
Environmental Compatibility and Genome Flexibility of Klebsiella oxytoca Isolated from Eight Species of Aquatic Animals


Abstract
1. Introduction
2. Materials and Methods
2.1. K. oxytoca Isolates and Cultural Conditions
2.2. Antibiotic Susceptibility and Heavy Metal Tolerance Assays
2.3. Growth Curve Assay
2.4. Swimming Mobility Analysis
2.5. Biofilm Formation Analysis
2.6. Genome Sequencing, Assembly, and Annotation
2.7. Comparative Genome Analysis
2.8. Statistical Analysis
3. Results and Discussion
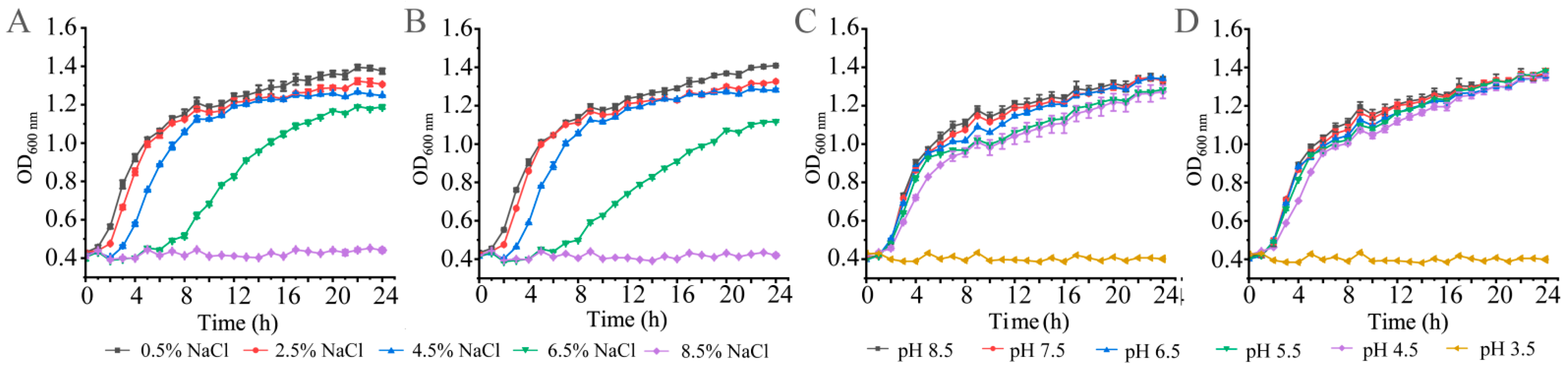
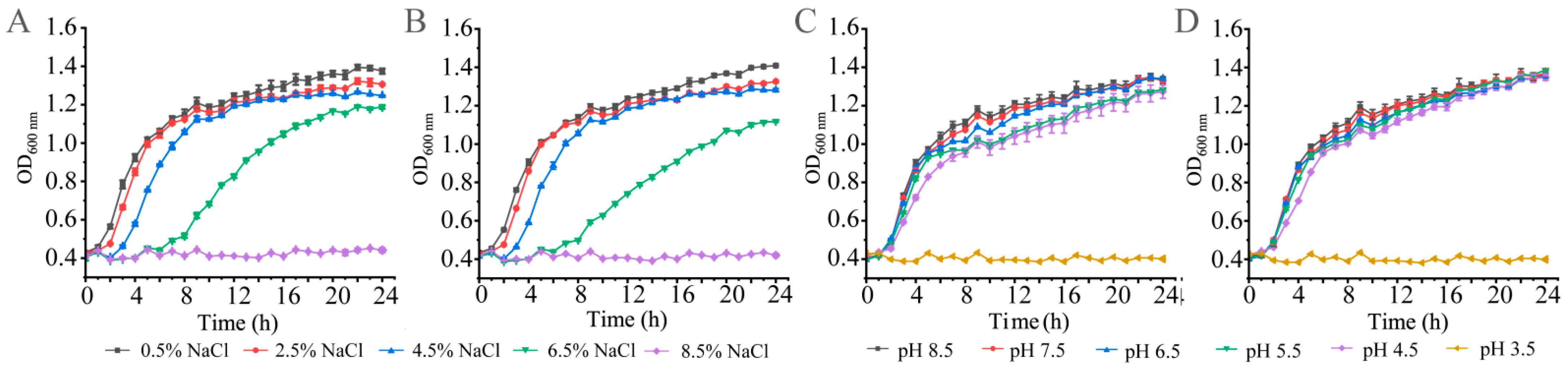
3.1. Growth of the K. oxytoca Isolates in Different NaCl and pH Conditions
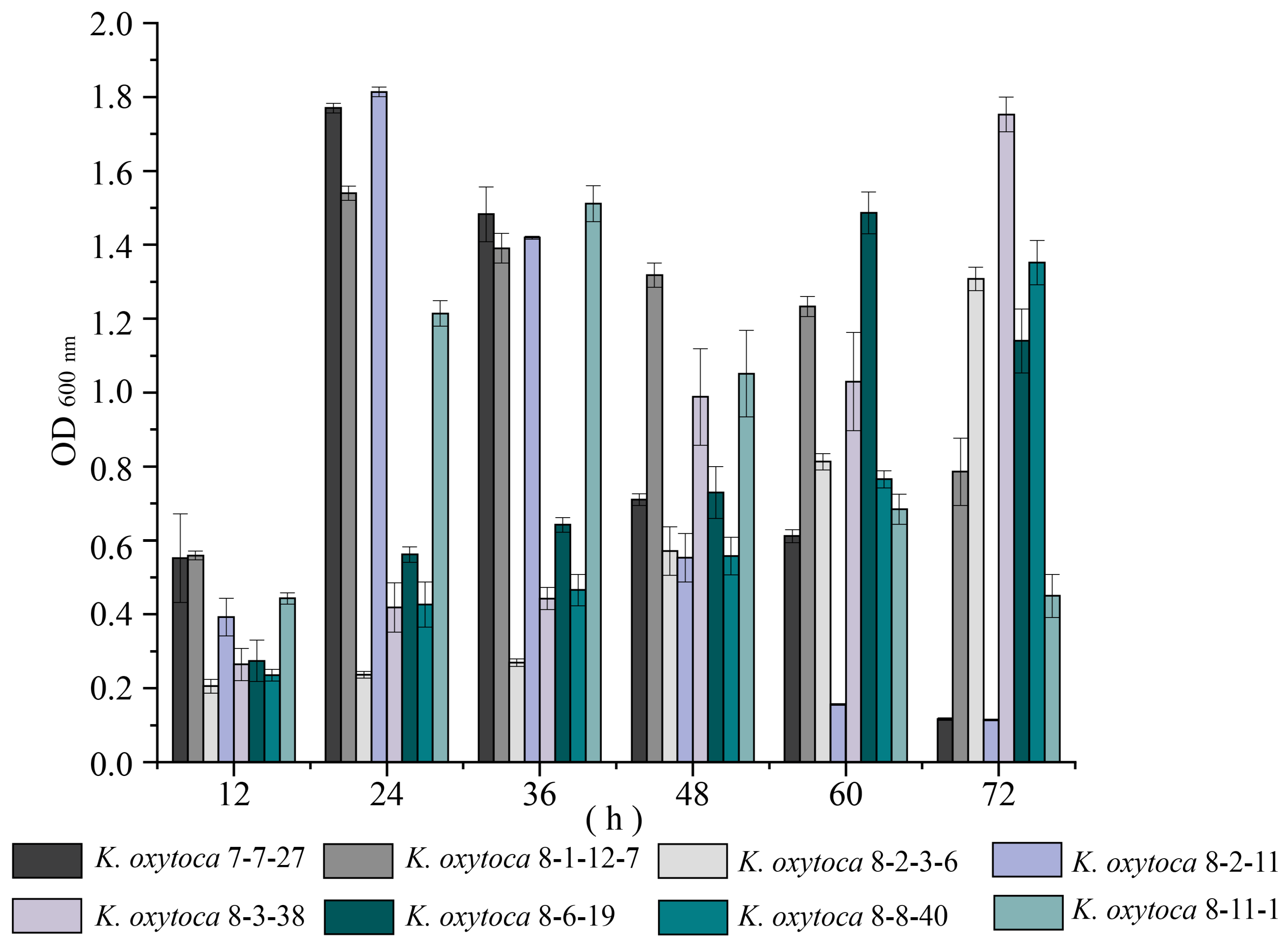
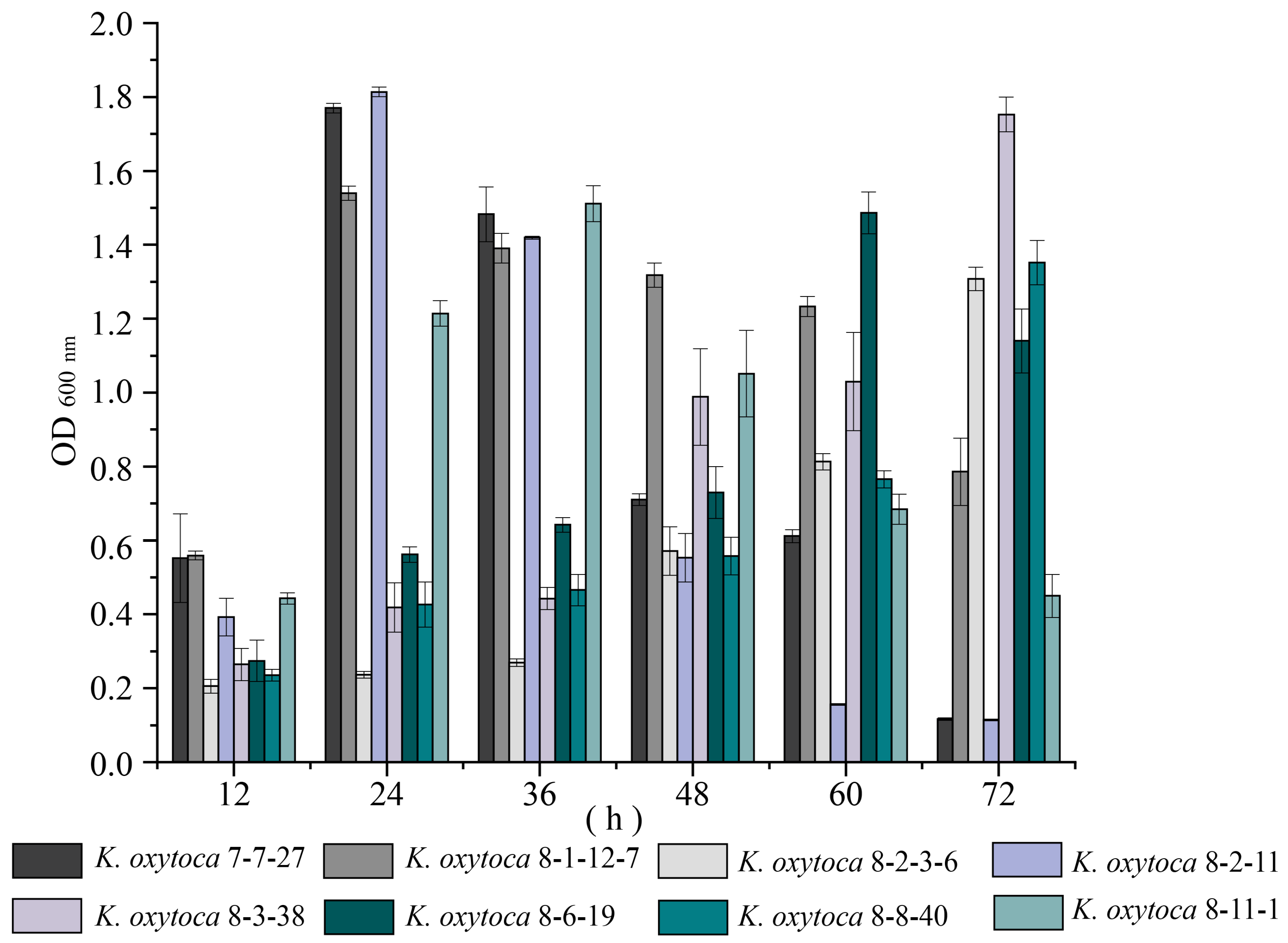
3.2. Biofilm Formation of K. oxytoca Isolates of Aquatic Animal Origins
3.3. Swimming Mobility of K. oxytoca Isolates of Aquatic Animal Origins
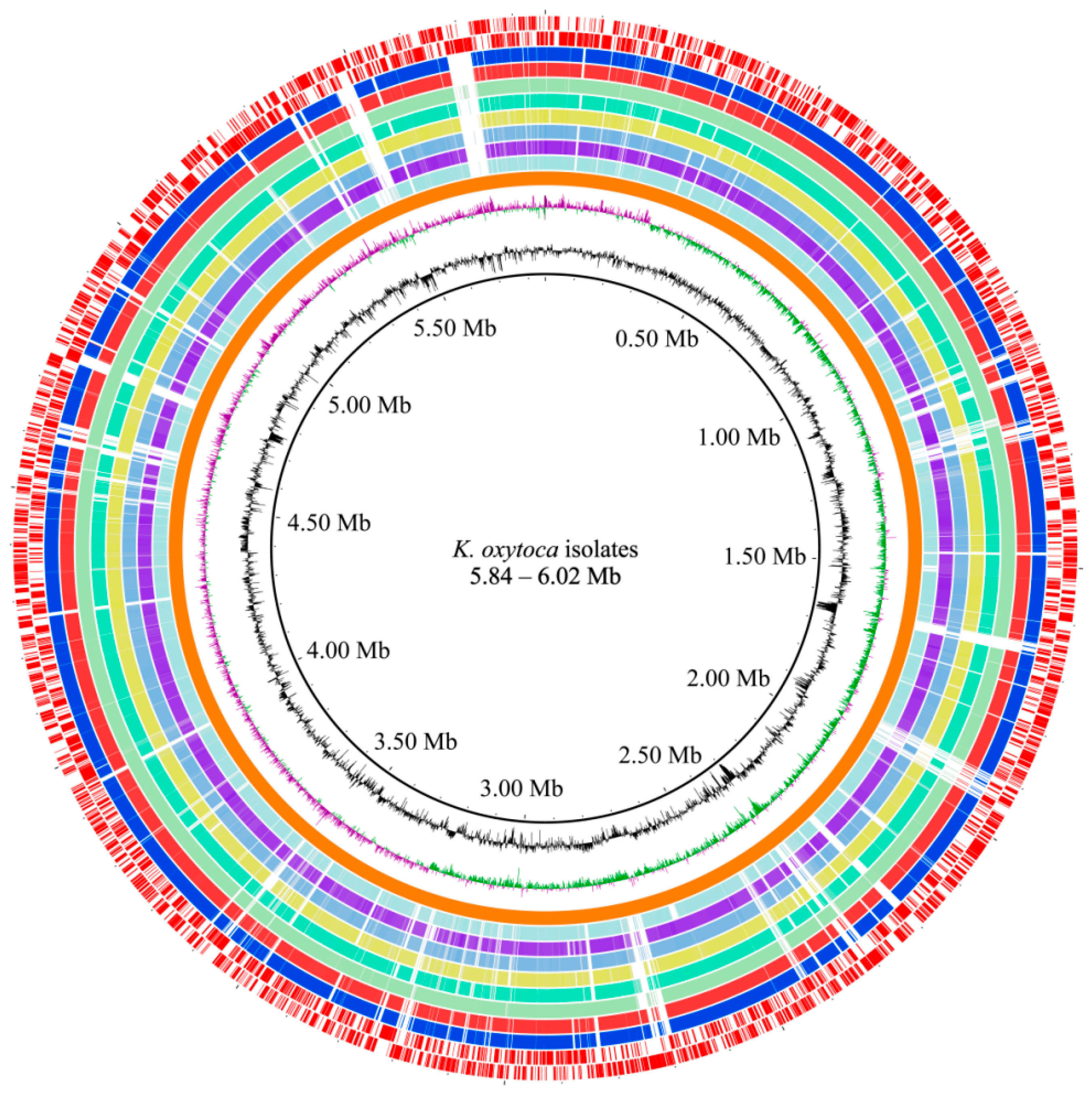
3.4. Genome Features of K. oxytoca Isolates of Aquatic Animal Origins
3.5. GIs
3.6. Putative MGEs
3.6.1. Prophages
3.6.2. Ins
3.6.3. ISs
3.7. CRISPR-Cas Arrays
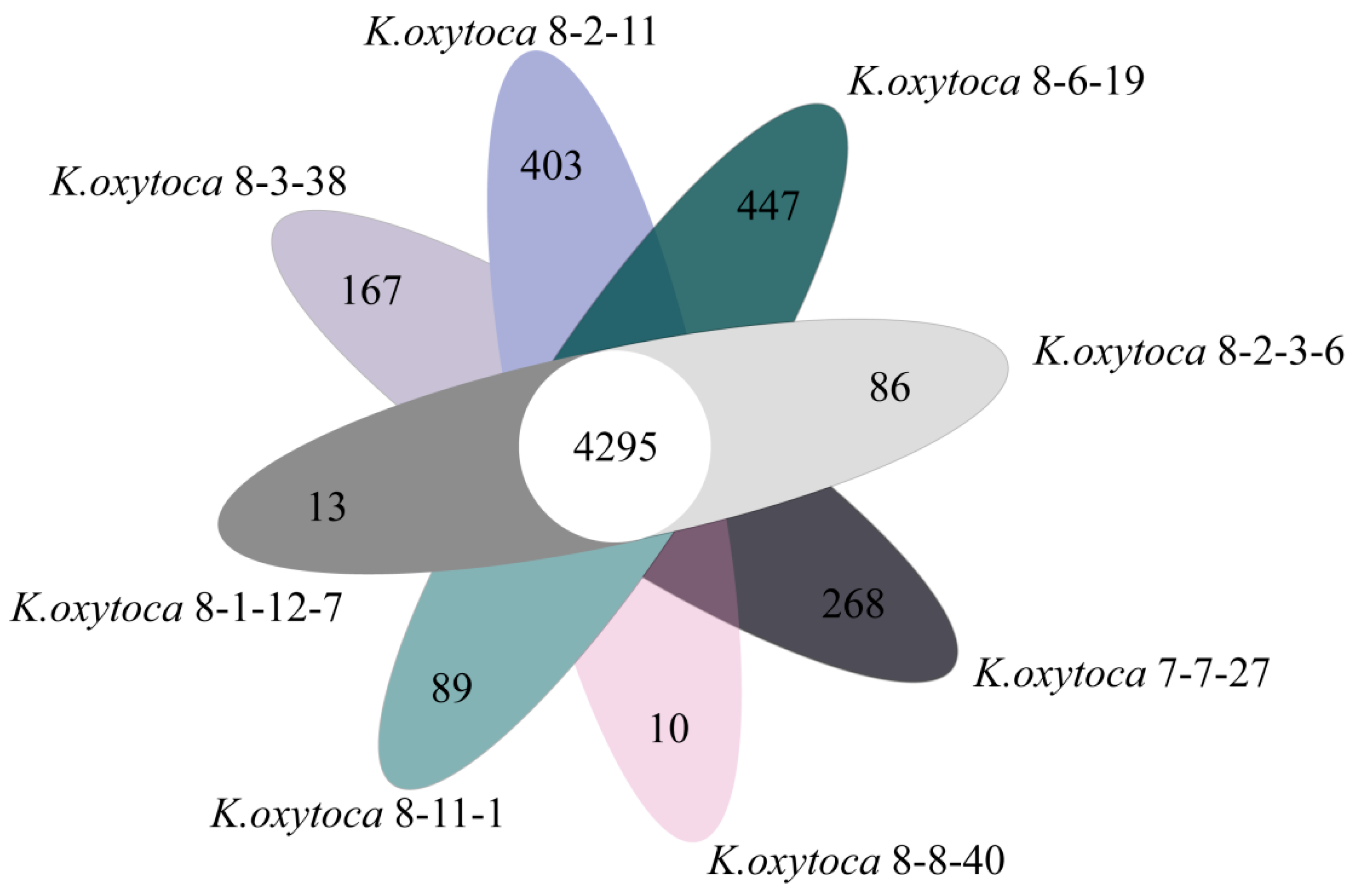
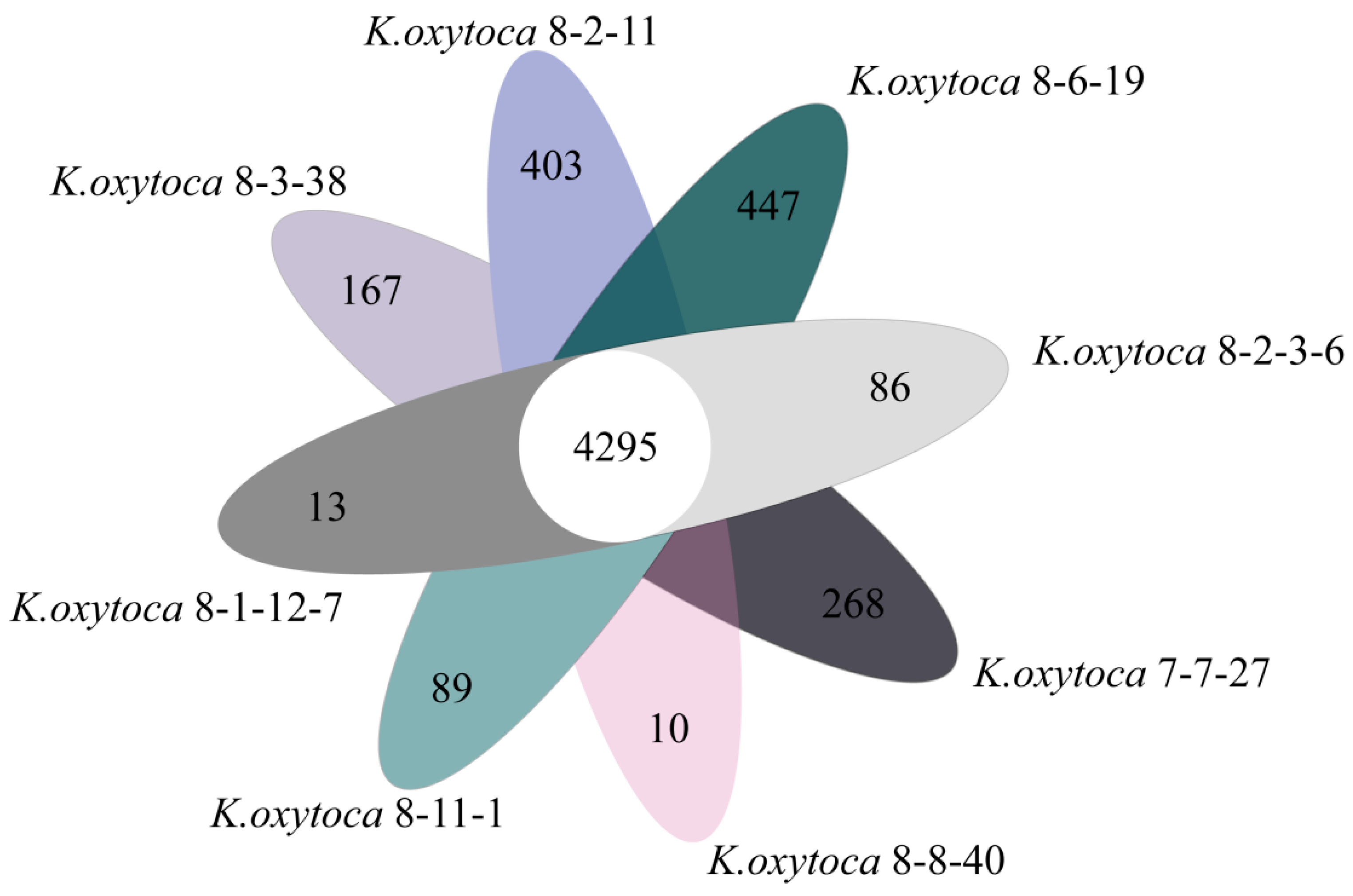
3.8. Strain-Specific Genes of the K. oxytoca Isolates of Aquatic Animal Origins
3.9. Putative Virulence-Associated Genes in the K. oxytoca Genomes
3.10. Antibiotic and Heavy Metal Resistance-Associated Genes in the K. oxytoca Genomes and Their Bacterial Resistance Phenotypes
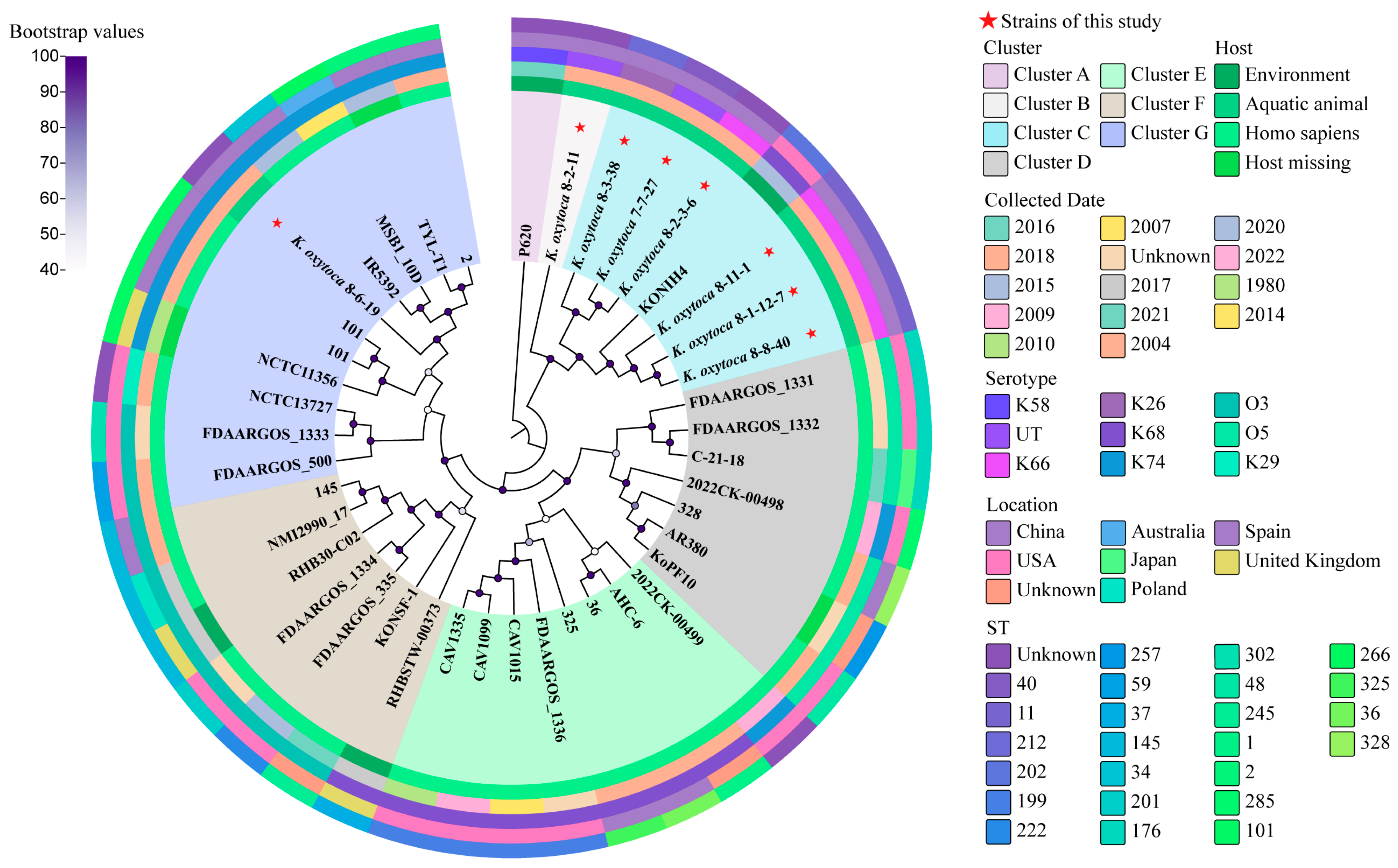
3.11. Phylogenetic Relatedness of the K. oxytoca Isolates of Aquatic Animal Origins
3.12. MLST of the K. oxytoca Isolates of Aquatic Animal Origins
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Power, J.T.; Calder, M.A. Pathogenic significance of Klebsiella oxytoca in acute respiratory tract infection. Thorax 1983, 38, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Herzog, K.A.; Schneditz, G.; Leitner, E.; Feierl, G.; Hoffmann, K.M.; Zollner-Schwetz, I.; Krause, R.; Gorkiewicz, G.; Zechner, E.L.; Högenauer, C. Genotypes of Klebsiella oxytoca isolates from patients with nosocomial pneumonia are distinct from those of isolates from patients with antibiotic-associated hemorrhagic colitis. J. Clin. Microbiol. 