
Population Genetic Structure and Biodiversity Conservation of a Relict and Medicinal Subshrub Capparis spinosa in Arid Central Asia



Abstract
:1. Introduction
2. Materials and Methods
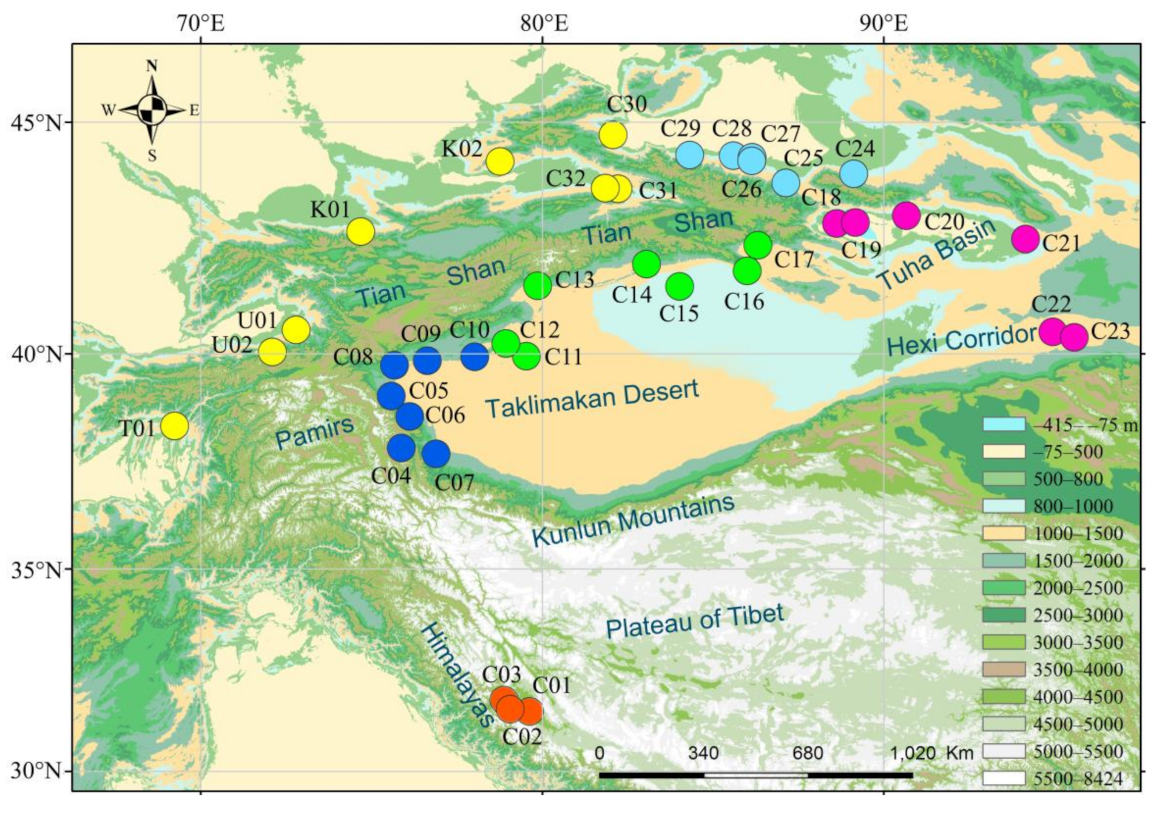
2.1. Sampling, DNA Extraction and dd-RAD Library Construction
2.2. Data Processing and SNP Calling
2.3. Genetic Structure Analysis
2.4. Genetic Diversity and IBD Analysis
3. Results
3.1. SNPs from dd-RAD Analysis of C. spinosa
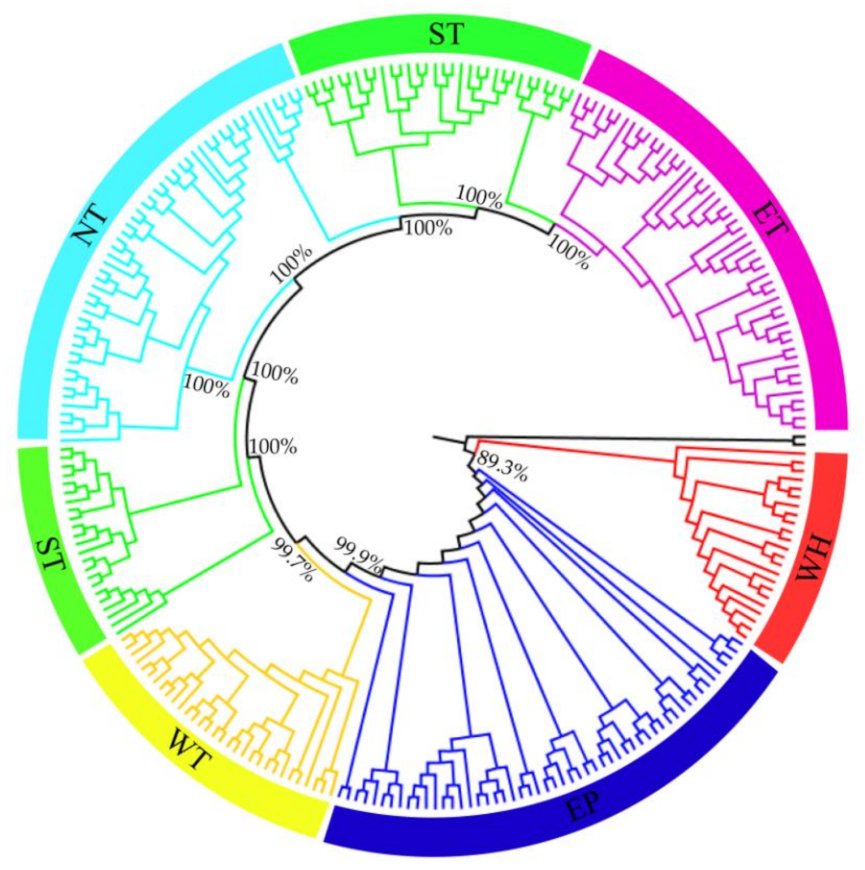
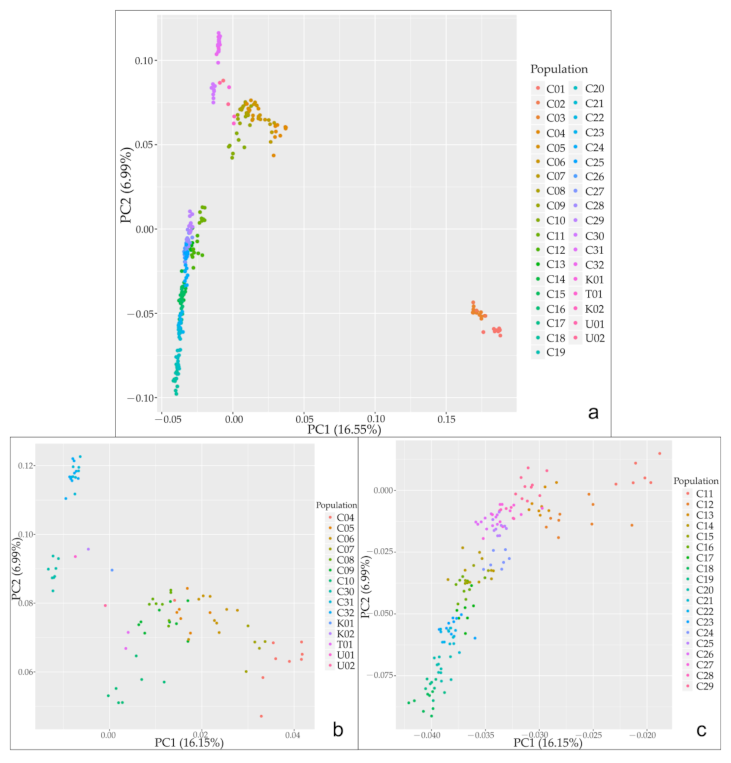
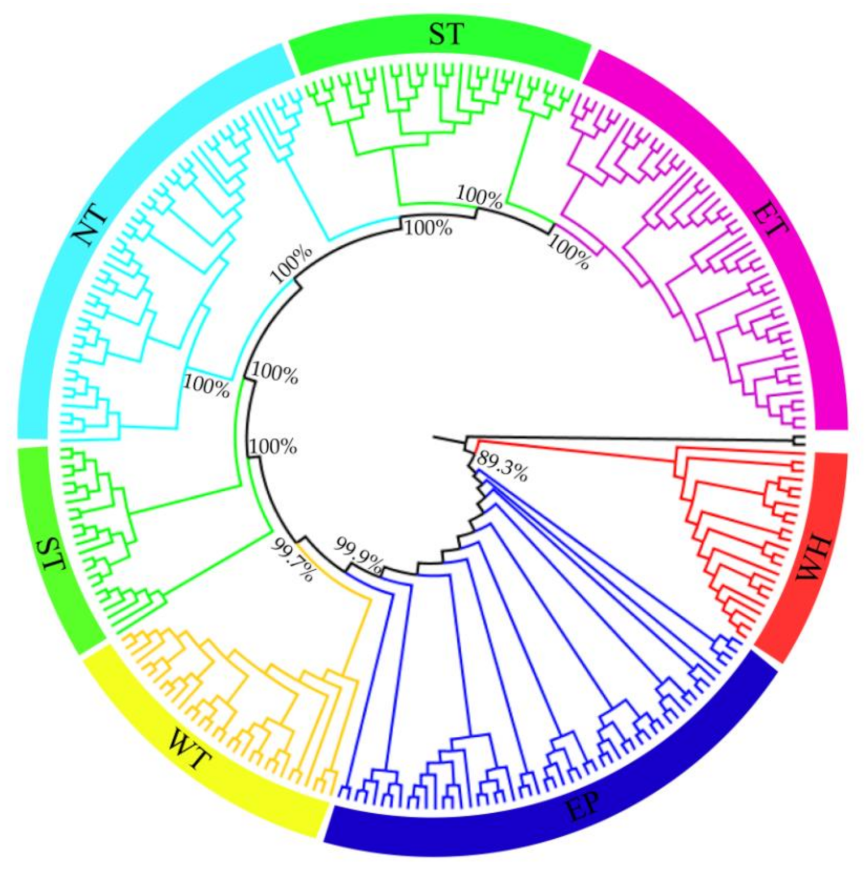
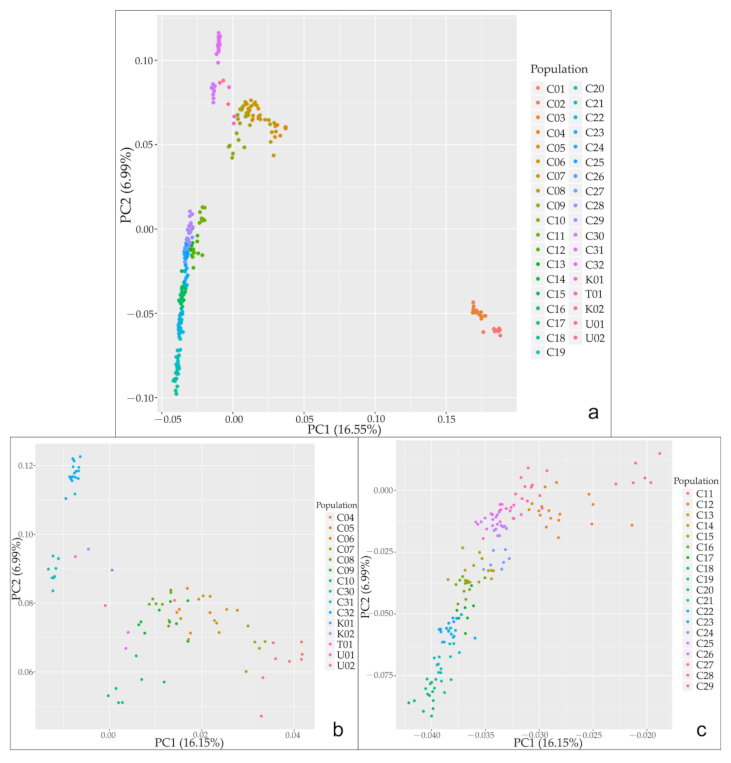
3.2. Population Genetic Structure of C. spinosa
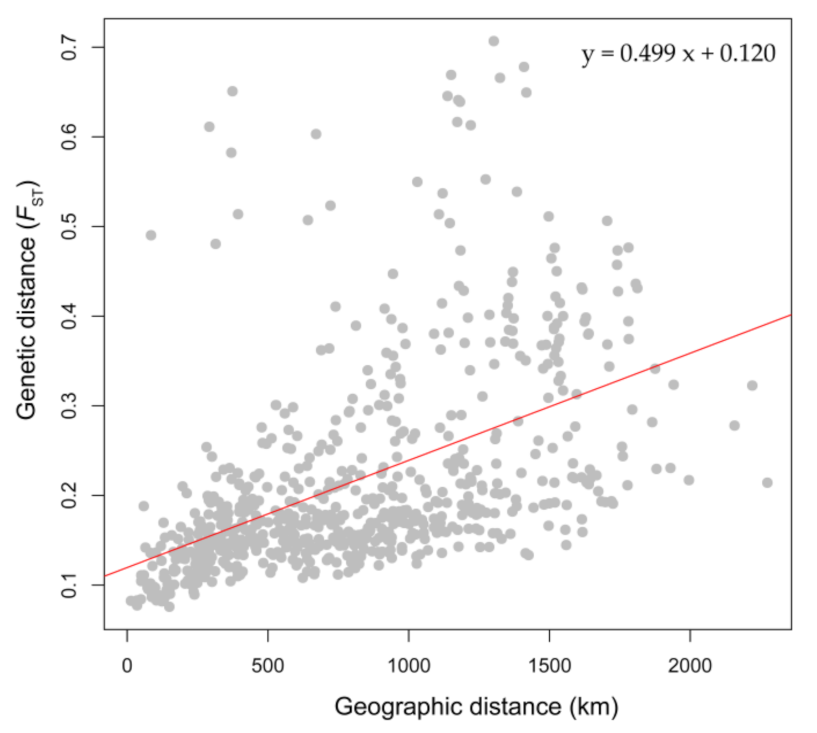
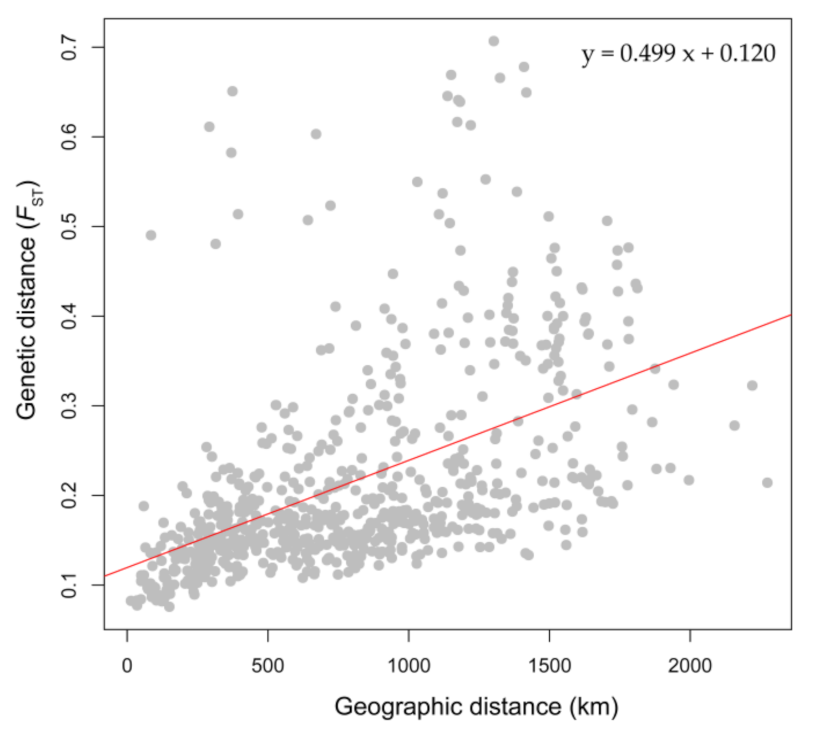
3.3. Genetic Diversity Pattern and IBD
4. Discussion
4.1. Relationships between Genetic Structure, Lineage Differentiation and Geographic Distribution
4.2. Genetic Diversity Pattern of Species Metapopulations
4.3. Implications for Conservation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, X.; Jiang, F.Q.; Wang, Y.J.; Li, Y.M.; Hu, R.J. Characteristics of the eco-geographical pattern in arid land of Central Asia. Arid Zone Res. 2013, 30, 385–390, (In Chinese with English Abstract). [Google Scholar]
- Yong, S.P.; Zhu, Z.Y. A certain fundamental characteristics of gobi desert vegetation in the Centre Asia. Acta Sci. Nat. Univ. Intramongol. 1992, 23, 235–244, (In Chinese with English Abstract). [Google Scholar]
- Wu, Z.Y.; Sun, H.; Zhou, Z.K.; Li, D.Z.; Peng, H. Floristics of Seed Plants from China; Science Press: Beijing, China, 2010. (In Chinese) [Google Scholar]
- Liu, Y.X. A study on origin and formation of the Chinese desert floras. Acta Phyt. Sin. 1995, 33, 131–143, (In Chinese with English Abstract). [Google Scholar]
- Sun, H.; Li, Z.M. Qinghai-Tibet Plateau uplift and its impact on Tethys Flora. Adv. Earth Sci. 2003, 18, 852–862. [Google Scholar]
