
Phylogeography of Ara militaris (Military Macaw): Implications for Conservation

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. DNA Extraction, PCR, and Sequencing
2.3. Sequences Alignment, Genetic Diversity, and Population Structure
2.4. Phylogenetic Reconstructions
2.5. Divergence Time Estimation
2.6. Contemporary and Paleo-Distribution Models: Niche Divergence Analyses
3. Results
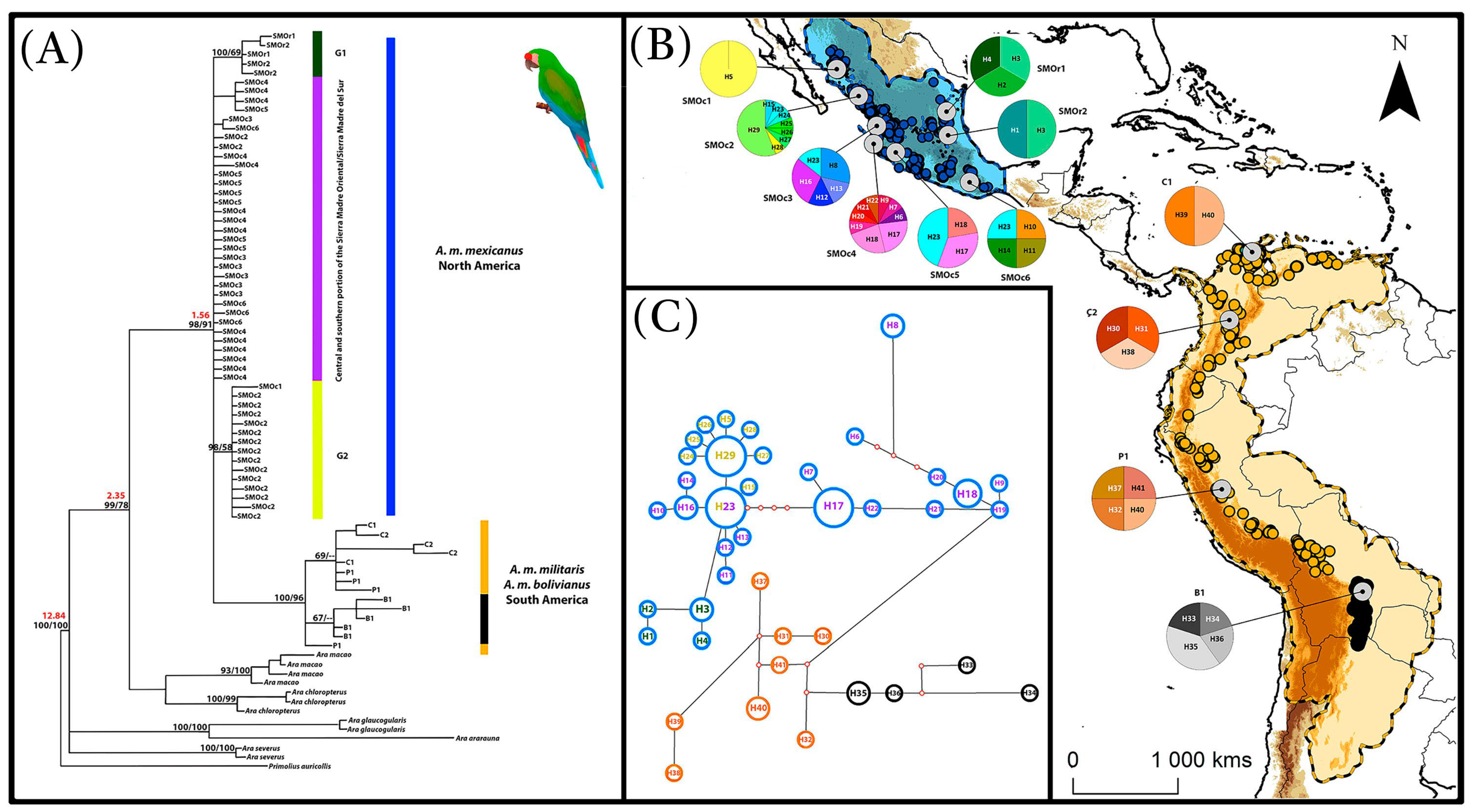
3.1. Haplotype Variation and Distribution
3.2. Phylogenetic Reconstruction and Divergence Time Estimation
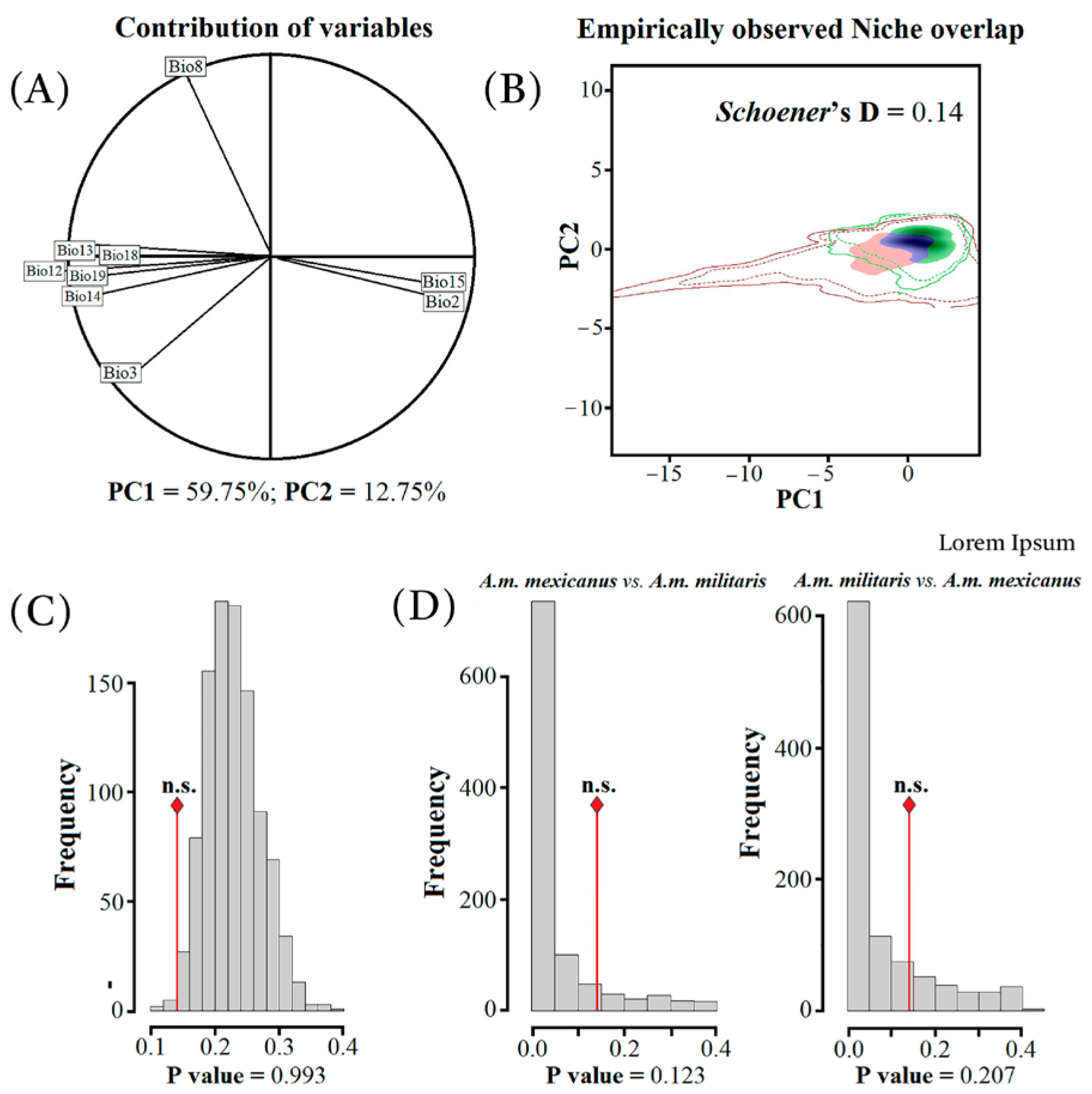
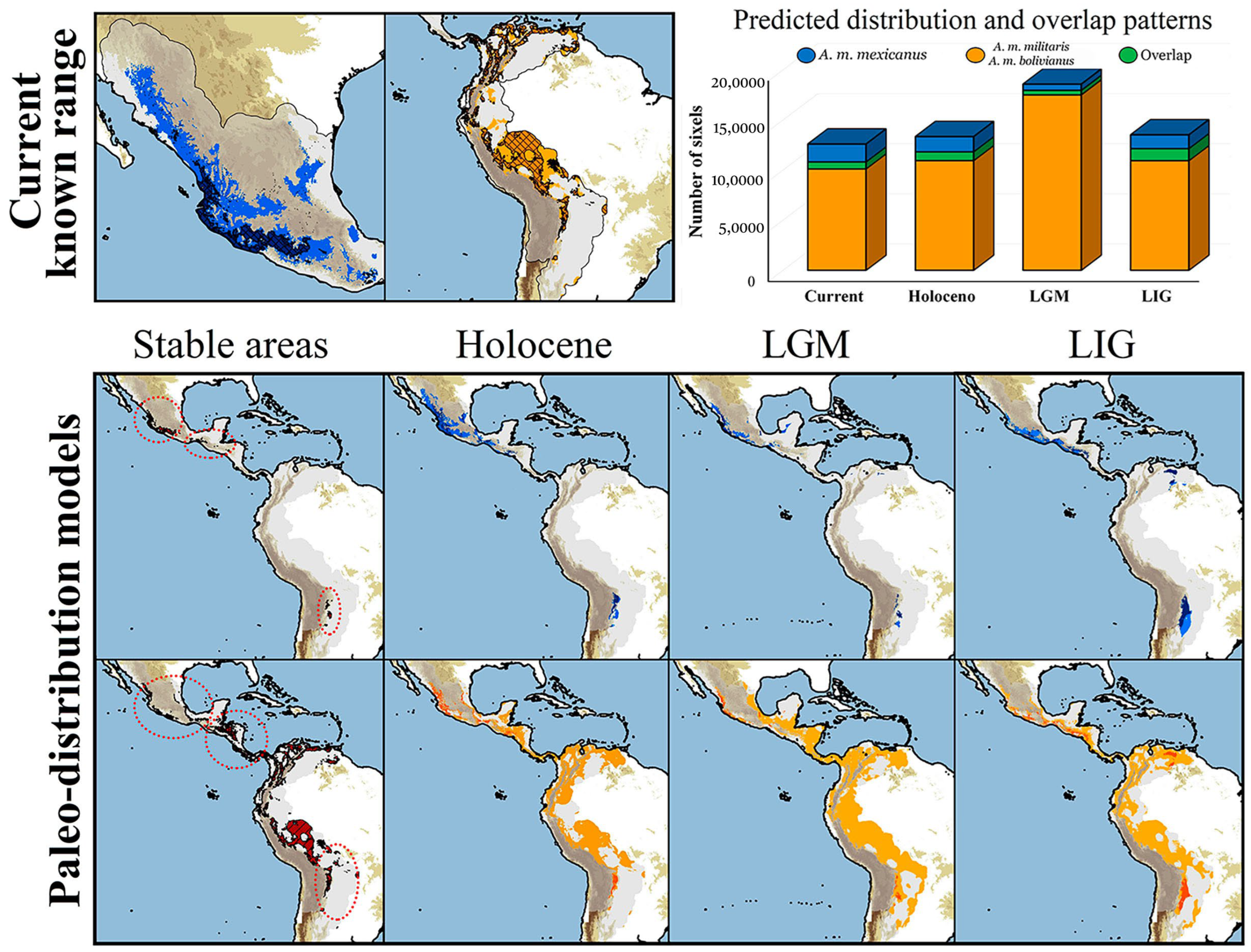
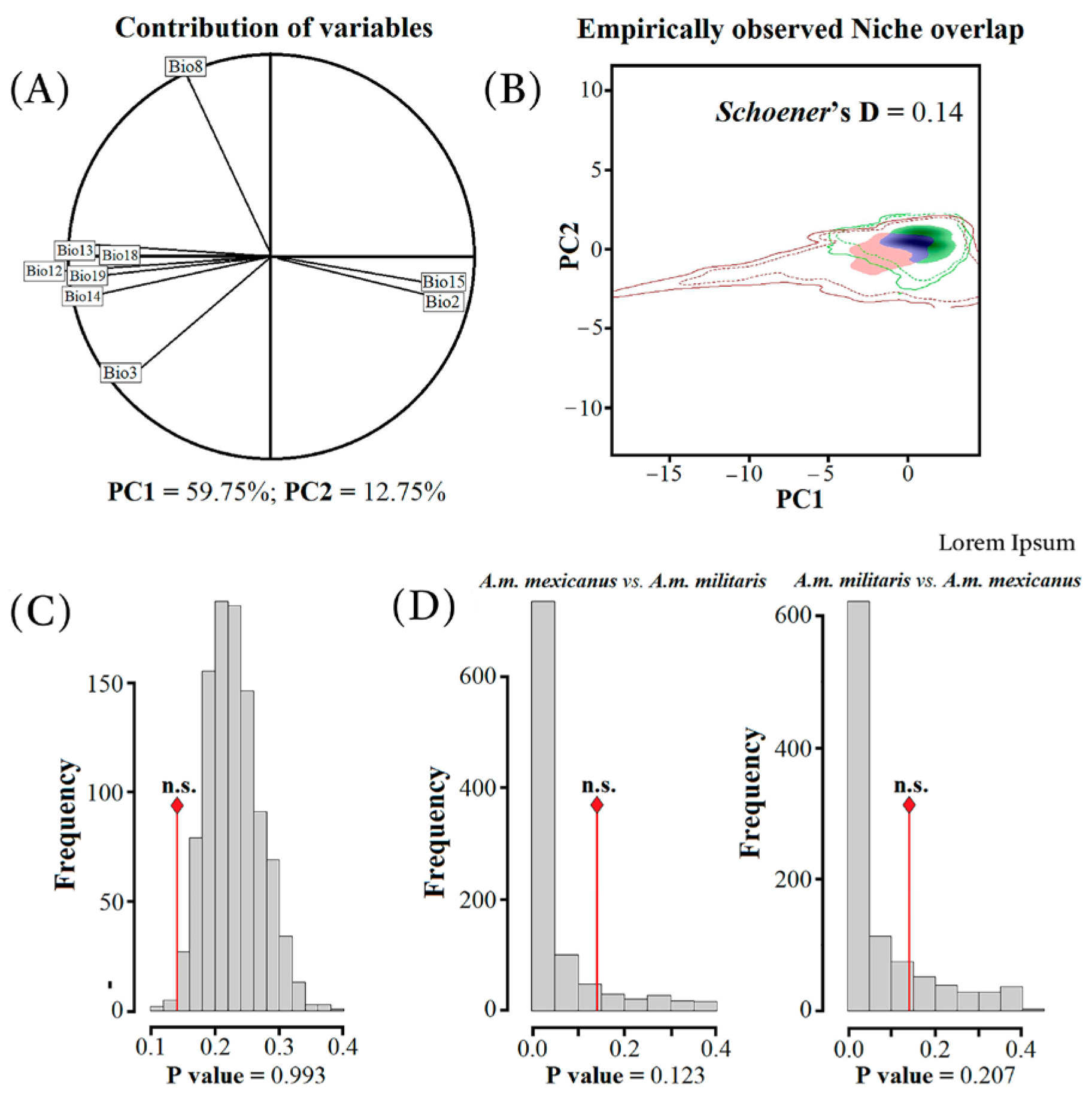
3.3. Niche Divergence Analyses and Paleo-Distributions Models
4. Discussion
4.1. Genetic Diversity and Structure
4.2. Phylogeography, Niche Divergence, and Paleo-Distributions Models
4.3. Conservation and Taxonomic Implications
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: Cambridge, MA, USA, 2000. [Google Scholar] [CrossRef]
- Moritz, C.; Patton, C.J.; Schneider, C.J.; Smith, T.B. Diversification of rainforest faunas: An integrated molecular approach. Ann. Rev. Ecol. Evol. Syst. 2000, 31, 533–563. [Google Scholar] [CrossRef]
- Caparroz, R.; Fernandes, G.H.; Berkunsky, I.; Garcia, R. The role of demography and climatic events in shaping the phylogeography of Amazona aestiva (Psittaciformes, Aves) and definition of management units for conservation. Divers. Distrib. 2009, 15, 459–468. [Google Scholar] [CrossRef]
- Sotelo-Muñoz, M.; Maldonado-Coelho, M.; Svensson-Coelho, M.; Dos Santos, S.S.; Miyaki, C.Y. Vicariance, dispersal, extinction and hybridization underlie the evolutionary history of Atlantic forest fire-eye antbirds (Aves: Thamnophilidae). Mol. Phylogenet. Evol. 2020, 148, 106820. [Google Scholar] [CrossRef] [PubMed]
