
Comparative Phylogeography of Southern African Bird Species Suggests an Ephemeral Speciation Model




Abstract
:1. Introduction
2. Materials and Methods
2.1. Focal Taxa
2.2. Genetic Data
2.3. Haplotype Network Estimation
2.4. Population and Demographic Analyses
2.5. Environmental Niche Models
3. Results
3.1. Genetic Data
3.2. Haplotype Networks
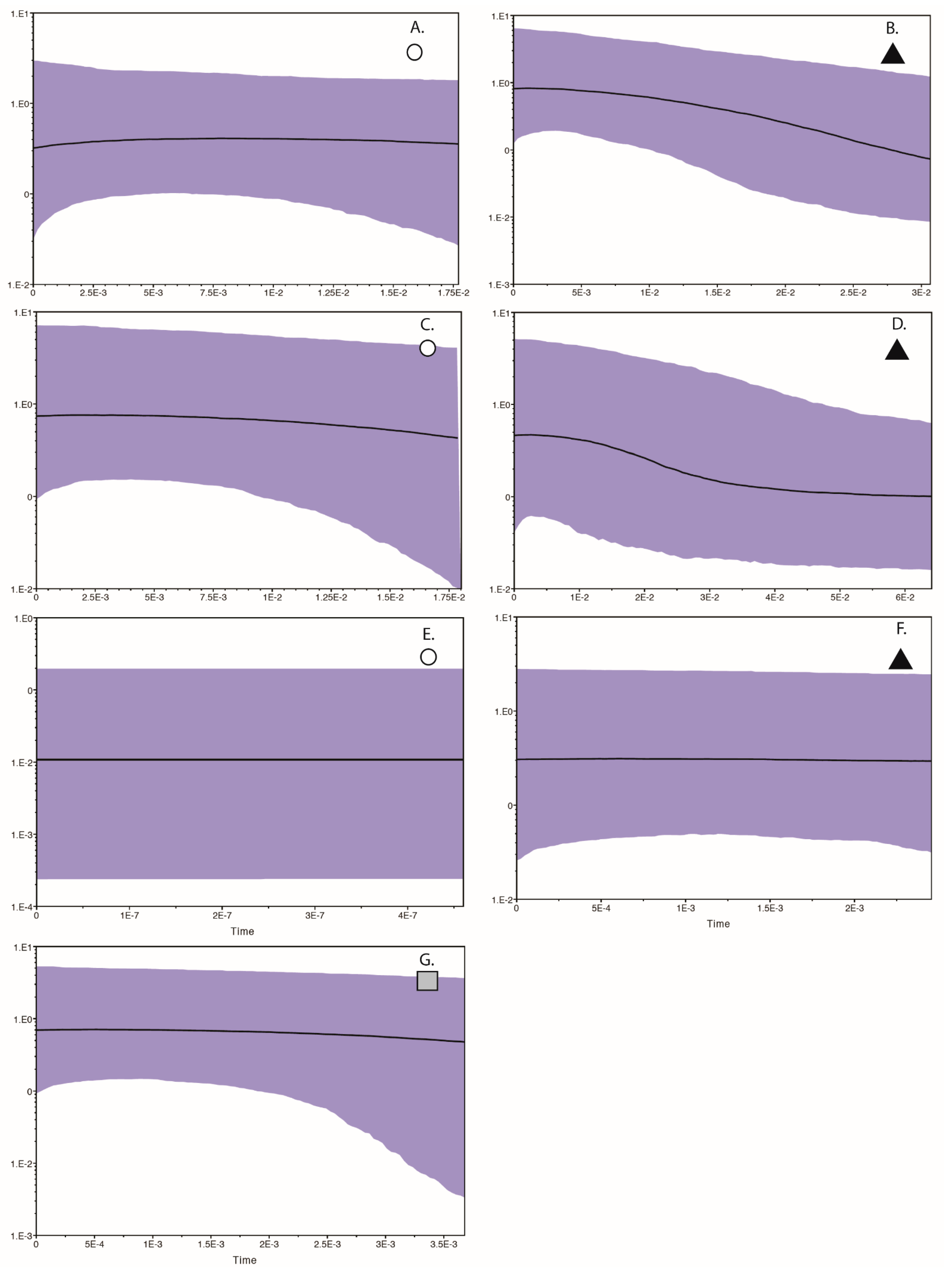
3.3. Population and Demographic Analyses
3.4. Environmental Niche Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hockey, P.A.R.; Dean, W.R.J.; Ryan, P.C. (Eds.) Roberts—Birds of Southern Africa, VIIth ed.; The Trustees of the John Voelcker Bird Book Fund: Cape Town, South Africa, 2005. [Google Scholar]
- Outlaw, R.K.; Voelker, G.; Outlaw, D.C. Molecular systematics and historical biogeography of the Rock-Thrushes (Muscicapidae: Monticola). Auk 2007, 124, 561–577. [Google Scholar] [CrossRef]
- Voelker, G. Dispersal, vicariance and clocks: Historical biogeography and speciation in a cosmopolitan passerine genus (Anthus: Motacillidae). Evolution 1999, 53, 1536–1552. [Google Scholar]
- Voelker, G. Systematics and historical biogeography of wagtails (Aves: Motacilla): Dispersal versus vicariance revisited. Condor 2002, 104, 725–739. [Google Scholar] [CrossRef]
- Voelker, G.; Bowie, R.C.K.; Wilson, B.; Anderson, C. Phylogenetic relationships and speciation patterns in an African savanna dwelling bird genus (Myrmecocichla). Biol. J. Linn. Soc. 2012, 106, 180–190. [Google Scholar] [CrossRef]
- Fuchs, J.; Crowe, T.M.; Bowie, R.C.K. Phylogeography of the fiscal shrike (Lanius collaris): A novel pattern of genetic structure across the arid zones and savannas of Africa. J. Biogeogr. 2011, 38, 2210–2222. [Google Scholar] [CrossRef]
- Fuchs, J.; Fjeldså, J.; Bowie, R.C.K. Diversification across major biogeographic breaks in the African Shining/Square-tailed Drongos complex (Passeriformes: Dicruridae). Zool. Scripta. 2017, 46, 27–41. [Google Scholar] [CrossRef]
- Colville, J.F.; Potts, A.J.; Bradshaw, P.L.; Measey, G.J.; Snijam, D.; Picker, M.D.; Proches, S.; Bowie, R.C.K.; Manning, J.C. Floristic and Faunal Cape biochoria: Do they exist? In Ecology, Evolution, and Conservation of A Megadiverse Region; Allsopp, N., Colville, J.F., Verboom, G.A., Eds.; Oxford University Press: Oxford, UK, 2014; pp. 73–93. [Google Scholar]
- Voelker, G.; Peñalba, J.V.; Huntley, J.W.; Bowie, R.C.K. Diversification in an Afro-Asian songbird clade (Erythropygia–Copsychus) reveals founder-event speciation via trans-oceanic dispersals and a southern to northern colonization pattern in Africa. Mol. Phylogenet. Evol. 2014, 73, 97–105. [Google Scholar] [CrossRef]
- Voelker, G.; Huntley, J.W.; Peñalba, J.V.; Bowie, R.C.K. Resolving taxonomic uncertainty and historical biogeographic patterns in Muscicapa flycatchers and their allies. Mol. Phylogenet. Evol. 2016, 94, 618–625. [Google Scholar] [CrossRef]
- Matthee, C.A.; Fleming, A.F. Population fragmentation in the southern rock agama, Agama atra: More evidence for vicariance in Southern Africa. Mol. Ecol. 2002, 11, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Barlow, A.; Baker, K.; Hendry, C.R.; Peppin, L.; Phelps, T.; Tolley, K.A.; Wüster, C.E.; Wüster, W. Phylogeography of the widespread African puff adder (Bitis arietans) reveals multiple Pleistocene refugia in southern Africa. Mol. Ecol. 2013, 22, 1134–1157. [Google Scholar] [CrossRef]
- Heinicke, M.P.; Turk, D.; Bauer, A.M. Molecular phylogeny reveals strong biogeographic signal and two new species in a Cape Biodiversity Hotspot endemic mini-radiation, the pygmy geckos (Gekkonidae: Goggia). Zootaxa 2017, 4312, 449–470. [Google Scholar] [CrossRef]
