
High Differentiation among Populations of Green Foxtail, Setaria viridis, in Taiwan and Adjacent Islands Revealed by Microsatellite Markers


Abstract
1. Introduction
2. Materials and Methods
2.1. Field Collections of S. viridis in Taiwan
2.2. Genotyping by Microsatellite
2.3. Genetic Diversity and Clustering Analysis
2.4. F-Statistics of Populations at 10 Collection Sites
3. Results
3.1. Molecular Diversity of Microsatellite Markers from the Individuals of Green Foxtail in Taiwan
3.2. Genetic Relationship of Green Foxtail Individuals Collected in Taiwan
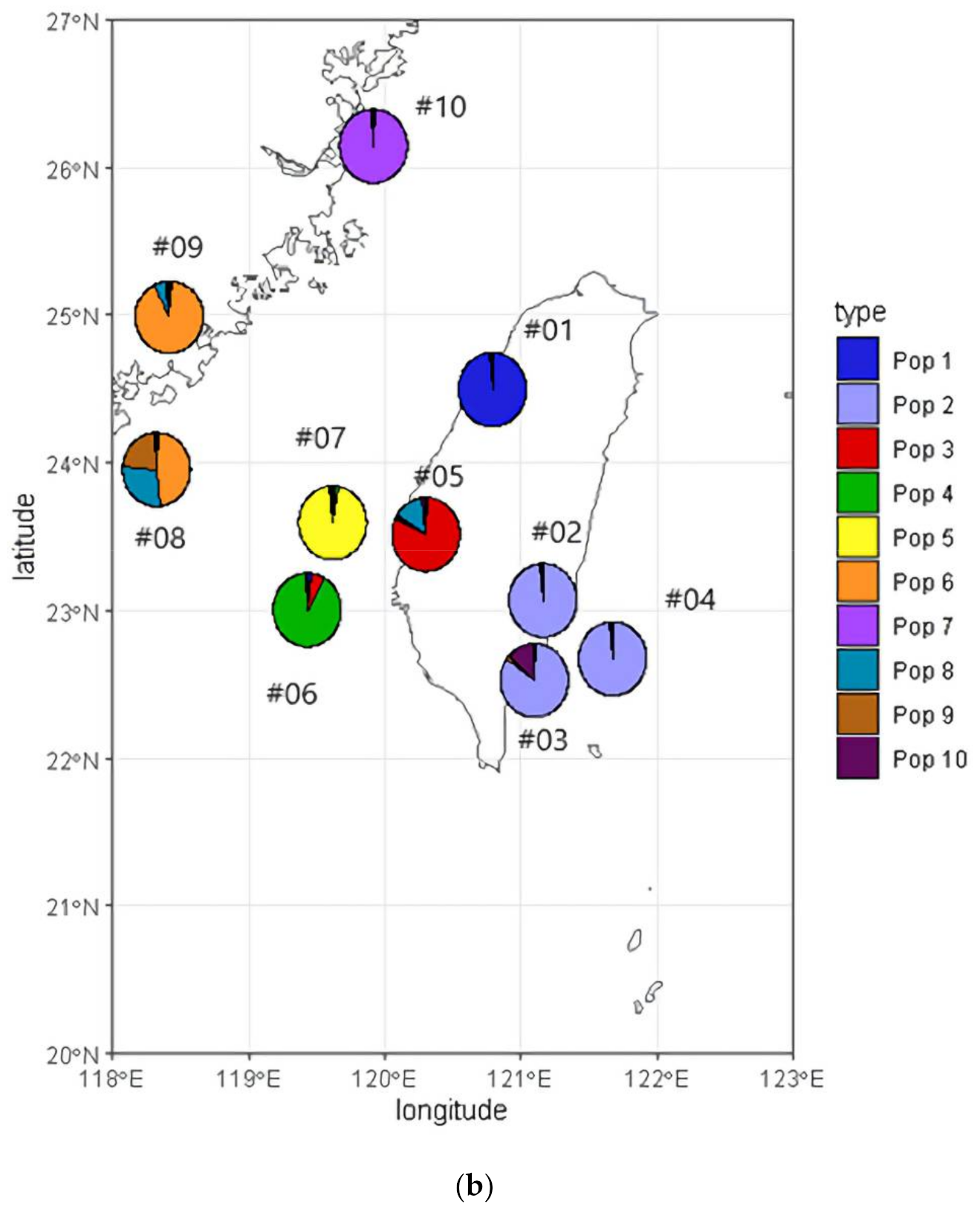
3.3. Population Structure and Geographic Distribution
3.4. Genetic Differentiation among the Populations of S. viridis in Taiwan
3.5. Pairwise FST Index of Collecting Populations in Taiwan
4. Discussion
4.1. Molecular Diversity of Microsatellite from the Individuals of Green Foxtail in Taiwan
4.2. Genetic Relationship and Structure of Green Foxtail Individuals among Different Parts of Taiwan
4.3. Genetic Differentiation and Possible Gene Flow among the Populations of Different Collection Sites
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brutnell, T.P.; Wang, L.; Swartwood, K.; Goldschmidt, A.; Jackson, D.; Zhu, X.G.; Kellogg, E.; Eck, J.V. Setaria viridis: A model for C4 photosynthesis. Plant Cell 2010, 22, 2537–2544. [Google Scholar] [CrossRef]
- Chen, C.H.; Lin, C.Y.; Kuoh, C.S. Panicoideae, Paniceae. In Grass Flora of Taiwan (2 of 3); Endemic Species Research Institute: Chichi, Taiwan, 2014; p. 158. [Google Scholar]
- Dekker, J. The foxtail (Setaria) species-group. Weed Sci. 2003, 51, 641–656. [Google Scholar] [CrossRef]
- Li, Y.; Wu, S.Z. Traditional maintenance and multiplication of foxtail millet (Setaria italica (L.) P. Beauv) landraces in China. Euphytica 1996, 87, 33–38. [Google Scholar] [CrossRef]
- Li, H.W.; Li, C.H.; Pao, W.K. Cytological and genetical studies of the interspecific cross of the cultivated foxtail millet, Setaria italica (L.) Beauv., and the green foxtail millet, S. viridis L. Agron. J. 1945, 9, 32–54. [Google Scholar] [CrossRef]
- Yang, X.; Wan, Z.; Perry, L.; Lu, H.; Wang, Q.; Zhao, C.; Li, J.; Xie, F.; Yu, J.; Cui, T.; et al. Early millet use in northern China. Proc. Natl. Acad. Sci. USA 2012, 109, 3726–3730. [Google Scholar] [CrossRef]
- Barton, L.; Newsome, S.D.; Chen, F.; Wang, H.; Guilderson, T.P.; Bettinger, R.L. Agricultural origins and the isotopic identity of domestication in northern china. Proc. Natl. Acad. Sci. USA 2009, 106, 5523–5528. [Google Scholar] [CrossRef]
- Austin, D.F. Foxtail millets (Setaria: Poaceae)—Abandoned food in two hemispheres. Econ. Bot. 2006, 60, 143–158. [Google Scholar] [CrossRef]
- Food and Agriculture Organization of the United Nations. FAOSTAT Statistical Database. Available online: http://www.fao.org/faostat/en/#data/QC/visualize (accessed on 15 October 2020).