- Pârâu, L.G.; Wink, M. Common patterns in the molecular phylogeography of western palearctic birds: A comprehensive review. J. Ornithol. 2021, 162, 937–959. [Google Scholar] [CrossRef]
- Irwin, D.E. Phylogeographic breaks without geographic barriers to gene flow. Evolution 2002, 56, 2383–2394. [Google Scholar] [CrossRef]
- Juniper, T.; Parr, M. Parrots: A Guide to the Parrots of the World; Yale University Press: New Haven, CO, USA, 1998. [Google Scholar]
- Caparroz, R.; Miyaki, C.Y.; Baker, A.J. Characterization of microsatellite loci in Blue-and-Gold Macaw, Ara ararauna (Psittaciforme: Aves). Mol. Ecol. Notes 2003, 3, 441–443. [Google Scholar] [CrossRef]
- Eberhard, J.R.; Bermingham, E. Phylogeny and biogeography of the Amazona ochrocephala (Ave: Psittacidae) complex. Auk 2004, 121, 318–332. [Google Scholar] [CrossRef]
- Ribas, C.C.; Tavares, E.S.; Yoshihara, C.Y.; Miyaki, C.Y. Phylogeny and biogeography of Yellow-headed and Blue-fronted parrots (Amazona ochrocephala and Amazona aestiva) with special reference to the South American taxa. Ibis 2007, 149, 564–574. [Google Scholar] [CrossRef]
- Ribas, C.C.; Miyaki, C.Y. Molecular systematics in Aratinga parakeets: Species limits and historical biogeography in the ‘solstitialis’ group, and the systematic position of Nandayus nenday. Mol. Phylogenet. Evol. 2004, 30, 663–675. [Google Scholar] [CrossRef]
- Russello, M.A.; Amato, G. A molecular phylogeny of Amazona: Implications for Neotropical parrot biogeography, taxonomy, and conservation. Mol. Phylogenet. Evol. 2004, 30, 421–437. [Google Scholar] [CrossRef]