- Lorenzen, E.D.; Heller, R.; Siegismund, H.R. Comparative phylogeography of African savannah ungulates. Mol. Ecol. 2012, 21, 3656–3670. [Google Scholar] [CrossRef]
- Smit, H.A.; Robinson, T.J.; van Vuuren, B.J. Coalescence methods reveal the impact of vicariance on the spatial genetic structure of Elephantulus edwardii (Afrotheria, Macroscelidea). Mol. Ecol. 2007, 16, 2680–2692. [Google Scholar] [CrossRef]
- Smit, H.A.; Watson, J.; van Vuuren, B.J. Relative importance of habitat connectivity in shaping the genetic profiles of two southern African elephant-shrews. J. Biogeogr. 2010, 37, 857–864. [Google Scholar] [CrossRef]
- Willows-Munro, S.; Matthee, C.A. Linking lineage diversification to climate and habitat heterogeneity: Phylogeography of the southern African shrew Myosorex Varius. J. Biogeogr. 2011, 38, 1976–1991. [Google Scholar] [CrossRef]
- Tolley, K.A.; Bowie, R.C.K.; Measey, G.J.; Price, B.W.; Forest, F. The shifting landscape of genes since the Pliocene: Terrestrial phylogeography in the Greater Cape Florisitic Region. In Ecology, Evolution, and Conservation of a Megadiverse Region; Allsopp, N., Colville, J.F., Verboom, G.A., Eds.; Oxford University Press: New York, NY, USA, 2014; pp. 142–163. [Google Scholar]
- Fjeldså, J.; Alström, P.; Bowie, R.C.K. Chapter 16: How new species evolve. In The Largest Avian Radiation: The Evolution of Perching Birds, or the Order Passeriformes; Fjeldså, J., Christidis, L., Ericson, P.G.P., Eds.; Lynx Edicions: Barcelona, Spain, 2020; pp. 327–337. [Google Scholar]
- Fjeldså, J.; Bowie, R.C.K. New perspectives on the origin and diversification of Africa’s forest avifauna. Afr. J. Ecol. 2008, 46, 235–247. [Google Scholar] [CrossRef]
- Huntley, J.W.; Keith, K.D.; Castellanos, A.A.; Musher, L.J.; Voelker, G. Underestimated and cryptic diversification patterns across Afro-tropical lowland forests. J. Biogeogr. 2019, 46, 381–391. [Google Scholar] [CrossRef]
- Wogan, G.O.U.; Voelker, G.; Oatley, G.; Bowie, R.C.K. Biome stability predicts population structure of a southern African aridland bird species. Ecol. Evol. 2020, 10, 4066–4081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- du Toit, N.; van Vuuren, B.J.; Matthee, S.; Matthee, C.A. Biome specificity of distinct genetic lineages within the four-striped mouse Rhabdomys pumilio (Rodentia: Muridae) from southern Africa with implications for taxonomy. Mol. Phylogenet. Evol. 2012, 65, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.-R.M.; Chimimba, C.T.; Bloomer, P. Bioregion heterogeneity correlates with extensive mitochondrial DNA diversity in the Namaqua rock mouse, Micaelamys namaquensis (Rodentia: Muridae) from southern Africa–evidence for a species complex. BMC Evol. Biol. 2012, 10, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, J.M.; Tolley, K.A. Diversification through ecological opportunity in dwarf chameleons. J. Biogeogr. 2017, 44, 834–847. [Google Scholar] [CrossRef]
- Evans, K.L.; Rodrigues, A.S.L.; Chown, S.L.; Gaston, K.J. Protected areas and regional avian species richness in South Africa. Biol. Lett. 2006, 2, 184–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rensburg, B.J.; Koleff, P.; Gaston, K.J.; Chown, S.L. Spatial congruence of ecological transition at the regional scale in South Africa. J. Biogeogr. 2004, 31, 843–854. [Google Scholar] [CrossRef]
- Van Rensburg, B.J.; Levin, N.; Kark, S. Spatial congruence between ecotones and range-restricted species: Implications for conservation biogeography at the subcontinental scale. Divers. Distrib. 2009, 15, 379–389. [Google Scholar] [CrossRef]