2014, 52, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Soto-Hernández, J.L.; Soto-Ramírez, A.; Pérez-Neri, I.; Angeles-Morales, V.; Cárdenas, G.; Barradas, V.A. Multidrug-resistant Klebsiella oxytoca ventriculitis, successfully treated with intraventricular tigecycline: A case report. Clin. Neurol. Neurosurg. 2020, 188, 105592. [Google Scholar] [CrossRef] [PubMed]
- Dago, T.R.; Zewudie, A.; Mamo, Y.; Feyissa, D.; Geleta, S. Multi-drug resistant post corneal repair Klebsiella oxytoca’s keratitis. Int. Med. Case. Rep. J. 2020, 13, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Högenauer, C.; Langner, C.; Beubler, E.; Lippe, I.T.; Schicho, R.; Gorkiewicz, G.; Krause, R.; Gerstgrasser, N.; Krejs, G.J.; Hinterleitner, T.A. Klebsiella oxytoca as a causative organism of antibiotic-associated hemorrhagic colitis. N. Engl. J. Med. 2006, 355, 2418–2426. [Google Scholar] [CrossRef] [PubMed]
- Paveglio, S.; Ledala, N.; Rezaul, K.; Lin, Q.; Zhou, Y.; Provatas, A.A.; Bennett, E.; Lindberg, T.; Caimano, M.; Matson, A.P. Cytotoxin-producing Klebsiella oxytoca in the preterm gut and its association with necrotizing enterocolitis. Emerg. Microbes. Infect. 2020, 9, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.C.; Hung, Y.P.; Lin, W.T.; Dai, W.; Huang, Y.L.; Ko, W.C. Risk factors and clinical impact of bacteremia due to carbapenem-nonsusceptible Enterobacteriaceae: A multicenter study in southern Taiwan. J. Microbiol. Immunol. Infect. 2021, 54, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Carrie, C.; Walewski, V.; Levy, C.; Alexandre, C.; Baleine, J.; Charreton, C.; Coche-Monier, B.; Caeymaex, L.; Lageix, F.; Lorrot, M.; et al. Klebsiella pneumoniae and Klebsiella oxytoca meningitis in infants. Epidemiological and clinical features. Arch. Pediatr. 2019, 26, 12–15. [Google Scholar] [CrossRef]