- Tavares, E.S.; Baker, A.J.; Pereira, S.L.; Miyaki, C.Y. Phylogenetic relationships and historical biogeography of Neotropical parrots (Psittaciformes: Psittacidae: Arini) inferred from mitochondrial and nuclear DNA sequences. Syst. Biol. 2006, 55, 454–470. [Google Scholar] [CrossRef]
- Ribas, C.C.; Miyaki, C.Y.; Cracraft, J. Phylogenetic relationships, diversification and biogeography in Neotropical Brotogeris parakeets. J. Biogeogr. 2009, 36, 1712–1729. [Google Scholar] [CrossRef]
- Avise, J.C.; Walker, D. Pleistocene phylogeography effects on avian populations and the speciation process. Proc. R. Soc. B Biol. Sci. 1998, 265, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Tavares, E.S.; Yamashita, C.; Miyaki, C.Y. Phylogenetic relationships among some Neotropical parrot genera (Psittacidae: Aves) based on mitochondrial sequences. Auk 2004, 121, 230–242. [Google Scholar] [CrossRef]
- Masello, J.F.; Quillfeldt, P.; Munimanda, G.K.; Klauke, N.; Segelbacher, G.; Schaefer, H.M.; Failla, M.; Cortés, M.; Moodley, Y. The high Andes, gene flow and a stable hybrid zone shape the genetic structure of a wide-ranging South American parrot. Front. Zool. 2011, 8, 1–16. [Google Scholar] [CrossRef]
- Presti, F.T.; Guedes, N.M.R.; Antas, P.T.Z.; Miyaki, C.Y. Population genetic structure in Hyacinth Macaws (Anodorhynchus hyacinthinus) and identification of the probable origin of confiscated individuals. J. Hered. 2015, 106, 491–502. [Google Scholar] [CrossRef]
- Bennett, P.M.; Owens, I.P.F. Variation in extinction risk among birds: Chance or evolutionary predisposition? Proc. R. Soc. 1997, 264, 401–408. [Google Scholar] [CrossRef]
- Forshaw, J.M. Parrots of the World; Princeton University Press: Princeton, NJ, USA, 2010. [Google Scholar]
- Bird Life’s Online World Bird Database: The Site for Bird Conservation. Available online: http://www.birdlife.org (accessed on 28 June 2023).