- Benabdelmouna, A.; Shi, Y.; Abirached-Darmency, M.; Darmency, H. Genomic in situ hybridization (GISH) discriminates between the A and the B genomes in diploid and tetraploid Setaria species. Genome 2001, 44, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Giussani, L.M.; Cota-Sánchez, J.H.; Zuloaga, F.O.; Kellogg, E.A. A molecular phylogeny of the grass subfamily Panicoideae (Poaceae) shows multiple origins of C4 photosynthesis. Am. J. Bot. 2001, 88, 1993–2012. [Google Scholar] [CrossRef] [PubMed]
- Doust, A.N.; Penly, A.M.; Jacobs, S.W.L.; Kellogg, E.A. Congruence, conflict, and polyploidization shown by nuclear and chloroplast markers in the monophyletic “bristle clade” (Paniceae, Panicoideae, Poaceae). Syst. Bot. 2007, 32, 531–544. [Google Scholar] [CrossRef]
- Harlan, J.R.; de Wet, J.M.J. Towards a rational classification of cultivated plants. Taxon 1971, 20, 509–517. [Google Scholar] [CrossRef]
- Darmency, H.; Zangre, G.R.; Pernes, J. The wild-weed-crop complex in Setaria: A hybridization study. Genetica 1987, 75, 103–107. [Google Scholar] [CrossRef]
- Mamidi, S.; Healey, A.; Huang, P.U.; Grimwood, J.; Jenkins, J.; Barry, K.; Sreedasyam, A.; Shu, S.; Lovell, J.T.; Feldman, M.; et al. A genome resource for green millet Setaria viridis enables discovery of agronomically valuable loci. Nat. Biotechnol. 2020, 38, 1203–1210. [Google Scholar] [CrossRef]
- Bennetzen, J.L.; Schmutz, J.; Wang, H.; Percifield, R.; Hawkins, J.; Pontaroli, A.C.; Estep, M.; Feng, L.; Vaughn, J.N.; Grimwood, J.; et al. Reference genome sequence of the model plant Setaria. Nat. Biotechnol. 2012, 30, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Brutnell, T.P.; Bennetzen, J.L.; Vogel, J.P. Brachypodium distachyon and Setaria viridis: Model genetic systems for the grasses. Annu. Rev. Plant Biol. 2015, 66, 465–485. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Shyu, C.; Coelho, C.P.; Cao, Y.; Brutnell, T.P. Setaria viridis as a model system to advance millet genetics and genomics. Front. Plant Sci. 2016, 7, 1781. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Brutnell, T.P. Setaria viridis and Setaria italica, model genetic systems for the Panicoid grasses. J. Exp. Bot. 2011, 62, 3031–3037. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, X.; Quan, Z.; Cheng, S.; Xu, X.; Pan, S.; Xie, M.; Zeng, P.; Yue, Z.; Wang, W.; et al. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat. Biotechnol. 2012, 30, 549–554. [Google Scholar] [CrossRef]
- Diao, X.; Schnable, J.; Bennetzen, J.L.; Li, J. Initiation of Setaria as a model plant. Front Agric. Sci. Eng. 2014, 1, 16–20. [Google Scholar]
- Doust, A.N.; Kellogg, E.A.; Devos, K.M.; Bennetzen, J.L. Foxtail millet: A sequence-driven grass model system. Plant Physiol. 2009, 149, 137–141. [Google Scholar] [CrossRef]
- Chen, S.L.; Phillips, S.M. Setaria P. Beauv. In Flora of Chine; Wu, Z.Y., Raven, P.R., Eds.; Science Press: Beijing, China, 2006; Volume 22, pp. 531–537. [Google Scholar]
- Lorenzi, H.J.; Jeffery, L.S. Weeds of the United States and Their Control; Van Nostrand Reinhold: New York, NY, USA, 1987. [Google Scholar]
- Defelice, M.S. Green Foxtail, Setaria viridis (L.) P. Beauv. Weed Technol. 2002, 16, 253–257. [Google Scholar] [CrossRef]