- Sabio, J.M.; López-Gómez, M.; Jiménez-Alonso, J. Spontaneous spondylodiscitis caused by Klebsiella oxytoca. Ann. Rheum. Dis. 2002, 61, 758–759. [Google Scholar] [CrossRef]
- Surani, A.; Slama, E.M.; Thomas, S.; Ross, R.W.; Cunningham, S.C. Raoultella ornithinolytica and Klebsiella oxytoca pyogenic liver abscess presenting as chronic cough. IDCases 2020, 20, e00736. [Google Scholar] [CrossRef]
- Gharavi, M.J.; Zarei, J.; Roshani-Asl, P.; Yazdanyar, Z.; Sharif, M.; Rashidi, N. Comprehensive study of antimicrobial susceptibility pattern and extended spectrum beta-lactamase (ESBL) prevalence in bacteria isolated from urine samples. Sci. Rep. 2021, 11, 578. [Google Scholar] [CrossRef] [PubMed]
- Yahya Abdulla, N.; Abduljabbar Jaloob Aljanaby, I.; Hayder Hasan, T.; Abduljabbar Jaloob Aljanaby, A. Assessment of ß-lactams and carbapenems antimicrobials resistance in Klebsiella oxytoca isolated from patients with urinary tract infections in Najaf, Iraq. Arch. Razi. Inst. 2022, 77, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Long, H.; Hu, Y.; Feng, Y.; McNally, A.; Zong, Z. Klebsiella oxytoca complex: Update on taxonomy, antimicrobial resistance, and virulence. Clin. Microbiol. Rev. 2022, 35, e0000621. [Google Scholar] [CrossRef] [PubMed]
- Abdurehman Damissie, A.; Abdurahman Musa, K. Isolation, assessments of risk factors, and antimicrobial susceptibility test of Klebsiella from gut of bee in and around Haramaya University bee farm, East Hararghe, Oromia regional state, Ethiopia. Vet. Med. Int. 2022, 2022, 9460543. [Google Scholar] [CrossRef]
- Ni, L.; Xu, Y.; Chen, L. First experimental evidence for the presence of potentially virulent Klebsiella oxytoca in 14 species of commonly consumed aquatic animals, and phenotyping and genotyping of K. oxytoca isolates. Antibiotics 2021, 10, 1235. [Google Scholar] [CrossRef] [PubMed]