- Oatley, G.; Voelker, G.; Crowe, T.M.; Bowie, R.C.K. A multi-locus phylogeny reveals a complex pattern of diversification related to climate and habitat heterogeneity in southern African white-eyes. Mol. Phylogenet. Evol. 2012, 64, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.M.; Lloyd, P.; Dean, W.R.J.; Brown, M.; Bowie, R.C.K. The Ecological and Geographic Context of Morphological and Genetic Divergence in an Understorey-Dwelling Bird. PLoS ONE 2014, 9, e85903. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.; Manzano, S.; Carr, A.S.; Cordova, C.; Ochando, J.; Bateman, M.D.; Carrión, J.S. A 14,000 year multi-proxy alluvial record of ecotone changes in a Fynbos-Succulent Karoo transition in South Africa. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2021, 569, 110331. [Google Scholar] [CrossRef]
- Huntley, B.; Collingham, Y.; Singarayer, J.S.; Valdes, P.J.; Barnard, P.; Midgely, G.F.; Altwegg, R.; Ohlemüller, R. Explaining patterns of avian diversity and endemicity: Climate and biomes of southern Africa over the last 140,000 years. J. Biogeogr. 2016, 43, 874–886. [Google Scholar] [CrossRef] [Green Version]
- Rosenblum, E.B.; Sarver, B.A.J.; Brown, J.W.; Des Roches, S.; Hardwick, K.M.; Hether, T.D.; Eastman, J.M.; Pennell, M.W.; Harmon, L.J. Goldilocks meets Santa Rosalia: An Ephemeral Speciation Model explains patterns of diversification across time scales. Evol. Biol. 2012, 39, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Quinn, T.W. The genetic legacy of Mother Goose—Phylogeographic patterns of lesser snow goose Chen caerulescens caerulescens maternal lineages. Mol. Ecol. 1992, 1, 105–117. [Google Scholar] [CrossRef]
- Hogner, S.; Laskemoen, T.; Lifjeld, J.T.; Porkert, J.; Kleven, O.; Albayrak, T.; Kabasakal, B.; Johnsen, A. Deep sympatric mitochondrial divergence without reproductive isolation in the common redstart Phoenicurus phoenicurus. Ecol. Evol. 2012, 2, 2974–2988. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Lacey, E.A.; Bach, B.H.; Bi, K.; Conroy, C.J.; Suvorov, A.; Bowie, R.C.K. Gut microbial diversity across a contact zone for California voles: Implications for lineage divergence of hosts and mito-nuclear mismatch in the assembly of the mammalian gut microbiome. Mol. Ecol. 2020, 29, 1873–1889. [Google Scholar] [CrossRef]
- Outlaw, R.K.; Voelker, G.; Bowie, R.C.K. Shall we chat? Evolutionary relationships in the genus Cercomela (Muscicapidae) and its relation to Oenanthe reveals extensive polyphyly among chats distributed in Africa, India and the Palearctic. Mol. Phylogenet. Evol. 2010, 55, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Clement, M.; Posada, D.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Excoffier, L.; Smouse, P.; Quattro, J. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Drummond, A.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.; Rambaut, A.; Shapiro, B.; Pybus, O. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Lerner, H.R.L.; Meyer, M.; James, H.F.; Hofreiter, M.; Fleischer, R.J. Multilocus resolution of phylogeny and timescale in the extant adapative radiation of Hawaiian Honeycreepers. Curr. Biol. 2011, 21, 1838–1844. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J. Tracer v1.5. 2007. Available online: http:/beast.bio.ed.ac.uk/Tracer (accessed on 9 September 2021).
- Phillips, S.J.; Anderson, R.; Schapire, R. Maximum entropy modeling of species geographic distributions. Ecol. Model. 2006, 190, 231–259. [Google Scholar] [CrossRef] [Green Version]
- Phillips, S.J.; Dudik, M. Modeling of species distributions with Maxent: New extensions and a comprehensive evaluation. Ecography 2008, 31, 161–175. [Google Scholar] [CrossRef]
- GBIF.org. Available online: http://doi.org/10.15468/dl.tplewp (accessed on 9 September 2015).
- GBIF.org. Available online: http://doi.org/10.15468/dl.mvegpj (accessed on 16 September 2015).
- GBIF.org. Available online: http://doi.org/10.15468/dl.nabqnh (accessed on 21 September 2016).