- Huang, P.; Feldman, M.; Schröder, S.; Bahri, B.A.; Diao, X.; Zhi, H.; Estep, M.; Baxter, I.; Devos, K.M.; Kellogg, E.A. Population genetics of Setaria viridis, a new model system. Mol. Ecol. 2014, 23, 4912–4925. [Google Scholar] [CrossRef]
- Ridley, H.N. The Dispersal of Plants throughout the World; Reeve: Ashford, UK, 1930; p. 744. [Google Scholar]
- Douglas, B.J.; Morrison, I.N.; Thomas, A.G.; Maw, M.G. The Biology of Canadian Weeds. 70. Setaria viridis (L.) Beauv. Can. J. Plant Sci. 1985, 65, 669–690. [Google Scholar] [CrossRef]
- Holm, L.G.; Plucknett, D.L.; Pancho, J.V.; Herberger, J.P. The World’s Worst Weeds: Distribution and Biology; Krieger Publishing: Malabar, FL, USA, 1991; p. 609. [Google Scholar]
- Holm, L.G.; Plucknett, D.L.; Pancho, J.V.; Herberger, J.P. The World’s Worst Weeds-Distribution and Biology; The East-West Food Institute: Honolulu, HI, USA, 1977. [Google Scholar]
- Wilson, R.G. Dissemination of weed seeds by surface irrigation water in Western Nebraska. Weed Sci. 1980, 28, 87–92. [Google Scholar] [CrossRef]
- Frankton, C.; Mulligan, G.A. Weeds of Canada; Canada Department of Agriculture, Queen’s Printer: Ottawa, ON, Canada, 1970; p. 948. [Google Scholar]
- Perrino, E.V.; Calabrese, G. Endangered segetal species in southern Italy: Distribution, conservation status, trends, actions and ethnobotanical notes. Genet. Resour. Crop Evol. 2018, 65, 2107–2134. [Google Scholar] [CrossRef]
- Rominger, J.M. Taxonomy of Setaria (Graminaceae) in North America; University of Illinois Press: Urbana, IL, USA, 1962. [Google Scholar]
- Yatskievych, G. Steyermarks’s Flora of Missouri; Missouri Botanical Garden Press: St. Louis, MO, USA, 1999; Volume 1, p. 991. [Google Scholar]
- Blackshaw, R.E.; Stobbe, E.H.; Sturko, A.R.W. Effect of seeding dates and densities of green foxtail (Setaria viridis) on the growth and productivity of spring wheat (Triticum aestivum). Weed Sci. 1981, 29, 212–217. [Google Scholar] [CrossRef]
- Gates, F.C. Weeds in Kansas; Kansas State Printing Plant: Topeka, KS, USA, 1941; p. 360. [Google Scholar]
- Hunt, H.V.; Linden, M.V.; Liu, X.; Motuzaite-Matuzeviciute, G.; Colledge, S.; Jones, M.K. Millets across Eurasia: Chronology and context of early records of the genera Panicum and Setaria from archaeological sites in the Old World. Veg. Hist. Archaeobot. 2008, 17, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Duke, J.A. Handbook of Edible Weeds; CRC Press: Boca Raton, FL, USA, 1992; p. 246. [Google Scholar]
- Wang, R.L.; Wendel, J.F.; Dekker, J.H. Weedy adaptation in Setaria spp. I. Isozyme analysis of genetic diversity and population genetic structure in Setaria viridis. Am. J. Bot. 1995, 82, 308–317. [Google Scholar] [CrossRef]
- Jia, G.; Shi, S.; Wang, C.; Niu, Z.; Chai, Y.; Zhi, H.; Diao, X. Molecular diversity and population structure of Chinese green foxtail [Setaria viridis (L.) Beauv.] revealed by microsatellite analysis. J. Exp. Bot. 2013, 64, 3645–3656. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, T.; Li, Y.; Darmency, H. Impact of transgene inheritance on the mitigation of gene flow between crops and their wild relatives: The example of foxtail millet. Genetics 2008, 180, 969–975. [Google Scholar] [CrossRef]