- Singh, L.; Cariappa, M.P.; Kaur, M. Klebsiella oxytoca: An emerging pathogen? Med. J. Armed. Forces. India. 2016, 72, S59–S61. [Google Scholar] [CrossRef] [PubMed]
- Kunhikannan, S.; Thomas, C.J.; Franks, A.E.; Mahadevaiah, S.; Kumar, S.; Petrovski, S. Environmental hotspots for antibiotic resistance genes. Microbiology Open 2021, 10, e1197. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Xie, L.; Chen, L. Survival and genome evolution signatures of Klebsiella pneumoniae isolates originated in seven species of aquatic animals. Diversity 2023, 15, 527. [Google Scholar] [CrossRef]
- Pal, A.; Bhattacharjee, S.; Saha, J.; Sarkar, M.; Mandal, P. Bacterial survival strategies and responses under heavy metal stress: A comprehensive overview. Crit. Rev. Microbiol. 2022, 48, 327–355. [Google Scholar] [CrossRef]
- Xu, Y.; Ni, L.; Guan, H.; Chen, D.; Qin, S.; Chen, L. First report of potentially pathogenic Klebsiella pneumoniae from serotype K2 in mollusk Tegillarca granosa and genetic diversity of Klebsiella pneumoniae in 14 species of edible aquatic animals. Foods 2022, 11, 4058. [Google Scholar] [CrossRef]
- Chen, D.; Li, X.; Ni, L.; Xu, D.; Xu, Y.; Ding, Y.; Xie, L.; Chen, L. First experimental evidence for the presence of potentially toxic Vibrio cholerae in snails, and virulence, cross-resistance and genetic diversity of the bacterium in 36 species of aquatic food animals. Antibiotics 2021, 10, 412. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Peng, X.; Xie, L.; Chen, L. Survival and genome diversity of Vibrio parahaemolyticus isolated from edible aquatic animals. Diversity 2022, 14, 350. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Y.; Yu, P.; Ren, S.; Zhu, Z.; Jin, Y.; Yan, J.; Peng, X.; Chen, L. Prophage-related gene VpaChn25_0724 contributes to cell membrane integrity and growth of Vibrio parahaemolyticus CHN25. Front. Cell. Infect. Microbiol. 2020, 10, 595709. [Google Scholar] [CrossRef] [PubMed]