- Collar, N.J.; Juniper, A.T. Dimensions and causes of the parrot conservation crisis. In New World Parrots in Crisis: Solutions from Conservation Biology; Beissinger, S.R., Snyder, N.F.R., Eds.; Smithsonian Institution: Washington, DC, USA, 1992; pp. 1–24. [Google Scholar]
- Olson, S.L.; Suárez, W. A fossil cranium of the Cuban Macaw Ara tricolor (Aves: Psittacidae) from Villa Clara Province, Cuba. Caribb. J. Sci. 2008, 44, 287–290. [Google Scholar] [CrossRef]
- Ríos-Muñoz, C.A. Navarro-Sigüenza, A.G. Efectos del cambio de uso de suelo en la disponibilidad hipotética de hábitat para los psitácidos de México. Ornitol. Neotrop. 2009, 20, 491–509. [Google Scholar]
- Rivera-Ortíz, F.A.; Oyama, K.; Ríos-Muñoz, C.A.; Solórzano, S.; Navarro-Sigüenza, A.G.; Arizmendi, M.C. Habitat characterization and modeling of the potential distribution of the Military Macaw (Ara militaris) in Mexico. Rev. Mex. Biodivers. 2013, 84, 1200–1215. [Google Scholar] [CrossRef]
- Landa, M.D.L.N.; Castro, J.C.M.; Monterrubio-Rico, T.C.; Lara-Cabrera, S.I.; Prieto-Torres, D.A. Predicting co-distribution patterns of parrots and woody plants under global changes: The case of the Lilac-crowned Amazon and Neotropical dry forests. J. Nat. Conserv. 2023, 71, 126323. [Google Scholar] [CrossRef]
- Nader, W.; Werner, D.; Wink, M. Genetic diversity of scarlet macaws Ara macao in reintroduction studies for threatened populations in Costa Rica. Biol. Conserv. 1999, 87, 269–272. [Google Scholar] [CrossRef]
- Caparroz, R.; Miyaki, C.Y.; Bampi, M.I.; Wajntal, A. Analysis of the genetic variability in a sample of the remaining group of Spix’s Macaw (Cyanopsitta spixii, Psittaciformes: Aves) by DNA fingerprinting. Biol. Conserv. 2001, 99, 307–311. [Google Scholar] [CrossRef]
- Eberhard, J.R.; Iñigo-Elias, E.E.; Enkerlin-Hoeflich, E.; Cun, E.P. Phylogeography of the Military Macaw (Ara militaris) and the Great Green Macaw (A. Ambiguus) based on MTDNA sequence data. Wilson J. Ornithol. 2015, 127, 661–669. [Google Scholar] [CrossRef]
- Dehasque, M. Applied Conservation Genomics of Military Macaws (Ara militaris). Ph.D. Thesis, Universiteit Antwerpen, Antwerp, Belgian, 2016. [Google Scholar]
- The Integrated Taxonomic Information System. Available online: http://www.catalogueoflife.org/annual-checklist/2010/info/about (accessed on 28 June 2023).
- Howell, S.N.G.; Webb, S. A Guide to the Birds of Mexico and Northern Central America; Oxford University Press: Oxford, UK, 1995; p. 337. [Google Scholar]
- Arbeláez-Cortés, E.; Roldán-Piña, D.; Navarro-Sigüenza, A.G. Multilocus phylogeography and morphology give insights into the recent evolution of a Mexican endemic songbird: Vireo hypochryseus. J. Avian Biol. 2014, 45, 253–263. [Google Scholar] [CrossRef]
- Arbeláez-Cortés, E.; Milá, B.; Navarro-Sigüenza, A.G. Multilocus analysis of intraspecific differentiation in three endemic bird species from the northern Neotropical dry forest. Mol. Phylogenet. Evol. 2014, 70, 362–377. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Gonzalez, L.A.; Cayetano, H.; Prieto-Torres, D.A.; Rojas-Soto, O.R.; Navarro-Sigüenza, A.G. The role of ecological and geographical drivers of lineage diversification in the Squirrel cuckoo Piaya cayana in Mexico: A mitochondrial DNA perspective. J. Ornithol. 2023, 164, 37–53. [Google Scholar] [CrossRef]
- IUCN. Red List of Threatened Species. Available online: https://www.iucnredlist.org/ (accessed on 24 June 2023).
- Rivera-Ortíz, F.A.; Solórzano, S.; Arizmendi, M.C.; Dávila-Aranda, P.; Oyama, K. Genetic diversity and structure of the Military Macaw (Ara militaris) in Mexico: Implications for conservation. Trop. Conserv. Sci. 2017, 10, 1–12. [Google Scholar] [CrossRef]
- Leeton, P.; Christidis, L. Feathers from Museum Bird Skins—A good source of DNA for Phylogenetic Studies. Condor 1993, 95, 465–466. [Google Scholar] [CrossRef]
- Murphy, S.A.; Double, M.C.; Legge, S.M. The phylogeography of palm cockatoos, Probosciger aterrimus, in the dynamic Australo-Papuan region. J. Biogeogr. 2007, 34, 1534–1545. [Google Scholar] [CrossRef]
- Miyaki, C.Y.; Matioli, S.R.; Burke, T.; Wajntal, A. Parrot evolution and paleogeographical events: Mitochondrial DNA evidence. Mol. Biol. Evol. 1998, 15, 544–551. [Google Scholar] [CrossRef]
- Groombridge, J.J.; Massey, J.G.; Bruch, J.C.; Malcolm, T.; Brosius, C.N.; Okada, M.M.; Sparklin, B.; Fretz, J.S.; VanderWerf, E.A. An attempt to recover the Po’ouli by translocation and an appraisal of recovery strategy for bird species of extreme rarity. Biol. Conserv. 2004, 118, 365–375. [Google Scholar] [CrossRef]
- Sorenson, M.D.; Ast, J.C.; Dimcheff, D.E.; Yuri, T.; Mindell, D.P. Primers for a PCRbased approach to mitochondrial genome sequencing in birds and other vertebrates. Mol. Phylogenet. Evol. 1999, 12, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Glenn, T.C.; Stephan, W.; Braun, M.J. Effects of a population bottleneck on whooping crane mitochondrial DNA variation. Conserv. Biol. 1999, 13, 1097–1107. [Google Scholar] [CrossRef]
- Abbott, C.L.; Double, M.C. Phylogeography of Shy and White-capped albatrosses inferred from mitochondrial DNA sequences: Implications for population history and taxonomy. Mol. Ecol. 2003, 12, 2747–2758. [Google Scholar] [CrossRef]
- Kundu, S.; Jones, C.G.; Prys-Jones, R.P.; Groombridge, J.J. The evolution of the Indian Ocean parrots (family Psittacidae): Extinction, adaptive radiation and eustasy. Mol. Phylogenet. Evol. 2012, 62, 296–305. [Google Scholar] [CrossRef]
- Zink, R.M.; Barrowclough, G.F. Mitochondrial DNA under siege in avian phylogeography. Mol. Ecol. 2008, 17, 2107–2121. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Rozas, J.; Sánchez-De, I.; Barrio, J.C.; Messeguer, X.; Rozas, R. DnaSP, DNA polymorphism analysis by coalescent and other methods. Bioinformatics 2003, 19, 2496–2497. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin 3.01: An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Dellicour, S.; Mardulyn, P. SPADS 1.0: A toolbox to perform spatial analyses on DNA sequence data sets. Mol. Ecol. Resour. 2014, 14, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Raymond, M.; Rousset, F. GENEPOP Version 1.2: Population genetics software for exact tests and ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Tavares, M.G.; Dias, L.A.S.; Borges, A.A.; Lopes, D.M.; Busse, A.H.P.; Costa, R.G.; Fernandes-Salomão, T.M.; Campos, L.A.O. Genetics divergence between population of the stingless bee uruçu amarela (Melipona rufiventris group, Hymenoptera, Meliponini): Is there a new Melipona species in the Brazilian state of Minas Gerais? Genetic Mol. Biol. 2007, 30, 667–675. [Google Scholar] [CrossRef]
- DeSalle, R.; Narechania, A.; Tessler, M. Multiple outgroups can cause random rooting in phylogenetics. Mol. Phylogenet. Evol. 2023, 184, 107806. [Google Scholar] [CrossRef]
- Lanfear, R.; Kokko, H.; Eyre-Walker, A. Population size and the rate of evolution. Trends Ecol. Evol. 2014, 29, 33–41. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A. Tracer MCMC Trace Analysis Tool; Univeristy of Oxford: Oxford, UK, 2003; Available online: https://tree.bio.ed.ac.uk/software/tracer/ (accessed on 15 January 2023).