- Pohl, R.W. The genus Setaria in Iowa. Iowa State. J. Sci. 1951, 25, 501–508. [Google Scholar]
- de Wet, J.M.J. The origin of weediness in plants. Proc. Okla. Acad. Sci. 1966, 1966, 14–17. [Google Scholar]
- Harlan, J.R. The possible role of weed races in the evolution of cultivated plants. Euphytica 1965, 14, 173–176. [Google Scholar] [CrossRef]
- Harlan, J.R.; de Wet, J.M.J.; Price, E.G. Comparative evolution of cereals. Evolution 1973, 27, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.Y.; Zhao, Z.H.; Yan, H.B.; Li, Y.; Zhu, X.H.; Shi, Y.S.; Song, Y.C.; Ma, Z.Y.; Darmency, H. Gene flow from cultivated herbicide-resistant foxtail millet to its wild relatives: A basis for risk assessment of the release of transgenic millet. Acta Agron. Sin. 2001, 27, 681–687. (in Chinese). [Google Scholar]
- Kuo, S.M.; Chen, Y.R.; Yin, S.Y.; Ba, Q.X.; Tsai, Y.C.; Kuo, W.H.J.; Lin, Y.R. Waxy allele diversification in foxtail millet (Setaria italica) landraces of Taiwan. PLoS ONE 2018, 13, e0210025. [Google Scholar] [CrossRef]
- Hirano, R.; Naito, K.; Fukunaga, K.; Watanabe, K.N.; Ohsawa, R.; Kawase, M. Genetic structure of landraces in foxtail millet (Setaria italica (L.) P. Beauv.) revealed with transposon display and interpretation to crop evolution of foxtail millet. Genome 2011, 54, 498–506. [Google Scholar] [CrossRef]
- Fukunaga, K.; Ichitani, K.; Kawase, M. Phylogenetic analysis of the rDNA intergenic spacer subrepeats and its implication for the domesticated history of foxtail millet, Setaria italica. Theor. Appl. Genet. 2006, 113, 261–269. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J.W. Allozyme diversity in plant species. In Plant Population Genetics, Breeding and Genetic Resources; Brown, A.H.D., Clegg, M.T., Kahler, A.L., Weir, B.S., Eds.; Sinauer: Sunderland, MA, USA, 1990; pp. 43–63. [Google Scholar]
- Wang, R.L.; Wendel, J.F.; Dekker, J.H. Weedy adaptation in Setaria spp. II. Genetic diversity and population genetic structure in S. glauca, S. geniculata, and S. faberii (Poaceae). Am. J. Bot. 1995, 82, 1031–1039. [Google Scholar] [CrossRef]
- Huang, X.; Kurata, N.; Wei, X.; Wang, Z.X.; Wang, A.; Zhao, Q.; Zhao, Y.; Liu, K.; Lu, H.; Li, W.; et al. A map of rice genome variation reveals the origin of cultivated rice. Nature 2012, 490, 497–501. [Google Scholar] [CrossRef]
- Wright, S.I.; Bi, I.V.; Schröder, S.G.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S. The effects of artificial selection on the maize genome. Science 2005, 308, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Bai, G.; Zhang, D.; Znu, C.; Xia, X.; Cheng, Z.; Shi, Z. Genetic diversity and population structure of elite foxtail millet (Setaria italica (L.) P. Beauv.) germplasm in China. Crop Sci. 2011, 51, 1655–1663. [Google Scholar] [CrossRef]
- Wang, C.; Chen, J.; Zhi, H.; Yang, L.; Li, W.; Wang, Y.; Li, H.; Zhao, B.; Chen, M.; Diao, X. Population genetics of foxtail millet and its wild ancestor. BMC Genet. 2010, 11, 90. [Google Scholar] [CrossRef]