- Fuchs, J.; Parra, J.L.; Goodman, S.M.; Raherilalao, M.J.; VanDerWal, J.; Bowie, R.C.K. Extending ecological niche models to the past 120, 000 years corroborates the lack of strong phylogeographic structure in the Crested Drongo (Dicrurus forficatus forficatus) on Madagascar. Biol. J. Linn. Soc. 2013, 108, 658–676. [Google Scholar] [CrossRef] [Green Version]
- Fielding, A.; Bell, J.A. review of methods for the assessment of prediction errors in conservation presence/absence models. Environ. Conserv. 1997, 24, 38–49. [Google Scholar] [CrossRef]
- VanDerWal, J.; Shoo, L.P.; Williams, S.E. New approaches to understanding late Quaternary climate fluctuations and refugial dynamics in Australian wet tropical rain forests. J. Biogeogr. 2009, 36, 291–301. [Google Scholar] [CrossRef]
- Sinclair, I.; Ryan, P. Birds of Africa, South of the Sahara; Struik Nature: Cape Town, South Africa, 2010. [Google Scholar]
- Huntley, B.; Midgley, G.F.; Barnard, P.; Valdes, P.J. Suborbital climatic variability and centres of biological diversity in the Cape region of southern Africa. J. Biogeogr. 2014, 41, 1338–1351. [Google Scholar] [CrossRef] [Green Version]