- Fang, X.M.; Lv, L.Q.; Yang, S.L.; Li, J.J.; An, Z.S.; Jiang, P.A.; Chen, X.L. Loess in Kunlun Mountain and its implications on desert development and Tibetan Plateau uplift in west China. Sci. China Ser. D 2002, 45, 289–299. [Google Scholar] [CrossRef]
- Guan, Q.Y.; Pan, B.T.; Li, N.; Zhang, J.D.; Xue, L.J. Timing and significance of the initiation of present day deserts in the northeastern Hexi Corridor, China. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2011, 306, 70–74. [Google Scholar]
- Zhang, L.Y.; Hai, Y. Plant communities excluded in the book of “The Vegetation and its Utilization in Xinjiang”: I. The desert plant communities. Arid Land Geogr. 2002, 25, 84–89. [Google Scholar]
- Su, Z.H.; Pan, B.R.; Zhang, M.L.; Shi, W. Conservation genetics and geographic patterns of genetic variation of endangered shrub Ammopiptanthus (Fabaceae) in northwestern China. Conserv. Genet. 2016, 17, 485–496. [Google Scholar] [CrossRef]
- Ma, S.M.; Nie, Y.B.; Jiang, X.L.; Xu, Z.; Ji, W.Q. Genetic structure of the endangered, relict shrub Amygdalus mongolica (Rosaceae) in arid northwest China. Aust. J. Botany 2019, 67, 128–139. [Google Scholar] [CrossRef]
- Zhang, H.X.; Wang, Q.; Jia, S.W. Genomic phylogeography of Gymnocarpos przewalskii (Caryophyllaceae): Insights into habitat fragmentation in arid Northwestern China. Diversity 2020, 12, 335. [Google Scholar] [CrossRef]
- Zhang, M.L.; Tucker, G.C. Capparaceae. In Flora of China; Wu, Z.Y., Raven, P.H., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2008; Volume 7, pp. 433–450. [Google Scholar]
- Wu, S.K. Capparidaceae. In Flora of Tibet; Wu, Z.Y., Ed.; Science Press: Beijing, China, 1985; Volume 2, pp. 322–323. (In Chinese) [Google Scholar]
- An, Z.X. Capparidaceae. In Flora Xinjiangensis; Mao, Z.M., Ed.; Xinjiang Science, Technology and Hygiene Publishing House: Urumqi, China, 1995; Volume 2, pp. 35–36. (In Chinese) [Google Scholar]
- Fici, S.; Gianguzzi, L. Diversity and conservation in wild and cultivated Capparis in Sicily. Bocconea 1997, 7, 437–443. [Google Scholar]