- Wang, C.; Jia, G.; Zhi, H.; Niu, Z.; Chai, Y.; Li, W.; Wang, Y.; Li, H.; Lu, P.; Zhao, B.; et al. Genetic diversity and population structure of Chinese foxtail millet [Setaria italica (L.) Beauv.] landraces. G3 (Bethesda) 2012, 2, 769–777. [Google Scholar] [CrossRef]
- Gupta, S.; Kumari, K.; Muthamilarasan, M.; Parida, S.K.; Prasad, M. Population structure and association mapping of yield contributing agronomic traits in foxtail millet. Plant Cell Rep. 2014, 33, 881–893. [Google Scholar] [CrossRef]
- Vetriventhan, M.; Upadhyaya, H.D.; Anandakumar, C.R.; Senthilvel, S.; Varshney, R.K.; Parzies, H.K. Population structure and linkage disequilibrium of ICRISAT foxtail millet (Setaria italica (L.) P. Beauv.) core collection. Euphytica 2014, 196, 423–435. [Google Scholar] [CrossRef]
- Fukunaga, K.; Wang, Z.; Kato, K.; Kawase, M. Geographical variation of nuclear genome RFLPs and genetic differentiation in foxtail millet, Setaria italica (L.) P. Beauv. Genet. Resour. Crop Evol. 2002, 49, 95–101. [Google Scholar] [CrossRef]
- Li, W.; Zhi, H.; Wang, Y.; Li, H.; Diao, X. Assessment of genetic relationship of foxtail millet with its wild ancestor and close relatives by ISSR markers. J. Integr. Agric. 2012, 11, 556–566. [Google Scholar] [CrossRef]
- Kim, S.; Plagnol, V.; Hu, T.T.; Toomajian, C.; Clark, R.M.; Ossowski, S.; Ecker, J.R.; Weigel, D.; Nordborg, M. Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat. Genet. 2007, 39, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Brachi, B.; Morris, G.P.; Borevitz, J.O. Genome-wide association studies in plants: The missing heritability is in the field. Genome Biol. 2011, 12, 232. [Google Scholar] [CrossRef]
- Honda, M. Monographia Poacearum Japonicarum, Bambusoideis Exclusis (Journal of the Faculty of Science: Section III, Botany); Imperial University of Tokyo: Tokyo, Japan, 1930; Volume 3, pp. 1–484. [Google Scholar]
- Tsang, C.H.; Li, K.T.; Hsu, T.F.; Tsai, Y.C.; Fang, P.H.; Hsing, Y.I.C. Broomcorn and foxtail millet were cultivated in Taiwan about 5000 years ago. Bot. Stud. 2017, 58, 3. [Google Scholar] [CrossRef] [PubMed]
- Thomson, D.; Dietzgen, R.G. Detection of DNA and RNA plant viruses by PCR and RT-PCR using a rapid virus release protocol without tissue homogenization. J. Virol. Methods 1995, 54, 85–95. [Google Scholar] [CrossRef]
- Zhang, S.; Tang, C.; Zhao, Q.; Li, J.; Yang, L.; Qie, L.; Fan, X.; Li, L.; Zhang, N.; Zhao, M.; et al. Development of highly polymorphic simple sequence repeat markers using genome-wide microsatellite variant analysis in Foxtail millet [Setaria italica (L.) P. Beauv.]. BMC Genom. 2014, 15, 78. [Google Scholar] [CrossRef]
- Hayden, M.J.; Nguyen, T.M.; Waterman, A.; Chalmers, K.J. Multiplex-Ready PCR: A new method for multiplexed SSR and SNP genotyping. BMC Genom. 2008, 9, 80. [Google Scholar] [CrossRef]
- Covarrubias-Pazaran, G.; Diaz-Garcia, L.; Schlautman, B.; Salazar, W.; Zalapa, J. Fragman: An R package for fragment analysis. BMC Genet. 2016, 17, 62. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.R-project.org/ (accessed on 10 November 2019).