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 21, 2688–2690. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Sykes, B.C.; Richards, M.B. Mitochondrial portraits of human populations using median networks. Genetics 1995, 141, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e934. [Google Scholar] [CrossRef] [PubMed]
- Rödder, D.; Engler, J.O. Quantitative metrics of overlaps in Grinnellian niches: Advances and possible drawbacks. Glob. Ecol. Biogeogr. 2011, 20, 915–927. [Google Scholar] [CrossRef]
- Navarro-Sigüenza, A.G.; Ortíz-Pulido, R.; Peterson, T. Un panorama breve de la historia de la ornitología mexicana. Ornitol. Neotrop. 2008, 19, 367–379. [Google Scholar]
- Aiello-Lammens, M.E.; Boria, R.A.; Radosavljevic, A.; Vilela, B.; Anderson, R.P. spThin: An R package for spatial thinning of species occurrence records for use in ecological niche models. Ecography 2015, 38, 541–545. [Google Scholar] [CrossRef]
- Soberon, J.; Peterson, A.T. Interpretation of models of fundamental ecological niches and species’ distributional areas. Biodivers. Inform. 2005, 2, 1–10. [Google Scholar] [CrossRef]
- Dinerstein, E.; Olson, D.; Joshi, A.; Vynne, C.; Burgess, N.D.; Wikramanayake, E.; Hahn, N.; Palminteri, S.; Hedao, P.; Noss, R.; et al. An ecoregion-based approach to protecting half the terrestrial realm. BioScience 2017, 67, 534–545. [Google Scholar] [CrossRef]
- Morrone, J.J. Biogeographical regionalization of the Neotropical region. Zootaxa 2014, 3782, 1–110. [Google Scholar] [CrossRef]
- Hijmans, R.J.; Cameron, S.E.; Parra, J.L.; Jones, P.G.; Jarvis, A. Very high-resolution interpolated climate surfaces for global land areas. J. R. Meteorol. Soc. 2005, 25, 1965–1978. [Google Scholar] [CrossRef]
- Wei, T.; Simko, V.; Levy, M.; Xie, Y.; Jin, Y.; Zemla, J. Package ‘corrplot’. Statistician 2017, 56, e24. [Google Scholar]
- Naimi, B. USDM: Uncertainty analysis for species distribution models. R package version 1.1. RDocumentation 2015, 1, 1–15. [Google Scholar]
- Warren, D.L.; Glor, R.E.; Turelli, M. Environmental niche equivalency versus conservatism: Quantitative approaches to niche evolution. Evolution 2008, 62, 2868–2883. [Google Scholar] [CrossRef] [PubMed]
- Strubbe, D.; Beauchard, O.; Matthysen, E. Niche conservatism among non-native vertebrates in Europe and North America. Ecography 2015, 38, 321–329. [Google Scholar] [CrossRef]
- Broennimann, O.; Fitzpatrick, M.C.; Pearman, P.B.; Petitpierre, B.; Pellissier, L.; Yoccoz, N.G.; Thuiller, W.; Fortin, M.J.; Randin, C.; Zimmermann, N.E.; et al. Measuring ecological niche overlap from occurrence and spatial environmental data. Glob. Ecol. Biogeogr. 2012, 21, 481–497. [Google Scholar] [CrossRef]
- Di Cola, V.; Broennimann, O.; Petitpierre, B.; Breiner, F.T.; d’Amen, M.; Randin, C.; Engler, R.; Pottier, J.; Pio, D.; Dubuis, A.; et al. Ecospat: An R package to support spatial analyses and modeling of species niches and distributions. Ecography 2017, 40, 774–787. [Google Scholar] [CrossRef]
- Phillips, S.J.; Anderson, R.P.; Schapire, R.E. Maximum entropy modeling of species geographic distributions. Ecol. Modell. 2006, 190, 231–259. [Google Scholar] [CrossRef]
- Cobos, M.E.; Peterson, A.T.; Barve, N.; Osorio-Olvera, L. Kuenm: An R package for detailed development of ecological niche models using Maxent. PeerJ 2019, 7, e6281. [Google Scholar] [CrossRef]
- Peterson, A.T.; Stewart, A.; Mohamed, K.I.; Araújo, M.B. Shifting global invasive potential of European plants with climate change. PLoS ONE 2008, 3, e2441. [Google Scholar] [CrossRef]
- Owens, H.L.; Campbell, L.P.; Dornak, L.L.; Saupe, E.E.; Barve, N.; Soberón, J.; Ingenloff, K.; Lira-Noriega, A.; Hensz, C.M.; Myers, C.E.; et al. Constraints on interpretation of ecological niche models by limited environmental ranges on calibration areas. Ecol. Modell. 2013, 263, 10–18. [Google Scholar] [CrossRef]
- Alkishe, A.A.; Peterson, A.T.; Samy, A.M. Climate change influences on the potential geographic distribution of the disease vector tick Ixodes ricinus. PLoS ONE 2017, 12, e0189092. [Google Scholar] [CrossRef]
- Castillo-Chora, V.D.J.; Sánchez-González, L.A.; Mastretta-Yanes, A.; Prieto-Torres, D.A.; Navarro-Sigüenza, A.G. Insights into the importance of areas of climatic stability in the evolution and maintenance of avian diversity in the Mesoamerican dry forests. Biol. J. Linn. Soc. 2021, 132, 741–758. [Google Scholar] [CrossRef]
- Warren, D.L.; Matzke, N.J.; Cardillo, M.; Baumgartner, J.B.; Beaumont, L.J.; Turelli, M.; Glor, R.E.; Huron, N.A.; Simoes, M.; Iglesias, T.L.; et al. ENMTools 1.0: An R package for comparative ecological biogeography. Ecography 2021, 44, 504–511. [Google Scholar] [CrossRef]
- Rodríguez-Robles, J.A.; Jezkova, T.; Leal, M. Genetic structuring in the threatened “Lagartijo del Bosque Seco” (Anolis cooki) from Puerto Rico. Mol. Phylogenet. Evol. 2008, 46, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Faria, P.J.; Guedes, N.M.R.; Yamashita, C.; Martuscelli, P.; Miyaki, C.Y. Genetic variation and population structure of the endangered Hyacinth Macaw (Anodorhynchus hyacinhinus): Implications for conservation. Biodivers. Conserv. 2008, 17, 765–779. [Google Scholar] [CrossRef]