- Matthee, C.; Robinson, T.J. Mitochondrial DNA differentiation among geographical populations of Pronolagus rupestris, Smith’s red rock rabbit (Mammalia: Lagomorpha). Heredity 1996, 76, 514–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, P.; Craig, A.J.F.K.; Hulley, P.E.; Essop, M.F.; Crowe, T.M. Ecology and genetics of hybrid zones in the southern African Pycnonotus bulbul species complex. Ostrich 1997, 68, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Oatley, G.; de Swardt, D.H.; Nuttall, R.J.; Crowe, T.M.; Bowie, R.C.K. Phenotypic and genotypic variation across a stable white-eye (Zosterops sp.) hybrid zone in central South Africa. Biol. J. Linn. Soc. 2017, 121, 670–684. [Google Scholar] [CrossRef]
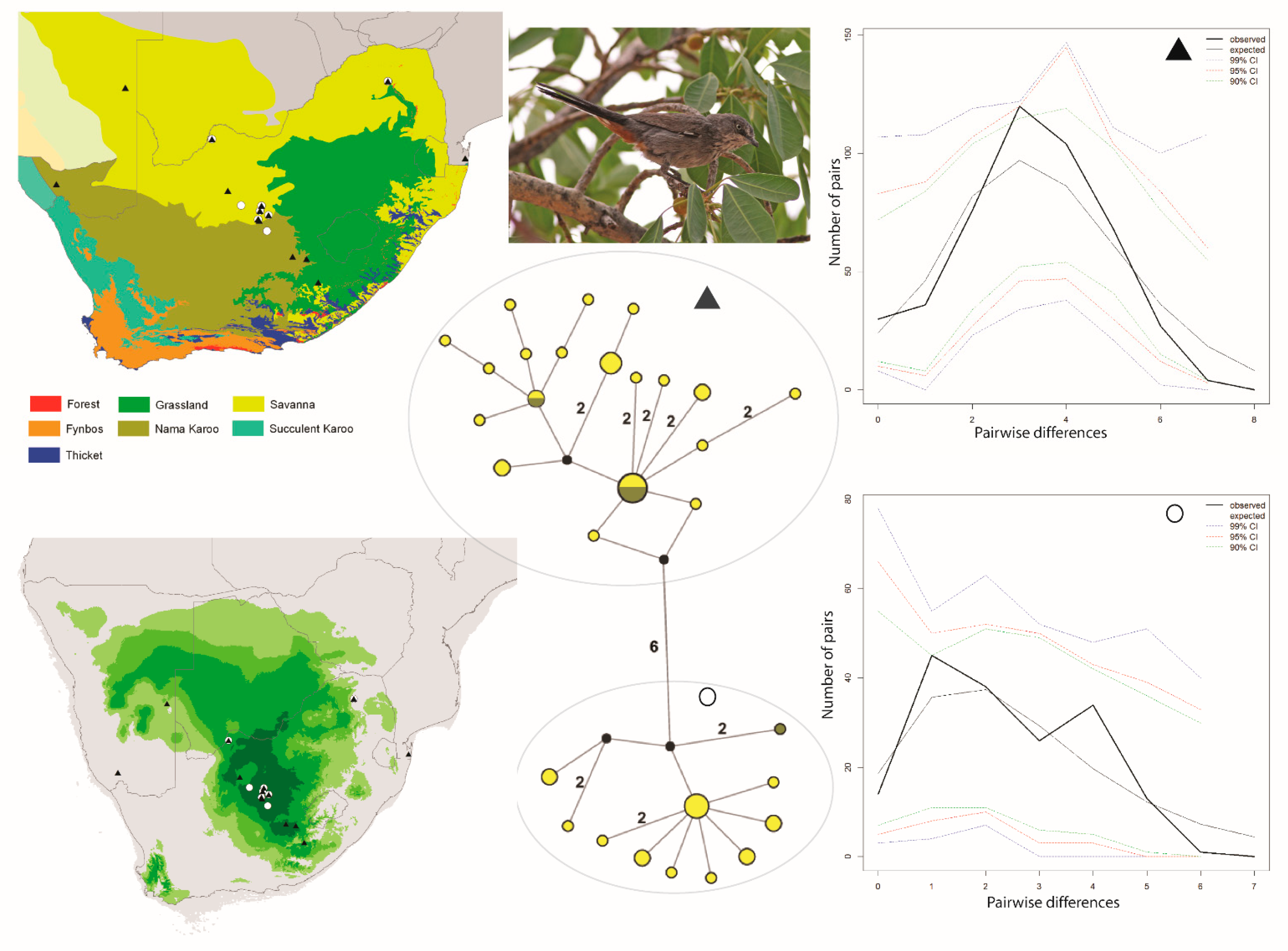
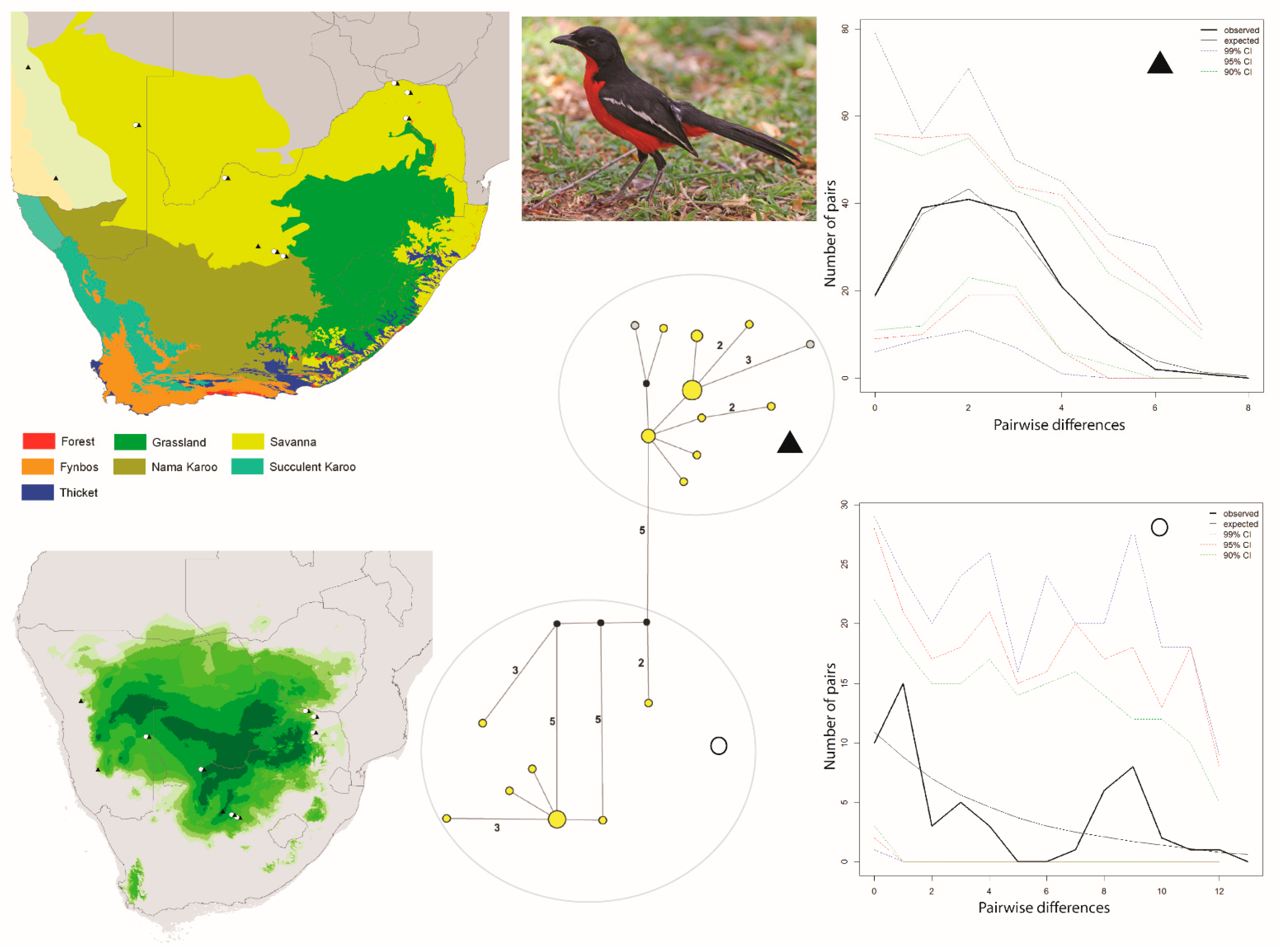
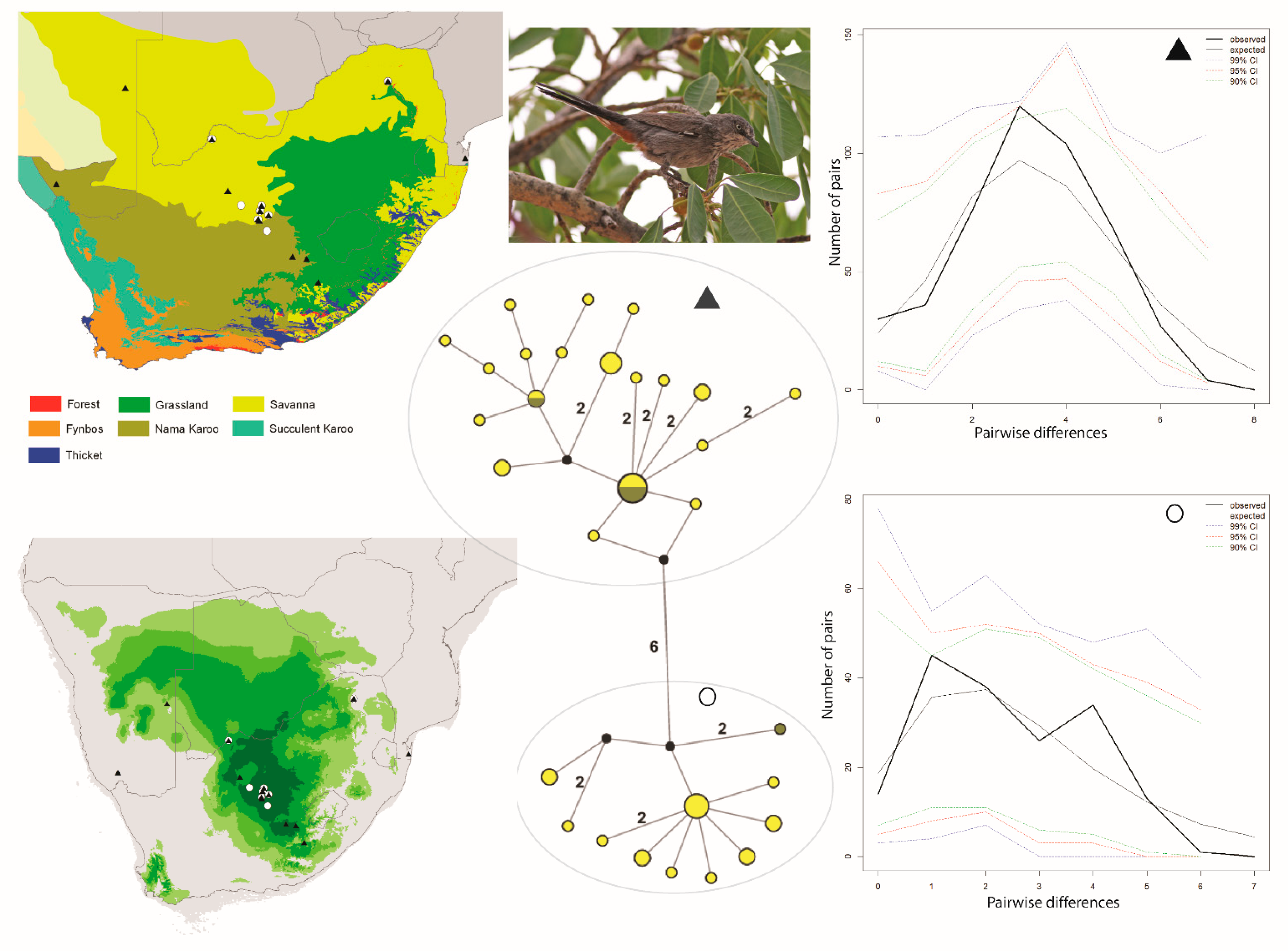
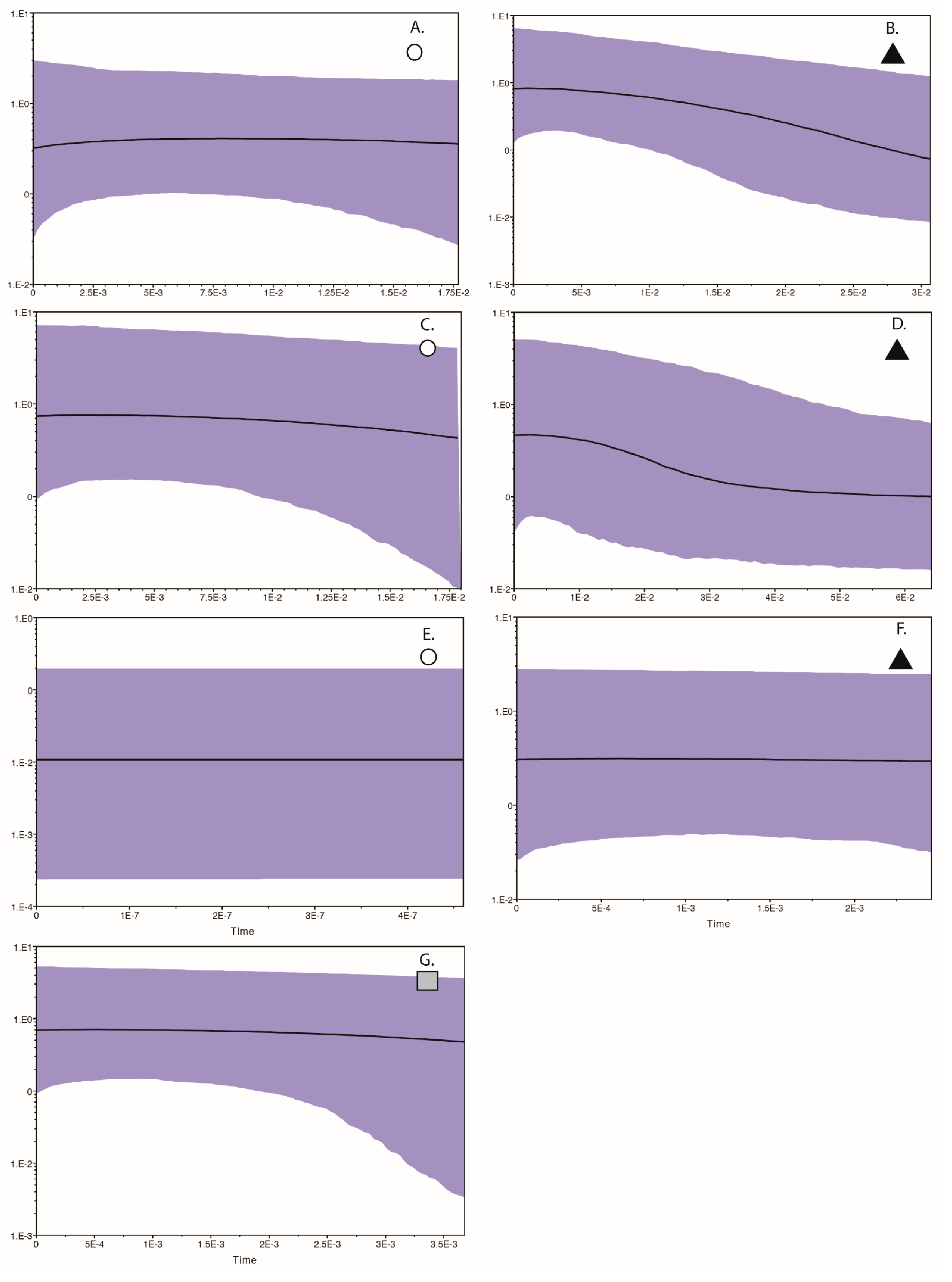
 and ▲, within southern Africa that are separated by up to 10 mutational steps; circle sizes in the networks correspond to the number of individuals with that haplotype (larger circles reflect more individuals with that haplotype), and black circles represent unsampled haplotypes. Other colors represent the biome (s) in which a haplotype was collected (top left); however, circle size has no relationship to the number of individuals from a given biome (if a haplotype is found in more than one biome)—in other words, just one individual from a specific biome would contribute that biome color to the circle. The geographic distribution of individuals in each haplotype cluster are depicted with respect to biome type (top left) and to the estimated species stability map (bottom left) is summed across all time slices, with darker shades of green indicating areas of higher stability. Mismatch distributions for each haplotype cluster depict demographic trends within each haplotype cluster (right, top, and bottom, and middle, bottom), where solid lines depict expected (black) and observed (black bold) pairwise differences, and dotted lines depict confidence intervals at 99% (blue), 95% (red), and 90% (green). Photo by Francesco Veronesi (https://www.flickr.com/photos/francesco_veronesi/4465034002/, accessed 10 January 2021).