- Saadaoui, E.; Khaldi, A.; Khouja, M.L.; Mohamed, E.G. Intraspecific variation of Capparis spinosa L. in Tunisia. J. Herbs Spices Med. Plants 2009, 15, 9–15. [Google Scholar] [CrossRef]
- Fici, S. A taxonomic revision of the Capparis spinosa group (Capparaceae) from the Mediterranean to Central Asia. Phytotaxa 2014, 174, 001–024. [Google Scholar] [CrossRef] [Green Version]
- Özbek, Ö.; Kara, A. Genetic variation in natural populations of Capparis from Turkey, as revealed by RAPD analysis. Plant Syst. Evol. 2013, 299, 1911–1933. [Google Scholar] [CrossRef]
- Al-Safadi, B.; Faouri, H.; Elias, R. Genetic diversity of some Capparis L. species growing in Syria. Braz. Arch. Biol. Technol. 2014, 57, 916–926. [Google Scholar] [CrossRef]
- Aichi-Yousfi, H.; Bahri, B.A.; Medini, M.; Rouz, S.; Rejeb, M.N.; Ghrabi-Gammar, Z. Genetic diversity and population structure of six species of Capparis in Tunisia using AFLP markers. Comptes Rendus Biol. 2016, 339, 442–453. [Google Scholar] [CrossRef]
- Mercati, F.; Fontana, I.; Gristina, A.S.; Martorana, A.; El Nagar, M.; De Michele, R.; Fici, S.; Carimi, F. Transcriptome analysis and codominant markers development in caper, a drought tolerant orphan crop with medicinal value. Sci. Rep. 2019, 9, 10411. [Google Scholar] [CrossRef] [Green Version]
- Futuyma, D.J. Evolutionary Biology, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 1986. [Google Scholar]
- Vellend, M.; Geber, M.A. Connections between species diversity and genetic diversity. Ecol. Lett. 2005, 8, 767–781. [Google Scholar] [CrossRef]
- Frankham, R. Genetics and extinction. Biol. Conserv. 2005, 126, 131–140. [Google Scholar] [CrossRef]
- Sakai, A.K.; Allendorf, F.W.; Holt, J.S.; Lodge, D.M.; Molofsky, J.; With, K.A.; Baughman, S.; Cabin, R.J.; Cohen, J.E.; Ellstrand, N.C.; et al. The population biology of invasive species. Annu. Rev. Ecol. Syst. 2001, 32, 305–332. [Google Scholar] [CrossRef] [Green Version]