- García, F.L. Un Enfoque Filogeográfico Para la Conservación de Poblaciones de Ara macao cyanoptera. Master’s Thesis, Instituto de Ecología, A.C., Xalapa, México, 2009. [Google Scholar]
- De Almeida, T.R.A.; Presti, F.T.; Cruz, V.P.; Wasko, A.P. Genetic analysis of the endangered Hyacinth Macaw (Anodorhynchus hyacinthinus) based on mitochondrial markers: Different conservation efforts are required for different populations. J. Ornithol. 2019, 160, 711–720. [Google Scholar] [CrossRef]
- Schmidt, K.L.; Aardema, M.L.; Amato, G. Genetic analysis reveals strong phylogeographical divergences within the Scarlet Macaw Ara macao. Ibis 2020, 162, 735–748. [Google Scholar] [CrossRef]
- Rivera-Arroyo, R.C.; Escalante-Pliego, P.; Aguilar-Torres, D.; Úbeda-Olivas, M.F. Phylogeography of the white-crowned parrot (Pionus senilis). Biota Neotrop. 2023, 22, 1382. [Google Scholar] [CrossRef]
- Marín-Togo, M.C.; Monterrubio-Rico, T.C.; Renton, K.; Rubio-Rocha, Y.; Macías-Caballero, C.; Ortega-Rodríguez, J.M.; Cancino-Murillo, R. Reduced current distribution of Psittacidae on the Mexican Pacific coast: Potential impacts of habitat loss and capture for trade. Biodivers. Conserv. 2012, 21, 451–473. [Google Scholar] [CrossRef]
- Hendrickson, S.L.; Bleiweiss, R.; Matheus, J.C. Low genetic variability in the geographically widespread Andean condor. Condor 2003, 105, 1–12. [Google Scholar] [CrossRef]
- Westemeier, R.L.; Brawn, J.D.; Simpson, S.A.; Esker, T.L.; Jansen, R.W.; Walk, J.W.; Kershner, E.L.; Bouzat, J.L.; Paige, K.N. Tracking the long-term decline and recovery of an isolated population. Science 1998, 282, 1695–1698. [Google Scholar] [CrossRef]
- Johnson, J.A.; Bellinger, M.R.; Toepfer, J.E.; Dunn, P. Temporal changes in allele frequencies and low effective population size in greater prairie-chickens. Mol. Ecol. 2004, 13, 2616–2630. [Google Scholar] [CrossRef] [PubMed]
- Barrowclough, G.F.; Gutiérrez, R.J.; Groth, J.G. Phylogeography of spotted owl (Strix occidentalis) populations based on mitochondrial DNA sequences: Gene flow, genetic structure, and a novel biogeographic pattern. Evolution 1999, 53, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Solórzano, S.; Baker, A.J.; Oyama, K. Conservation priorities for resplendent quetzals based on analysis of mitochondrial DNA control region sequences. Condor 2004, 106, 449–456. [Google Scholar] [CrossRef]
- Mock, K.E.; Theimer, T.C.; Rhodes, O.E.; Greenberg, D.L.; Keim, P. Genetic variation across the historical range of the Wild Turkey. Mol. Ecol. 2002, 11, 643–657. [Google Scholar] [CrossRef]
- Proudfoot, G.A.; Honeycutt, R.; Slack, R. Mitochondrial DNA variation and phylogeography of the Ferruginous Pygmy-Owl (Glaucidium brasilianum). Conserv. Genet. 2006, 7, 1–12. [Google Scholar] [CrossRef]
- Firestone, K.B.; Elphinstone, M.S.; Sherwin, W.B.; Houlden, B.A. Phylogeographical population structure of tiger quolls Dasyurus maculatus (Dasyuridae: Marsupialia), an endangered carnivorous marsupial. Mol. Ecol. 1999, 8, 1613–1625. [Google Scholar] [CrossRef]
- Birky, C.W.; Fuerst, P.; Maruyama, T. Organelle gene diversity under migration, mutation, and drift: Equilibrium expectations, approach to equilibrium, effects of heteroplasrnic cells, and comparison to nuclear genes. Genetics 1989, 121, 613–627. [Google Scholar] [CrossRef]
- Perrin, N.; Mazalov, V. Local competition, inbreeding, and the evolution of sex-biased dispersal. Am. Nat. 2000, 155, 116–127. [Google Scholar] [CrossRef]
- Bonaccorso, E.; Peterson, A.T.; Navarro-Sigüenza, A.G.; Fleischer, R.C. Molecular systematics and evolution of the Cyanocorax jays. Mol. Phylogenet. Evol. 2010, 54, 897–909. [Google Scholar] [CrossRef]
- Rivera-Ortiz, F.A.; Contreras-González, A.M.; Soberanes-González, C.A.; Valiente-Banuet, A.; Arizmendi, M.C. Seasonal abundance and breeding chronology of the Military Macaw (Ara militaris) in a semi-arid region of central Mexico. Neotrop. Ornithol. 2008, 19, 255–263. [Google Scholar]
- Contreras-González, A.M.; Rivera-Ortiz, F.A.; Soberanes-González, C.; Valiente-Banuet, A.; Arizmendi, M.C. Feeding ecology of Military Macaw (Ara militaris) in a semiarid region of central Mexico. Wilson J. Ornithol. 2009, 121, 384–391. [Google Scholar] [CrossRef]
- Barhoum, D.N.; Burns, K.J. Phylogenetic relationships of the Wrentit based on mitochondrial cytochrome b sequences. Condor 2002, 104, 740–749. [Google Scholar] [CrossRef]
- Pennington, R.T.; Prado, D.E.; Pendry, C.A. Neotropical seasonally dry forests and Quaternary vegetation changes. J. Biogeogr. 2000, 27, 261–273. [Google Scholar] [CrossRef]
- Oswald, J.A.; Steadman, D.W. The changing diversity and distribution of dry forest passerine birds in northwestern Peru since the last ice age. Auk 2015, 132, 836–862. [Google Scholar] [CrossRef]
- Pennington, R.T.; Lavin, M.; Prado, D.E.; Pendry, C.A.; Pell, S.K.; Butterworth, C.A. Historical climate change and speciation: Neotropical seasonally dry forest plants show patterns of both Tertiary and Quaternary diversification. Phillos. Trans. R. Soc. 2004, 359, 515–538. [Google Scholar] [CrossRef]