- Nei, M. Analysis of gene diversity in subdivided populations. Proc. Nat. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef]
- Adamack, A.T.; Gruber, B. Popgen Report: Simplifying basic population genetic analyses in R. Methods Ecol. Evol. 2014, 5, 384–387. [Google Scholar] [CrossRef]
- Clark, L.V.; Jasieniuk, M. POLYSAT: An R package for polyploid microsatellite analysis. Mol. Ecol. Resour. 2011, 11, 562–566. [Google Scholar] [CrossRef]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Francis, R.M. pophelper: An R package and web app to analyze and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software Structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer Publishing Company: New York, NY, USA, 2009. [Google Scholar]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Wright, S. The genetical structure of populations. Ann. Eugen. 1951, 15, 323–354. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Wright, S. Isolation by distance. Genetics 1943, 28, 114–138. [Google Scholar]
- Matsuoka, Y.; Mitchell, S.E.; Kresovich, S.; Goodman, M.; Doebley, J. Microsatellites in Zea-variability, patterns of mutations, and use for evolutionary studies. Theor. Appl. Genet. 2002, 104, 436–450. [Google Scholar] [CrossRef]
- Tautz, D.; Renz, M. Simple sequences are ubiquitous components of eukaryotic genomes. Nucleic Acids Res. 1984, 12, 4127–4138. [Google Scholar] [CrossRef]
- Kumar, P.; Gupta, V.K.; Misra, A.K.; Modi, D.R.; Pandey, B.K. Potential of molecular markers in plant biotechnology. Plant OMIC 2009, 2, 141–162. [Google Scholar]
- Zhao, Y.; Basak, S.; Fleener, C.E.; Egnin, M.; Sacks, E.J.; Prakash, C.S.; He, G. Genetic diversity of Miscanthus sinensis in US naturalized populations. GCB Bioenergy 2017, 9, 965–972. [Google Scholar] [CrossRef]
- Ecker, G.; Zalapa, J.; Auer, C. Switchgrass (Panicum virgatum L.) genotypes differ between coastal sites and inland road corridors in the Northeastern US. PLoS ONE 2015, 10, e0130414. [Google Scholar] [CrossRef] [PubMed]
- Schröder, S.; Bahri, B.A.; Eudy, D.M.; Layton, D.J.; Kellogg, E.A.; Devos, K.M. Genetic diversity and origin of North American green foxtail [Setaria viridis (L.) Beauv.] accessions. Genet. Resour. Crop Evol. 2017, 64, 367–378. [Google Scholar] [CrossRef]
- Fang, X.; Dong, K.; Wang, X.; Liu, T.; He, J.; Ren, R.; Zhang, L.; Liu, R.; Liu, X.; Li, M.; et al. A high density genetic map and QTL for agronomic and yield traits in Foxtail millet [Setaria italica (L.) P. Beauv.]. BMC Genom. 2016, 17, 336. [Google Scholar] [CrossRef]
- Gupta, S.; Kumari, K.; Sahu, P.P.; Vidapu, S.; Prasad, M. Sequence-based novel genomic microsatellite markers for robust genotyping purposes in foxtail millet [Setaria italica (L.) P. Beauv]. Plant Cell Rep. 2012, 31, 323–337. [Google Scholar] [CrossRef]
- Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 1978, 89, 583–590. [Google Scholar]
- Muraya, M.M.; de Villiers, S.; Parzies, H.K.; Mutegi, E.; Sagnard, F.; Kanyenji, B.M.; Kiambi, D.; Geiger, H.H. Genetic structure and diversity of wild sorghum populations (Sorghum spp.) from different eco-geographical regions of Kenya. Theor. Appl. Genet. 2011, 123, 571–583. [Google Scholar] [CrossRef]
- Hsieh, C.F.; Shen, C.F. Introduction to the flora of Taiwan, 1: Geography, geology, climate, and soils. In Flora of Taiwan, 2nd ed.; Department of Botany, National Taiwan University: Taipei, Taiwan, 1994; Volume 1, pp. 1–3. [Google Scholar]
- Jusuf, M.; Pernes, J. Genetic variability of foxtail millet (Setaria italica P. Beauv.). Theor. Appl. Genet. 1985, 71, 385–391. [Google Scholar] [CrossRef]