- Pulliam, R.H. Sources, sinks, and population regulation. Am. Nat. 1988, 132, 652–661. [Google Scholar] [CrossRef]
- Heinrichs, J.A.; Lawler, J.J.; Schumaker, N.H. Intrinsic and extrinsic drivers of source–sink dynamics. Ecol. Evol. 2016, 6, 892–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, S. Isolation by distance. Genetics 1943, 28, 114–138. [Google Scholar] [CrossRef] [PubMed]
- Walters, S. Landscape pattern and productivity effects on source–sink dynamics of deer populations. Ecol. Model. 2001, 143, 17–32. [Google Scholar] [CrossRef]
- Crutzen, P.J. Geology of mankind. Nature 2002, 415, 23. [Google Scholar] [CrossRef]
- Lewis, S.L.; Maslin, M.A. Defining the Anthropocene. Nature 2015, 519, 171–180. [Google Scholar] [CrossRef]
- Meng, H.H.; Zhou, S.S.; Li, L.; Tan, Y.H.; Li, J.W.; Li, J. Confict between biodiversity conservation and economic growth: Insight into rare plants in tropical China. Biodivers. Conserv. 2019, 28, 523–537. [Google Scholar] [CrossRef]
- Hanski, I. Metapopulation dynamics. Nature 1998, 396, 41–49. [Google Scholar] [CrossRef]
- Nunes, F.; Norris, R.D.; Knowlton, N. Implications of isolation and low genetic diversity in peripheral populations of an amphi-Atlantic coral. Mol. Ecol. 2009, 18, 4283–4297. [Google Scholar] [CrossRef]
- Myers, N.; Mittermeier, R.A.; Mittermeier, C.G.; da Fonseca, G.A.B.; Kent, J. Biodiversity hotspots for conservation priorities. Nature 2000, 403, 853–858. [Google Scholar] [CrossRef]
- Kessler, M. Patterns of diversity and range size of selected plant groups along an elevational transect in the Bolivian Andes. Biodivers. Conserv. 2001, 10, 1897–1921. [Google Scholar] [CrossRef]