- Oswald, J.A.; Overcast, I.; Mauck, W.M., III; Andersen, M.J.; Smith, B.T. Isolation with asymmetric gene flow during the nonsynchronous divergence of dry forest birds. Mol. Ecol. 2017, 26, 1386–1400. [Google Scholar] [CrossRef]
- Prado, D.E.; Gibbs, P.E. Patterns of species distributions in the dry seasonal forests of South America. Ann. Mo. Bot. Gard. 1993, 80, 902–927. [Google Scholar] [CrossRef]
- Wilkinson, G.S.; Fleming, T.H. Migration and evolution of lesser long-nosed bats Leptonycteris curasoae, inferred from mitochondrial DNA. Mol. Ecol. 1996, 5, 329–339. [Google Scholar] [CrossRef]
- Fjeldsâ, J.; Krabbe, N.; Ridgely, R.S. Great Green Macaw Ara ambigua collected in northwest Ecuador, with taxonomic comments on Ara militaris. Bull. Br. Ornithol. Club. 1987, 107, 28–31. [Google Scholar]
- Hinkelmann, C.; Schuchmann, K.L. Phylogeny of the hermit hummingbirds (Trochilidae: Phaethornithinae). Stud. Neotrop. 1997, 32, 142–163. [Google Scholar] [CrossRef]
- Ferrusquía-Villafranca, I.; Arroyo-Cabrales, J.; Martínez-Hernández, E.; Gama-Castro, J.; Ruiz-Gonzalez, J.; Polaco, O.J.; Johnson, E. Pleistocene mammals of Mexico: A critical review of regional chronofaunas, climate change response and biogeographic provinciality. Quatern. Int. 2010, 217, 53–104. [Google Scholar] [CrossRef]
- Sirkin, L. Late Quaternary stratigraphy and environments of the West Mexican coastal plain. Palynology 1985, 9, 3–25. [Google Scholar] [CrossRef]
- Cornejo-Romero, A.; Vargas-Mendoza, C.F.; Aguilar-Martínez, G.F.; Medina-Sánchez, J.; Rendón-Aguilar, B.; Valverde, P.L.; Zavala-Hurtado, J.A.; Serrato, A.; Rivas-Arancibia, S.; Pérez-Hernández, M.A.; et al. Alternative glacial-interglacial refugia demographic hypotheses tested on Cephalocereus columna-trajani (Cactaceae) in the intertropical Mexican drylands. PLoS ONE 2017, 12, e0175905. [Google Scholar] [CrossRef]
- Cordova, C.E. Lacustrine Change in the Late Quaternary. In The Lakes of the Basin of Mexico: Dynamics of a Lacustrine System and the Evolution of a Civilization; Cordova, C.E., Ed.; Springer: Stillwater, OK, USA, 2022; pp. 123–140. [Google Scholar] [CrossRef]
- Arbelaez-Cortes, E.; Navarro-Sigueenza, A.G. Molecular evidence of the taxonomic status of western Mexican populations of Phaethornis longirostris (Aves: Trochilidae). Zootaxa 2013, 3716, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Perini, C.R.; Sosa, V.I.; Koda, V.E.; Silva, H.; Risso, A.A.; Vasconcelos, W.N.; Gonçalves, C.F.; Ugalde, G.A.; Machado, D.N.; Bevilacqua, C.B.; et al. Genetic structure of two Plusiinae species suggests recent expansion of Chrysodeixis includens in the American continent. Agric. For. Entomol. 2021, 23, 250–260. [Google Scholar] [CrossRef]
- Luo, Y.; Li, S. Global expansion of a solitary-social tropical spitting spider shaped by multiple long-distance dispersals. Ecography 2023, 3, e06632. [Google Scholar] [CrossRef]
- Moritz, C. Defining ‘evolutionary significant units’ for conservation. Trends Ecol. Evol. 1994, 9, 373–375. [Google Scholar] [CrossRef]
- Moritz, C. Applications of mitochondrial DNA analysis in conservation: Critical review. Mol. Ecol. 1994, 3, 401–411. [Google Scholar] [CrossRef]
- Frankham, R.; Ballou, J.D.; Briscoe, D.A. Introduction to Conservation Genetics; Cambridge University Press: Cambridge, UK, 2010; pp. 161–181. [Google Scholar]
- Avise, J.C.; Ball, M.R. Principles of geneological concordance in species concepts and biological taxonomy. In Oxford Surveys in Evolutionary Biology; Futuyma, D.J., Antonovics, J., Eds.; Oxford University Press: Oxford, UK, 1990; pp. 45–67. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Regions (Subspecies) | Locality (State/Department) Abbreviation | Location | Altitude (m) | Precipitation (mm) | Temperature (°C) | Vegetation |
|---|---|---|---|---|---|---|
| Mexico (A. m. mexicanus) | La Sierrita (Sonora) LS º | 26° 52′ 48″ N 108° 34′ 12″ W | 800–1200 | 60 | 22 | Tropical deciduous forest |
| Cósala (Sinaloa) C º | 24° 24′ 44″ N 106° 41′ 22″ W | 500–1800 | 250 | 24 | Tropical deciduous forest | |
| Mirador del Águia (Nayarit) MA º | 21° 30′ 28″ N 104° 55′ 47″ W | 600–1200 | 1121 | 21 | Tropical subdeciduous forest | |
| Barranca del Oro (Jalisco) T + | 21° 50′ 53″ N 105° 07′ 44″ O | 600–800 | 1000 | 28 | Tropical deciduous forest | |
| Quimichis (Nayarit) Q + | 22° 15′ 49″ N 105° 34′ 55″O | 100–300 | 1200 | 26 | Tropical deciduous forest | |
| Los Mazos (Jalisco) LM + | 19° 33′ 57″ N 103° 30′ 25″ O | 1400–1800 | 78 | 21 | Tropical deciduous forest | |
| Las Peñas (Jalisco) LP + | 19° 41′ 58″ N 103° 26′ 41″ O | 1500–1800 | 690 | 27 | Tropical deciduous forest | |
| Volcán de Colima (Jalisco) VC + | 19° 28′ 15″ N 103° 56′ 19″ O | 700–2200 | 800 | 27 | Tropical subdeciduous forest | |
| Bahia Balderas (Jalisco) BB + | 20° 34′ 10″ N 104° 59′ 22″ O | 10–600 | 930 | 28 | Tropical subdeciduous forest | |
| El Tuito (Jalisco) ET º | 20° 17′ 35″ N 105° 23′ 6.4″ W | 0–400 | 800 | 26 | Tropical subdeciduous forest | |
| Sta. Tecomavaca (Oaxaca) ST º | 17° 51′ 43″ N 97° 02′ 40″ W | 660–820 | 400 | 22 | Tropical deciduous forest | |