and ▲, within southern Africa that are separated by up to 10 mutational steps; circle sizes in the networks correspond to the number of individuals with that haplotype (larger circles reflect more individuals with that haplotype), and black circles represent unsampled haplotypes. Other colors represent the biome (s) in which a haplotype was collected (top left); however, circle size has no relationship to the number of individuals from a given biome (if a haplotype is found in more than one biome)—in other words, just one individual from a specific biome would contribute that biome color to the circle. The geographic distribution of individuals in each haplotype cluster are depicted with respect to biome type (top left) and to the estimated species stability map (bottom left) is summed across all time slices, with darker shades of green indicating areas of higher stability. Mismatch distributions for each haplotype cluster depict demographic trends within each haplotype cluster (right, top, and bottom, and middle, bottom), where solid lines depict expected (black) and observed (black bold) pairwise differences, and dotted lines depict confidence intervals at 99% (blue), 95% (red), and 90% (green). Photo by Francesco Veronesi (https://www.flickr.com/photos/francesco_veronesi/4465034002/, accessed 10 January 2021).
and ▲, within southern Africa that are separated by up to 10 mutational steps; circle sizes in the networks correspond to the number of individuals with that haplotype (larger circles reflect more individuals with that haplotype), and black circles represent unsampled haplotypes. Other colors represent the biome (s) in which a haplotype was collected (top left); however, circle size has no relationship to the number of individuals from a given biome (if a haplotype is found in more than one biome)—in other words, just one individual from a specific biome would contribute that biome color to the circle. The geographic distribution of individuals in each haplotype cluster are depicted with respect to biome type (top left) and to the estimated species stability map (bottom left) is summed across all time slices, with darker shades of green indicating areas of higher stability. Mismatch distributions for each haplotype cluster depict demographic trends within each haplotype cluster (right, top, and bottom, and middle, bottom), where solid lines depict expected (black) and observed (black bold) pairwise differences, and dotted lines depict confidence intervals at 99% (blue), 95% (red), and 90% (green). Photo by Francesco Veronesi (https://www.flickr.com/photos/francesco_veronesi/4465034002/, accessed 10 January 2021).