- Liu, J.; Moller, M.; Provan, J.; Gao, L.M.; Poudel, R.M.; Li, D.Z. Geological and ecological factors drive cryptic speciation of yews in a biodiversity hotspot. New Phytol. 2013, 199, 1093–1108. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Mazuecos, M.; Jiménez-Mejías, P.; Rotllan-Puig, X.; Vargas, P. Narrow endemics to Mediterranean islands: Moderate genetic diversity but narrow climatic niche of the ancient, critically endangered Naufraga (Apiaceae). Perspect. Plant Ecol. Evol. Syst. 2014, 16, 190–202. [Google Scholar] [CrossRef]
- Millar, C.I.; Libby, W.J. Strategies for conserving clinal, ecotypic, and disjunct population diversity in widespread species. In Genetics and Conservation of Rare Plants; Falk, D.A., Holsinger, K.E., Eds.; Oxford University Press: New York, NY, USA, 1991; Volume 10, pp. 149–170. [Google Scholar]
- Brumfield, R.T.; Beerli, P.; Nickerson, D.A.; Edwards, S.V. The utility of single nucleotide polymorphisms in inferences of population history. Trends Ecol. Evol. 2003, 18, 249–256. [Google Scholar] [CrossRef]
- Morin, P.A.; Luikart, G.; Wayne, R.K.; The SNP Workshop Group. SNPs in ecology, evolution and conservation. Trends Ecol. Evol. 2004, 19, 208–216. [Google Scholar] [CrossRef]
- Leaché, A.D.; Oaks, J.R. The utility of single nucleotide polymorphism (SNP) data in phylogenetics. Annu. Rev. Ecol. Evol. Syst. 2017, 48, 69–84. [Google Scholar] [CrossRef]
- Zhang, H.X.; Li, X.S.; Wang, J.C.; Zhang, D.Y. Insights into the aridification history of Central Asian Mountains and international conservation strategy from the endangered wild apple tree. J. Biogeogr. 2021, 48, 332–344. [Google Scholar] [CrossRef]
- Davey, J.W.; Blaxter, M.L. RADSeq: Next-generation population genetics. Brief. Funct. Genom. 2011, 9, 416–423. [Google Scholar] [CrossRef]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [Green Version]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 2016, 17, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.L.; Gardner, E.M.; Meng, H.H.; Deng, M.; Xu, G.B. Land bridges in the Pleistocene contributed to flora assembly on the continental islands of South China: Insights from the evolutionary history of Quercus