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial biofilms: From the natural environment to infectious diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Josenhans, C.; Suerbaum, S. The role of motility as a virulence factor in bacteria. Int. J. Med. Microbiol. 2002, 291, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Hsiao, W.W.; Brinkman, F.S. Detecting genomic islands using bioinformatics approaches. Nat. Rev. Microbiol. 2010, 8, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Rezaei Javan, R.; Ramos-Sevillano, E.; Akter, A.; Brown, J.; Brueggemann, A.B. Prophages and satellite prophages are widespread in Streptococcus and may play a role in pneumococcal pathogenesis. Nat. Commun. 2019, 10, 4852. [Google Scholar] [CrossRef]
- Engelstädter, J.; Harms, K.; Johnsen, P.J. The evolutionary dynamics of integrons in changing environments. ISME J. 2016, 10, 1296–1307. [Google Scholar] [CrossRef]
- Sabbagh, P.; Rajabnia, M.; Maali, A.; Ferdosi-Shahandashti, E. Integron and its role in antimicrobial resistance: A literature review on some bacterial pathogens. Iran. J. Basic. Med. Sci. 2021, 24, 136–142. [Google Scholar] [CrossRef]
- Ahmadian, L.; Haghshenas, M.R.; Mirzaei, B.; Bazgir, Z.N.; Goli, H.R. Distribution and molecular characterization of resistance gene cassettes containing class 1 integrons in multi-drug resistant (MDR) clinical isolates of Pseudomonas aeruginosa. Infect. Drug. Resist. 2020, 13, 2773–2781. [Google Scholar] [CrossRef]
- Ghaly, T.M.; Chow, L.; Asher, A.J.; Waldron, L.S.; Gillings, M.R. Evolution of class 1 integrons: Mobilization and dispersal via food-borne bacteria. PLoS ONE 2017, 12, e0179169. [Google Scholar] [CrossRef] [PubMed]
- Patricia, S.; Edith, G.; Mick, C. Bacterial insertion sequences: Their genomic impact and diversity. FEMS Microbiol. Rev. 2015, 38, 865–891. [Google Scholar] [CrossRef]