- Slatkin, M.; Barton, N.M. A comparison of three indirect methods for estimating average levels of gene flow. Evolution 1989, 43, 1349–1368. [Google Scholar] [CrossRef]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Upadhyaya, H.D.; Dwivedi, S.L.; Baum, M.; Varshney, R.K.; Udupa, S.M.; Gowda, C.L.L. Genetic structure, diversity, and allelic richness in composite collection and reference set in chickpea (Cicer arietinum L.). BMC Plant Biol. 2008, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.E.; Pernès, J. Genetic diversity of foxtail millet (S. italica). In Genetic Differenciation and Dispersal in Plants; Jacquard, P., Ed.; NATO ASI series; Springer: Berlin/Heidelberg, Germany, 1985; Volume G5, pp. 113–128. [Google Scholar]
- Ohadi, S.; Littlejohn, M.; Mesgaran, M.; Rooney, W.; Bagavathiannan, M. Surveying the spatial distribution of feral sorghum (Sorghum bicolor L.) and its sympatry with johnsongrass (S. halepense) in South Texas. PLoS ONE 2018, 13, e0195511. [Google Scholar]
- Sezen, U.U.; Barney, J.N.; Atwater, D.Z.; Pederson, G.A.; Pederson, J.F.J.; Chandler, M.; Cox, T.S.; Cox, S.; Dotray, P.; Kopec, D.; et al. Multi-Phase US Spread and Habitat Switching of a Post-Columbian Invasive, Sorghum halepense. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Kono, Y.; Chung, K.F.; Chen, C.H.; Hoshi, Y.; Setoguchi, H.; Chou, C.H.; Oginuma, K.; Peng, C.I. Intraspecific karyotypic polymorphism is highly concordant with allozyme variation in Lysimachia mauritiana (Primulaceae: Myrsinoideae) in Taiwan: Implications for the colonization history and dispersal patterns of coastal plants. Ann. Bot. 2012, 110, 1119–1135. [Google Scholar] [CrossRef] [PubMed]
- Le Thierry d’Ennequin, M.; Panaud, O.; Toupance, B.; Sarr, A. Assessment of genetic relationships between Setaria italica and its wild relative S. viridis using AFLP markers. Theor. Appl. Genet. 2000, 100, 1061–1066. [Google Scholar] [CrossRef]
- Barrett, S.C.H.; Shore, J.S. Isozyme variation in colonizing plants. In Isozymes in Plant Biology; Soltis, D.E., Soltis, P.S., Eds.; Dioscorides: Portland, OR, USA, 1989; pp. 106–126. [Google Scholar]
- Barrett, S.C.H. Genetics and evolution of agricultural weeds. In Weed Management in Agro-Ecosystems: Ecological Approach; Altieri, M., Liebman, M., Eds.; CRC Press: Boca Raton, FL, USA, 1988; pp. 58–75. [Google Scholar]
- Rice, K.; Jain, S.K. Plant population genetics and evolution in disturbed environments. In The Ecology of Natural Disturbance and Patch Dynamics; Pickett, S.T.A., White, P.A., Eds.; Academic Press: New York, NY, USA, 1985; pp. 287–303. [Google Scholar]
- Hartl, D.L.; Clark, A.G. Principles of Population Genetics; Sinauer Associates: Sunderland, MA, USA, 2007. [Google Scholar]
- Perrino, E.V.; Wagensommer, R.P. Crop Wild Relatives (CWR) Priority in Italy: Distribution, Ecology, In Situ and Ex Situ Conservation and Expected Actions. Sustainability 2021, 13, 1682. [Google Scholar] [CrossRef]
- Castañeda-Álvarez, N.; Khoury, C.; Achicanoy, H.; Bernau, V.; Dempewolf, H.; Eastwood, R.J.; Guarino, L.; Harker, R.H.; Jarvis, A.; Maxted, N.; et al. Global conservation priorities for crop wild relatives. Nat. Plants 2016, 2, 16022. [Google Scholar] [CrossRef]
- Jarvis, D.I.; Brown, A.H.; Cuong, P.H.; Collado-Panduro, L.; Latournerie-Moreno, L.; Gyawali, S.; Tanto, T.; Sawadogo, M.; Mar, I.; Sadiki, M.; et al. A global perspective of the richness and evenness of traditional crop-variety diversity maintained by farming communities. Proc. Natl. Acad. Sci. USA 2008, 105, 5326–5331. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Site (Township/County) | Latitude | Longitude | Sample Size |
|---|---|---|---|---|
| #01 | Tongluo, Miaoli | 24°29′48.00″ N | 120°47′17.00″ E | 16 |
| #02 | Haiduan, Taitung | 23°4′9.00″ N | 121°9′33.00″ E | 8 |
| #03 | Taitung, Taitung | 22°43′36.00″ N | 121°5′48.00″ E | 16 |
| #04 | Ludao, Taitung | 22°40′25.00″ N | 121°28′17.00″ E | 9 |