championii. Mol. Phylogenet. Evol. 2019, 132, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.N.; Zhu, S.S.; Chen, J.; Comes, H.P.; Wang, I.J.; Chen, L.Y.; Sakaguchi, S.; Qiu, Y.X. Genomic insights into historical population dynamics, local adaptation, and climate change vulnerability of the East Asian Tertiary relict Euptelea (Eupteleaceae). Evol. Appl. 2020, 13, 2038–2055. [Google Scholar] [CrossRef] [PubMed]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Res. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Meng, H.H.; Gao, X.Y.; Huang, J.F.; Zhang, M.L. Plant phylogeography in arid Northwest China: Retrospectives and perspectives. J. Syst. Evol. 2015, 53, 33–46. [Google Scholar] [CrossRef]
- Mims, M.C.; Hauser, L.; Goldberg, C.S.; Olden, J.D. Distance, and metapopulation dynamics of the Arizona Treefrog (Hyla wrightorum) in an isolated portion of its range. PLoS ONE 2016, 11, e0160655. [Google Scholar] [CrossRef] [Green Version]
- Frankham, R.; Ballou, J.D.; Briscoe, D.A. Introduction to Conservation Genetics; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Beebee, T.J.C. Conservation genetics of amphibians. Heredity 2005, 95, 423–427. [Google Scholar] [CrossRef]
- Tero, N.; Aspi, J.; Siikamäki, P.; Jäkäläniemi, A.; Tuomi, J. Genetic structure and gene flow in a metapopulation of an endangered plant species, Silene tatarica. Mol. Ecol. 2003, 12, 2073–2085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, L.M.; Mosseler, A.; Rajora, O.P. Extensive long-distance pollen dispersal in a fragmented landscape maintains genetic diversity in white spruce. J. Hered. 2007, 98, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Howe, R.W.; Davis, G.J.; Mosca, G.J. The demographic significance of sink populations. Biol. Conserv. 1991, 57, 239–255. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Pop. Locality | Ho | He | Pi |
|---|---|---|---|---|
| Group WH | (Western Himalayas) | 0.0455 | 0.0648 | 0.0671 |