and ▲, within southern Africa that are separated by up to 10 mutational steps; circle sizes in the networks correspond to the number of individuals with that haplotype (larger circles reflect more individuals with that haplotype), and black circles represent unsampled haplotypes. Other colors represent the biome (s) in which a haplotype was collected (top left); however, circle size has no relationship to the number of individuals from a given biome (if a haplotype is found in more than one biome)—in other words, just one individual from a specific biome would contribute that biome color to the circle. The geographic distribution of individuals in each haplotype cluster are depicted with respect to biome type (top left) and to the estimated species stability map (bottom left) is summed across all time slices, with darker shades of green indicating areas of higher stability. Mismatch distributions for each haplotype cluster depict demographic trends within each haplotype cluster (right, top, and bottom, and middle, bottom), where solid lines depict expected (black) and observed (black bold) pairwise differences, and dotted lines depict confidence intervals at 99% (blue), 95% (red), and 90% (green). Photo by Francesco Veronesi (https://www.flickr.com/photos/francesco_veronesi/4465034002/, accessed 10 January 2021).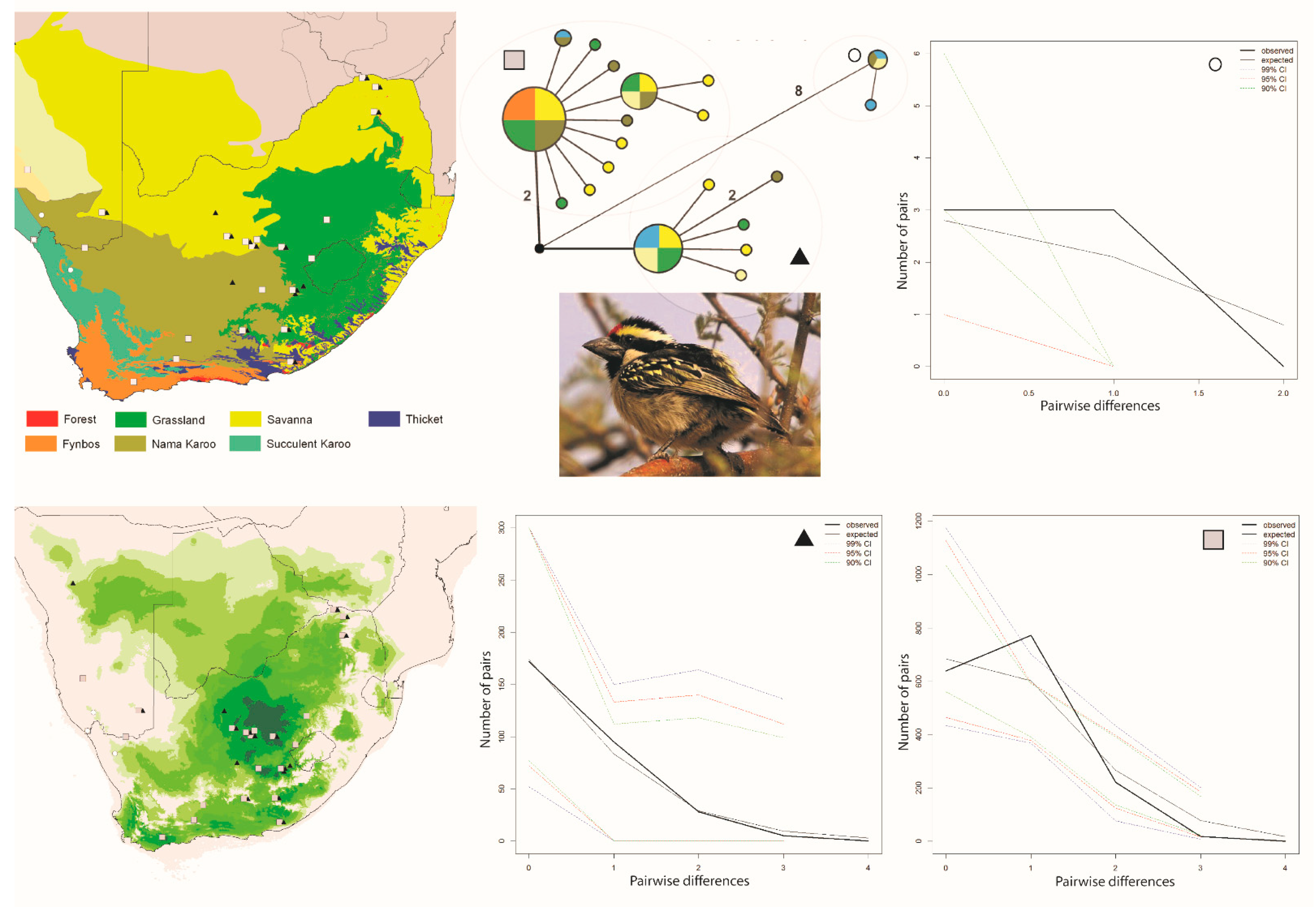

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | n | π | θ(S) | Tajima’s D | Fu’s Fs | SSD (D) | Harpending’s R (D) | SSD (S) | Harpending’s R (S) |
|---|---|---|---|---|---|---|---|---|---|
| Curruca subcoerulea | |||||||||
| O | 19 | 2.374 | 4.292 | −1.670 (0.031) * | −26.159 (0.000) * | 0.012 (0.050) * | 0.062 (0.050) * | 0.012 (0.250) | 0.062 (0.210) |
| ▲ | 31 | 3.213 | 6.008 | −1.640 (0.027) * | −26.192 (0.000) * | 0.008 (0.010) * | 0.034 (0.020) * | 0.006 (0.180) | 0.034 (0.210) |
| Laniarius atrococcineus | |||||||||
| O | 11 | 3.964 | 5.804 | −1.420 (0.069) | −8.199 (0.000) * | 0.046 (0.610) | 0.084 (0.720) | 0.040 (0.620) | 0.084 (0.780) |
| ▲ | 19 | 2.269 | 4.006 | −1.608 (0.052) * | −26.476 (0.000) * | 0.001 (0.940) | 0.030 (0.580) | 0.001 (0.870) | 0.030 (0.590) |
| Tricholaema leucomelas | |||||||||
| O | 4 | 5.000 | 0.545 | −0.612 (0.391) | −4.644 (0.000) * | 0.0220 (0.000) * | 0.250 (1.000) | 0.022 (0.200) | 0.250 (1.000) |
| ▲ | 25 | 0.553 | 1.589 | −1.949 (0.009) * | −3.402 × 1038 (0.000) * | <0.000 (1.000) | 0.122 (0.970) | 0.002 (0.450) | 0.122 (0.810) |
| | 58 | 0.768 | 2.376 | −9.204 (0.010) * | −3.402 × 1038 (0.000) * | 0.078 (0.020) * | 0.277 (0.030) * | 0.013 (0.000) * | 0.134 (0.110) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voelker, G.; Wogan, G.O.U.; Huntley, J.W.; Bowie, R.C.K. Comparative Phylogeography of Southern African Bird Species Suggests an Ephemeral Speciation Model. Diversity 2021, 13, 434. https://doi.org/10.3390/d13090434
Voelker G, Wogan GOU, Huntley JW, Bowie RCK. Comparative Phylogeography of Southern African Bird Species Suggests an Ephemeral Speciation Model. Diversity. 2021; 13(9):434. https://doi.org/10.3390/d13090434
Chicago/Turabian StyleVoelker, Gary, Guinevere O. U. Wogan, Jerry W. Huntley, and Rauri C. K. Bowie. 2021. "Comparative Phylogeography of Southern African Bird Species Suggests an Ephemeral Speciation Model" Diversity 13, no. 9: 434. https://doi.org/10.3390/d13090434
APA StyleVoelker, G., Wogan, G. O. U., Huntley, J. W., & Bowie, R. C. K. (2021). Comparative Phylogeography of Southern African Bird Species Suggests an Ephemeral Speciation Model. Diversity, 13(9), 434. https://doi.org/10.3390/d13090434