D. Relationships Among the Bryophytes and Vascular Plants: A Case Study in Deep-Time Reconstruction. Diversity 2024, 16, 426. https://doi.org/10.3390/d16070426
Qiu Y-L, Mishler BD. Relationships Among the Bryophytes and Vascular Plants: A Case Study in Deep-Time Reconstruction. Diversity. 2024; 16(7):426. https://doi.org/10.3390/d16070426
Chicago/Turabian StyleQiu, Yin-Long, and Brent D. Mishler. 2024. "Relationships Among the Bryophytes and Vascular Plants: A Case Study in Deep-Time Reconstruction" Diversity 16, no. 7: 426. https://doi.org/10.3390/d16070426
APA StyleQiu, Y.-L., & Mishler, B. D. (2024). Relationships Among the Bryophytes and Vascular Plants: A Case Study in Deep-Time Reconstruction. Diversity, 16(7), 426. https://doi.org/10.3390/d16070426