| C01 | Zabrang, Tibet, China | 0.0394 | 0.0429 | 0.0465 |
| C02 | Sarang, Tibet, China | 0.0512 | 0.0448 | 0.0594 |
| C03 | Diya, Tibet, China | 0.0528 | 0.0637 | 0.0687 |
| Group EP | (Eastern Pamirs) | 0.1169 | 0.2283 | 0.2318 |
| C04 | Taxkorgan, Xinjiang, China | 0.1109 | 0.1822 | 0.2003 |
| C05 | Akto, Xinjiang, China | 0.1154 | 0.1665 | 0.1932 |
| C06 | Yengisar, Xinjiang, China | 0.1168 | 0.1805 | 0.1989 |
| C07 | Yecheng, Xinjiang, China | 0.1220 | 0.1420 | 0.1703 |
| C08 | Wuqia, Xinjiang, China | 0.1026 | 0.1665 | 0.1858 |
| C09 | Artux, Xinjiang, China | 0.1234 | 0.1946 | 0.2134 |
| C10 | Jiashi, Xinjiang, China | 0.1273 | 0.1991 | 0.2234 |
| Group WT | (Western Tianshan) | 0.1025 | 0.1914 | 0.1965 |
| K02 | Almaty, Kazakhstan | 0.0932 | 0.0466 | 0.0932 |
| K01 | Bishkek, Kyrgyzstan | 0.0561 | 0.0281 | 0.0561 |
| U01 | Osh, Kyrgyzstan | 0.1057 | 0.0529 | 0.1057 |
| U02 | Batken, Kyrgyzstan | 0.1115 | 0.0558 | 0.1115 |
| T01 | Nurek, Khatlon, Tajikistan | 0.0940 | 0.0769 | 0.1154 |
| C30 | Bole, Xinjiang, China | 0.1150 | 0.1693 | 0.1866 |
| C31 | Yining, Xinjiang, China | 0.1021 | 0.1367 | 0.1539 |
| C32 | Gongliu, Xinjiang, China | 0.1020 | 0.1475 | 0.1602 |
| Group ST | (southern piedmont of Tianshan) | 0.1125 | 0.2223 | 0.2255 |
| C11 | Tumxuk, Xinjiang, China | 0.0911 | 0.1372 | 0.1585 |
| C12 | Kalpin, Xinjiang, China | 0.1178 | 0.1840 | 0.2042 |
| C13 | Wensu, Xinjiang, China | 0.1145 | 0.1739 | 0.1945 |
| C14 | Baicheng, Xinjiang, China | 0.1208 | 0.1745 | 0.1885 |
| C15 | Lunnan, Xinjiang, China | 0.0956 | 0.1321 | 0.1555 |
| C16 | Korla, Xinjiang, China | 0.1135 | 0.1714 | 0.1897 |
| C17 | Hejing, Xinjiang, China | 0.1178 | 0.1691 | 0.1904 |
| Group NT | (northern piedmont of Tianshan) | 0.1206 | 0.2214 | 0.2245 |
| C24 | Jimsar, Xinjiang, China | 0.1155 | 0.1623 | 0.1827 |
| C25 | Urumqi, Xinjiang, China | 0.1120 | 0.1850 | 0.2020 |
| C26 | Manas, Xinjiang, China | 0.1288 | 0.1783 | 0.1925 |