- Deveau, H.; Garneau, J.E.; Moineau, S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol. 2010, 64, 475–493. [Google Scholar] [CrossRef] [PubMed]
- Horvath, P.; Barrangou, R. CRISPR/Cas, the immune system of bacteria and archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Iwadare, T.; Kimura, T.; Sugiura, A.; Takei, R.; Kamakura, M.; Wakabayashi, S.I.; Okumura, T.; Hara, D.; Nakamura, A.; Umemura, T. Pyogenic liver abscess associated with Klebsiella oxytoca: Mimicking invasive liver abscess syndrome. Heliyon 2023, 9, e21537. [Google Scholar] [CrossRef] [PubMed]
- Araújo, B.F.; Ferreira, M.L.; Campos, P.A.; Royer, S.; Gonçalves, I.R.; da Fonseca Batistão, D.W.; Fernandes, M.R.; Cerdeira, L.T.; Brito, C.S.; Lincopan, N.; et al. Hypervirulence and biofilm production in KPC-2-producing Klebsiella pneumoniae CG258 isolated in Brazil. J. Med. Microbiol. 2018, 67, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Arena, F.; Henrici De Angelis, L.; Pieralli, F.; Di Pilato, V.; Giani, T.; Torricelli, F.; D’Andrea, M.M.; Rossolini, G.M. Draft genome sequence of the first hypermucoviscous Klebsiella quasipneumoniae subsp. quasipneumoniae isolate from a bloodstream infection. Genome Announc. 2015, 3, e00952-15. [Google Scholar] [CrossRef]
- Galvani, C.; Terry, J.; Ishiguro, E.E. Purification of the RelB and RelE proteins of Escherichia coli: RelE binds to RelB and to ribosomes. J. Bacteriol. 2001, 183, 2700–2703. [Google Scholar] [CrossRef]
- Kawano, M.; Aravind, L.; Storz, G. An antisense RNA controls synthesis of an SOS-induced toxin evolved from an antitoxin. Mol. Microbiol. 2007, 64, 738–754. [Google Scholar] [CrossRef]
- Zheng, H.Y.; Yan, L.; Yang, C.; Wu, Y.R.; Qin, J.L.; Hao, T.Y.; Yang, D.J.; Guo, Y.C.; Pei, X.Y.; Zhao, T.Y.; et al. Population genomics study of Vibrio alginolyticus. Hereditas 2021, 43, 350–361. [Google Scholar] [CrossRef]