| #05 | Liujiao, Chiayi | 23°30′56.00″ N | 120°17′56.00″ E | 10 |
| #06 | Qimei, Penghu | 23°12′18.00″ N | 119°25′37.00″ E | 15 |
| #07 | Huxi, Penghu | 23°35′40.00″ N | 119°36′47.00″ E | 11 |
| #08 | Jinning, Kinmen | 24°27′20.00″ N | 118°19′8.00″ E | 16 |
| #09 | Jinsha, Kinmen | 24°29′36.00″ N | 118°24′44.00″ E | 24 |
| #10 | Nangan, Lienchiang | 26°8′45.00″ N | 119°54′47.00″ E | 16 |
| Total | 141 |
| Markers | Na | Ne | Ho | He | PIC |
|---|---|---|---|---|---|
| SICAAS1015 | 14 | 9.18 | 0.000 | 0.89 | 0.88 |
| SICAAS1065 | 16 | 7.79 | 0.015 | 0.87 | 0.86 |
| SICAAS2084 | 8 | 5.16 | 0.000 | 0.81 | 0.78 |
| SICAAS3090 | 10 | 6.06 | 0.022 | 0.83 | 0.81 |
| SICAAS5005 | 2 | 1.26 | 0.000 | 0.21 | 0.18 |
| SICAAS5081 | 4 | 2.19 | 0.000 | 0.54 | 0.48 |
| SICAAS6052 | 2 | 1.37 | 0.000 | 0.27 | 0.30 |
| SICAAS7002 | 7 | 4.35 | 0.017 | 0.77 | 0.73 |
| SICAAS7008 | 4 | 1.98 | 0.000 | 0.49 | 0.44 |
| SICAAS7090 | 2 | 1.41 | 0.000 | 0.29 | 0.27 |
| SICAAS8025 | 4 | 2.12 | 0.016 | 0.53 | 0.41 |
| SICAAS9121 | 2 | 1.41 | 0.000 | 0.29 | 0.26 |
| SICAAS9130 | 4 | 1.98 | 0.000 | 0.50 | 0.45 |
| Average | 6.1 | 3.558 | 0.0054 | 0.561 | 0.527 |
| Collecting Site | SS | Na | Ne | Ho | He | FIS | FST | FIT | Nm |
|---|---|---|---|---|---|---|---|---|---|
| #01 | 16 | 13 | 13.00 | 0.00 | 0.00 | 0.9896 | 0.4895 | 0.9947 | 0.26 |
| #02 | 8 | 13 | 13.00 | 0.00 | 0.00 | 0.9901 | 0.4995 | 0.9950 | 0.25 |
| #03 | 16 | 19 | 14.25 | 0.01 | 0.12 | 0.9841 | 0.3954 | 0.9904 | 0.38 |
| #04 | 9 | 14 | 13.60 | 0.00 | 0.03 | 0.9904 | 0.4783 | 0.9950 | 0.27 |
| #05 | 10 | 22 | 18.63 | 0.00 | 0.16 | 0.9921 | 0.3540 | 0.9949 | 0.46 |
| #06 | 15 | 22 | 14.94 | 0.00 | 0.11 | 0.9905 | 0.4476 | 0.9948 | 0.31 |
| #07 | 11 | 20 | 15.57 | 0.00 | 0.12 | 0.9915 | 0.3984 | 0.9949 | 0.38 |
| #08 | 16 | 29 | 21.82 | 0.02 | 0.27 | 0.9723 | 0.2525 | 0.9793 | 0.74 |
| #09 | 24 | 18 | 15.04 | 0.00 | 0.07 | 0.9901 | 0.4277 | 0.9943 | 0.33 |
| #10 | 16 | 19 | 16.34 | 0.02 | 0.14 | 0.9653 | 0.3943 | 0.9790 | 0.38 |
| Average | 18.9 | 15.619 | 0.005 | 0.102 |
| #01 | #02 | #03 | #04 | #05 | #06 | #07 | #08 | #09 | |
|---|---|---|---|---|---|---|---|---|---|
| #02 | 0.47 | ||||||||
| #03 | 0.54 | 0.22 | |||||||
| #04 | 0.55 | 0.28 | 0.17 | ||||||
| #05 | 0.36 | 0.47 | 0.46 | 0.51 | |||||
| #06 | 0.57 | 0.52 | 0.56 | 0.52 | 0.50 | ||||
| #07 | 0.58 | 0.59 | 0.58 | 0.62 | 0.50 | 0.50 | |||
| #08 | 0.41 | 0.38 | 0.42 | 0.44 | 0.32 | 0.49 | 0.40 | ||
| #09 | 0.52 | 0.49 | 0.55 | 0.55 | 0.42 | 0.59 | 0.50 | 0.14 | |
| #10 | 0.51 | 0.52 | 0.55 | 0.58 | 0.46 | 0.59 | 0.54 | 0.32 | 0.45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, W.-H.; Chen, Y.-C.; Liao, H.-C.; Lin, Y.-R.; Chen, C.-H. High Differentiation among Populations of Green Foxtail, Setaria viridis, in Taiwan and Adjacent Islands Revealed by Microsatellite Markers. Diversity 2021, 13, 159. https://doi.org/10.3390/d13040159
Hsieh W-H, Chen Y-C, Liao H-C, Lin Y-R, Chen C-H. High Differentiation among Populations of Green Foxtail, Setaria viridis, in Taiwan and Adjacent Islands Revealed by Microsatellite Markers. Diversity. 2021; 13(4):159. https://doi.org/10.3390/d13040159
Chicago/Turabian StyleHsieh, Wei-Hsun, Yen-Chiun Chen, Hsien-Chun Liao, Yann-Rong Lin, and Chih-Hui Chen. 2021. "High Differentiation among Populations of Green Foxtail, Setaria viridis, in Taiwan and Adjacent Islands Revealed by Microsatellite Markers" Diversity 13, no. 4: 159. https://doi.org/10.3390/d13040159
APA StyleHsieh, W.-H., Chen, Y.-C., Liao, H.-C., Lin, Y.-R., & Chen, C.-H. (2021). High Differentiation among Populations of Green Foxtail, Setaria viridis, in Taiwan and Adjacent Islands Revealed by Microsatellite Markers. Diversity, 13(4), 159. https://doi.org/10.3390/d13040159