| C27 | Shihezi, Xinjiang, China | 0.1161 | 0.1873 | 0.2046 |
| C28 | Shawan, Xinjiang, China | 0.1298 | 0.1692 | 0.1940 |
| C29 | Wusu, Xinjiang, China | 0.1198 | 0.1915 | 0.2069 |
| Group ET | (Eastern Tianshan) | 0.0999 | 0.1910 | 0.1944 |
| C18 | Toksun, Xinjiang, China | 0.0973 | 0.1270 | 0.1455 |
| C19 | Turpan, Xinjiang, China | 0.1060 | 0.1479 | 0.1631 |
| C20 | Shanshan, Xinjiang, China | 0.1059 | 0.1549 | 0.1699 |
| C21 | Hami, Xinjiang, China | 0.0920 | 0.1310 | 0.1473 |
| C22 | Dunhuang, Gansu, China | 0.1029 | 0.1286 | 0.1516 |
| C23 | Guazhou, Gansu, China | 0.0942 | 0.1595 | 0.1749 |
| Source of Variation | df | Sum of Squares | Variance Components | Percentage of Variation (%) | Φ-Statistics |
|---|---|---|---|---|---|
| Among groups | 5 | 99,228.136 | 212.66396 | 27.36 | ΦCT = 0.27 ** |
| Among populations within groups | 29 | 39,934.919 | 59.33213 | 7.63 | ΦSC = 0.11 ** |
| Within populations | 491 | 248,090.120 | 505.27519 | 65.01 | ΦST = 0.35 ** |
| Total | 525 | 387,253.175 | 777.27128 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Zhang, H.-X. Population Genetic Structure and Biodiversity Conservation of a Relict and Medicinal Subshrub Capparis spinosa in Arid Central Asia. Diversity 2022, 14, 146. https://doi.org/10.3390/d14020146
Wang Q, Zhang H-X. Population Genetic Structure and Biodiversity Conservation of a Relict and Medicinal Subshrub Capparis spinosa in Arid Central Asia. Diversity. 2022; 14(2):146. https://doi.org/10.3390/d14020146
Chicago/Turabian StyleWang, Qian, and Hong-Xiang Zhang. 2022. "Population Genetic Structure and Biodiversity Conservation of a Relict and Medicinal Subshrub Capparis spinosa in Arid Central Asia" Diversity 14, no. 2: 146. https://doi.org/10.3390/d14020146
APA StyleWang, Q., & Zhang, H.-X. (2022). Population Genetic Structure and Biodiversity Conservation of a Relict and Medicinal Subshrub Capparis spinosa in Arid Central Asia. Diversity, 14(2), 146. https://doi.org/10.3390/d14020146