- Gual-de-Torrella, A.; Delgado-Valverde, M.; Pérez-Palacios, P.; Oteo-Iglesias, J.; Rojo-Molinero, E.; Macià, M.D.; Oliver, A.; Pascual, Á.; Fernández-Cuenca, F. Prevalence of the fimbrial operon mrkABCD, mrkA expression, biofilm formation and effect of biocides on biofilm formation in carbapenemase-producing Klebsiella pneumoniae isolates belonging or not belonging to high-risk clones. Int. J. Antimicrob. Agents 2022, 60, 106663. [Google Scholar] [CrossRef] [PubMed]
- Nagano, K. FimA fimbriae of the periodontal disease-associated bacterium Porphyromonas gingivalis. Yakugaku Zasshi 2013, 133, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Cerceo, E.; Deitelzweig, S.B.; Sherman, B.M.; Amin, A.N. Multidrug-resistant gram-negative bacterial infections in the hospital setting: Overview, implications for clinical practice, and emerging treatment options. Microb. Drug. Resist. 2016, 22, 412–431. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.L.; Bundle, S.F.; Kresge, M.E.; Eggers, C.H.; Samuels, D.S. aadA confers streptomycin resistance in Borrelia burgdorferi. J. Bacteriol. 2003, 185, 6723–6727. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Lei, T.; Zhou, Y.; Dai, Y.; Yang, Z.; Luo, H. EBR-5, a novel variant of metallo-β-lactamase EBR from multidrug-resistant Empedobacter stercoris. Microbiol. Spectr. 2023, 12, e0003923. [Google Scholar] [CrossRef]
- Sadek, M.; Poirel, L.; Nordmann, P.; Nariya, H.; Shimamoto, T.; Shimamoto, T. Draft genome sequence of an mcr-1/IncI2-carrying multidrug-resistant Escherichia coli B1:ST101 isolated from meat and meat products in Egypt. J. Glob. Antimicrob. Resist. 2020, 20, 41–42. [Google Scholar] [CrossRef]
- Freeman, Z.N.; Dorus, S.; Waterfield, N.R. The KdpD/KdpE two-component system: Integrating K⁺ homeostasis and virulence. PLoS Pathog. 2013, 9, e1003201. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Feature | K. oxytoca Isolate | |||||||
|---|---|---|---|---|---|---|---|---|