| El Cielo (Tamaulipas) EC º | 23° 04′ 22″ N 99° 09′ 24″ W | 700–1400 | 1800 | 18 | Tropical subdeciduous forest | |
| Sta. Cocos (Querétaro) SC º | 21° 18′ 37″ N 99° 40′ 4″ W | 700–1800 | 400 | 22 | Tropical deciduous forest | |
| Colombia (A. m. militaris) | Sta. Marta (La Magdalena) SM + | 11° 9′ 41″ N 73° 42′ 26″ O | 200–800 | 1000 | 24 | Tropical deciduous forest |
| Bogotá * (Cundinamarca) B + | 06° 17′ 20″ N 74° 37′ 36″ O | 1200 | 2000 | 25 | Tropical subdeciduous forest | |
| Peru (A. m. militaris) | Loreta (Loreto) L + | 04° 11′ 35″ S 74° 12′ 24″ O | 100–300 | 4000 | 25 | Tropical subdeciduous forest |
| Bolivia (A. m. bolivianus) | Sara (Santa Cruz) S + | 14° 48′ 41″ S 62° 07′ 24″ O | 200–400 | 1500 | 27 | Tropical subdeciduous forest |
| Subspecies | Populations Abbreviation | Locality | N | P | Cyt-b Haplotypes | π | h | D | Fs |
|---|---|---|---|---|---|---|---|---|---|
| A. m. mexicanus | Mexico-Sierra Madre Occidental/Sierra Madre del Sur 1 SMOc1 | LS | 1 | 1 | Hap5 | — | — | — | — |
| Mexico-Sierra Madre Occidental/Sierra Madre del Sur 2 SMOc2 | C | 16 | 8 | Hap 15, Hap 23, Hap 24, Hap 25, Hap 26, Hap 27, Hap 28, Hap 29. | 0.0022 | 0.7000 | −1.6238 | −4.5029 | |
| Mexico-Sierra Madre Occidental/Sierra Madre del Sur 3 SMOc3 | Q, MA | 6 | 5 | Hap 12, Hap 13, Hap 16, Hap 17, Hap 23. | 0.0017 | 0.9333 | −1.2331 | 5.8783 | |
| Mexico-Sierra Madre Occidental/Sierra Madre del Sur 4 SMOc4 | LM, LP, VC | 13 | 9 | Hap 6, Hap 7, Hap 9, Hap 17, Hap 18, Hap 19, Hap 20, Hap 21, Hap 22. | 0.0018 | 0.9231 | 0.6005 | −1.6903 | |
| Mexico-Sierra Madre Occidental/Sierra Madre del Sur 5 SMOc5 | BB, ET, BO | 8 | 3 | Hap 8, Hap 17, Hap 23. | 0.0011 | 0.7143 | 0.3335 | 22.7426 | |
| Mexico-Sierra Madre Occidental/Sierra Madre del Sur 6 SMOc6 | ST | 4 | 4 | Hap 10, Hap 11, Hap 14, Hap 23. | 0.0040 | 0.9736 | 0.6501 | −1.6221 | |
| Mexico-Sierra Madre Oriental 1 SMOr1 | SC | 2 | 2 | Hap 1, Hap 3. | 0.0051 | 0.9843 | 0.0000 | 1.0986 | |
| Mexico-Sierra Madre Oriental 2 SMOr2 | EC | 3 | 3 | Hap 2, Hap 3, Hap 4. | 0.0034 | 0.9801 | 0.0000 | −0.6933 | |
| Total subspecies | — | 53 | 35 | 29 | 0.0024 ± 00.0002 | 0.7760 ± 0.0417 | — | — | |
| A. m. militaris | Colombia-1 C1 | SM | 2 | 2 | Hap 39, Hap 40. | 0.0045 | 0.9765 | 0.0000 | 1.3862 |
| Colombia-2 * C2 | B | 3 | 3 | Hap 30, Hap 31, Hap 38. | 0.0284 | 0.9856 | 0.0000 | 0.7032 | |
| Peru-1 P1 | L | 4 | 4 | Hap 32, Hap 37, Hap 40, Hap 41. | 0.0147 | 0.9887 | −0.3144 | −0.5683 | |
| Total subspecies | — | 9 | 9 | 8 | 0.0158 ± 0.0084 | 0.9836 ± 0.0032 | — | — | |
| A. m. bolivianus | Bolivia-1 B1 | S | 5 | 4 | Hap 33, Hap 34, Hap 35, Hap 36. | 0.0097 | 0.9000 | −0.8429 | 4.7699 |
| Overall | — | 67 | 48 | 41 | 0.0063 ± 0.0007 | 0.8382 ± 0.0257 | — | — |
| Source of Variation | Percentage of Variation | |
|---|---|---|
| Among groups * | 45.33 | FCT = 0.8071 |
| Among population within groups | 12.40 | FSC = 0.2208 |
| Within populations | 42.26 | FST = 0.7525 |
| Subspecies | A. m. mexicanus | A. m. miliatris | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A. m. mexicanus | Populations | SMOc1 | SMOc2 | SMOc3 | SMOc4 | SMOc5 | SMOc6 | SMOr1 | SMOr2 | C1 | C2 + | P1 |
| SMOc1 | — | |||||||||||
| SMOc2 | 0.0606 | — | ||||||||||
| SMOc3 | 0.1397 | 0.0146 | — | |||||||||
| SMOc4 | 0.0986 | 0.0008 | 0.8828 | — | ||||||||
| SMOc5 | 0.3008 | 0.0001 | 0.9990 | 0.4775 | — | |||||||
| SMOc6 | 0.2002 | 0.0009 | 0.5175 | 0.7568 | 0.9990 | — | ||||||
| SMOr1 | 0.3594 | 0.0097 | 0.0429 | 0.0263 | 0.9990 | 0.0634 | — | |||||
| SMOr2 | 0.2422 | 0.0009 | 0.0087 | 0.0039 | 0.9824 | 0.0166 | 0.6943 | — | ||||
| A. m. miliatris | C1 | 0.3428 | 0.0068 | 0.0341 | 0.0175 | 0.0097 | 0.0498 | 0.3339 | 0.0869 | — | ||
| C2 + | 0.9990 | 0.0058 | 0.0351 | 0.0068 | 0.0019 | 0.1298 | 0.4013 | 0.4111 | 0.4111 | — | ||
| P1 | 0.5869 | 0.0705 | 0.0058 | 0.0019 | 0.1488 | 0.0585 | 0.3447 | 0.0537 | 0.6523 | 0.0771 | — | |
| A. m. bolivianus | B1 | 0.3389 | 0.0187 | 0.0048 | 0.0115 | 0.0540 | 0.0195 | 0.0430 | 0.0176 | 0.0430 | 0.0195 | 0.0127 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivera-Ortíz, F.A.; Sanabria-Urbán, S.; Prieto-Torres, D.A.; Navarro-Sigüenza, A.G.; Arizmendi, M.d.C.; Oyama, K. Phylogeography of Ara militaris (Military Macaw): Implications for Conservation. Diversity 2023, 15, 1035. https://doi.org/10.3390/d15101035
Rivera-Ortíz FA, Sanabria-Urbán S, Prieto-Torres DA, Navarro-Sigüenza AG, Arizmendi MdC, Oyama K. Phylogeography of Ara militaris (Military Macaw): Implications for Conservation. Diversity. 2023; 15(10):1035. https://doi.org/10.3390/d15101035
Chicago/Turabian StyleRivera-Ortíz, Francisco A., Salomón Sanabria-Urbán, David A. Prieto-Torres, Adolfo G. Navarro-Sigüenza, María del C. Arizmendi, and Ken Oyama. 2023. "Phylogeography of Ara militaris (Military Macaw): Implications for Conservation" Diversity 15, no. 10: 1035. https://doi.org/10.3390/d15101035
APA StyleRivera-Ortíz, F. A., Sanabria-Urbán, S., Prieto-Torres, D. A., Navarro-Sigüenza, A. G., Arizmendi, M. d. C., & Oyama, K. (2023). Phylogeography of Ara militaris (Military Macaw): Implications for Conservation. Diversity, 15(10), 1035. https://doi.org/10.3390/d15101035