| 7-7-27 | 8-1-12-7 | 8-2-3-6 | 8-2-11 | 8-3-38 | 8-6-19 | 8-8-40 | 8-11-1 | |
| Genome size (bp) | 6,018,771 | 5,896,043 | 5,837,340 | 5,927,746 | 5,942,002 | 5,878,009 | 5,905,709 | 5,851,997 |
| G + C (%) | 55.97 | 56 | 56.02 | 55.41 | 56.06 | 55.07 | 55.99 | 55.98 |
| DNA Scaffold | 90 | 36 | 46 | 34 | 40 | 74 | 49 | 35 |
| Total Predicted Gene | 5601 | 5423 | 5369 | 5480 | 5479 | 5461 | 5431 | 5398 |
| Protein-Coding Gene | 5595 | 5417 | 5367 | 5474 | 5476 | 5457 | 5426 | 5395 |
| RNA Gene | 210 | 238 | 222 | 222 | 224 | 236 | 256 | 207 |
| Genes Assigned to COG | 4856 | 4799 | 4764 | 4796 | 4813 | 4713 | 4807 | 4774 |
| Genes with Unknown Function | 80 | 73 | 69 | 32 | 73 | 62 | 85 | 79 |
| GI | 11 | 12 | 12 | 10 | 19 | 15 | 13 | 13 |
| Prophage | 6 | 3 | 1 | 1 | 4 | 2 | 3 | 4 |
| In | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| IS | 7 | 1 | 0 | 2 | 1 | 4 | 1 | 0 |
| CRISPR | 12 | 13 | 6 | 7 | 7 | 14 | 10 | 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, S.; Gu, T.; Ou, Y.; Wang, Y.; Xie, L.; Chen, L. Environmental Compatibility and Genome Flexibility of Klebsiella oxytoca Isolated from Eight Species of Aquatic Animals. Diversity 2024, 16, 30. https://doi.org/10.3390/d16010030
Sun S, Gu T, Ou Y, Wang Y, Xie L, Chen L. Environmental Compatibility and Genome Flexibility of Klebsiella oxytoca Isolated from Eight Species of Aquatic Animals. Diversity. 2024; 16(1):30. https://doi.org/10.3390/d16010030
Chicago/Turabian StyleSun, Shuo, Tingting Gu, Yafei Ou, Yongjie Wang, Lu Xie, and Lanming Chen. 2024. "Environmental Compatibility and Genome Flexibility of Klebsiella oxytoca Isolated from Eight Species of Aquatic Animals" Diversity 16, no. 1: 30. https://doi.org/10.3390/d16010030
APA StyleSun, S., Gu, T., Ou, Y., Wang, Y., Xie, L., & Chen, L. (2024). Environmental Compatibility and Genome Flexibility of Klebsiella oxytoca Isolated from Eight Species of Aquatic Animals. Diversity, 16(1), 30. https://doi.org/10.3390/d16010030