Genetic Structure and Phylogeography of Tuber magnatum Populations

 , , ,
, , ,  , and
, and
Abstract

1. Introduction
2. Materials and Methods
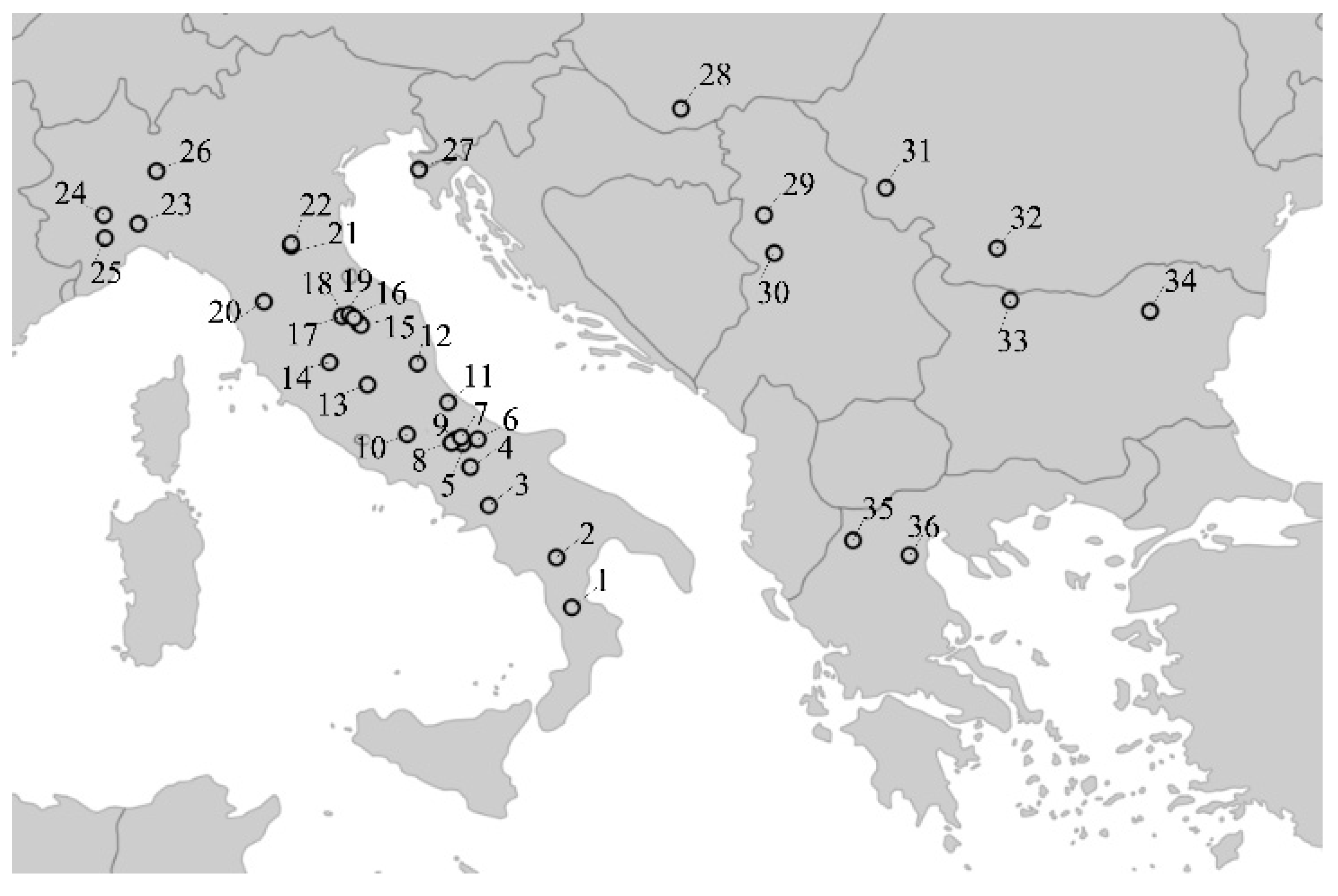
2.1. Sample Source and DNA Analysis
2.2. Genetic Diversity Data and Population Structure Analyses
3. Results
3.1. Genetic Diversity of T. magnatum Populations
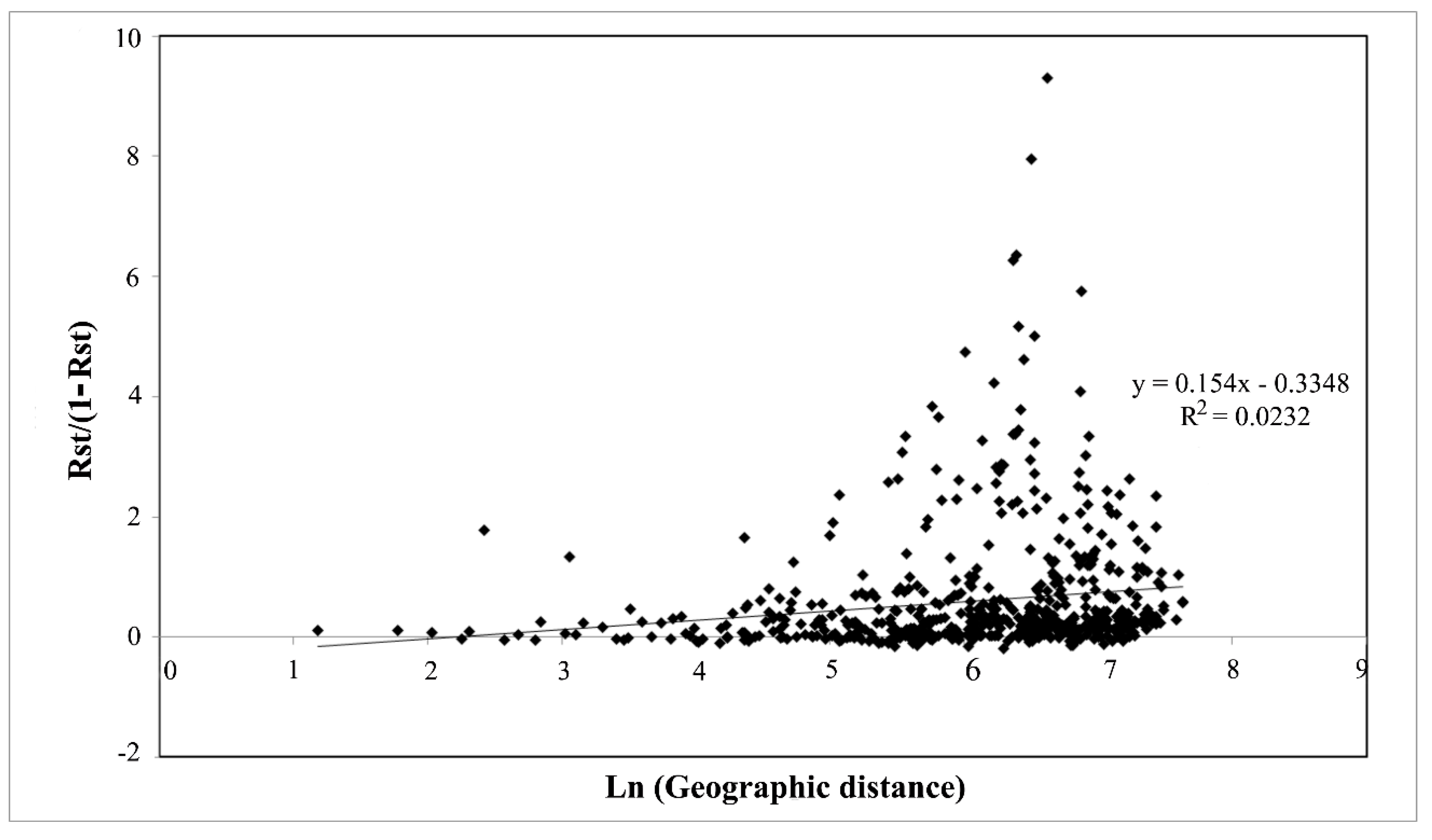
3.2. The Balkan/Pannonian and Italian Populations Were Genetically Differentiated
4. Discussion
4.1. New Insights into Genetic Diversity of White Truffle
4.2. Large Scale Sampling Reveals a Phylogeographic Structure in T. magnatum
4.3. Genetic Structure and Conservation Implications
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smith, S.E.; Read, D.J. Mycorrhizal Symbioses, 3rd ed.; Academic Press: Cambridge, MA, USA, 2008. [Google Scholar]
- Trappe, J.M.; Molina, R.; Luoma, D.L.; Cázares, E.; Pilz, D.; Smith, J.E.; Castellano, M.A.; Miller, S.L.; Trappe, M.J. Diversity, Ecology and Conservation of Truffle Fungi in Forests of the Pacific Northwest; Gen. Tech. Rep. PNW-GTR-772; USDA Forest Service, Pacific Northwest Research Station: Portland, OR, USA, 2009.
- Murat, C.; Díez, J.; Luis, P.; Delaruelle, C.; Dupré, C.; Chevalier, G.; Bonfante, P.; Martin, F. Polymorphism at the ribosomal DNA ITS and its relation to postglacial re-colonization routes of the Perigord truffle Tuber melanosporum. New Phytol. 2004, 164, 401–411. [Google Scholar] [CrossRef]
- Rubini, A.; Paolocci, F.; Riccioni, C.; Vendramin, G.G.; Arcioni, S. Genetic and phylogeographic structures of the symbiotic fungus Tuber magnatum. Appl. Environ. Microbiol. 2005, 71, 6584–6589. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Riccioni, C.; Belfiori, B.; Rubini, A.; Passeri, V.; Arcioni, S.; Paolocci, F. Tuber melanosporum outcrosses: Analysis of the genetic diversity within and among its natural populations under this new scenario. New Phytol. 2008, 180, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Merényi, Z.; Varga, T.; Geml, J.; Orczán, Á.K.; Chevalier, G.; Bratek, Z. Phylogeny and phylogeography of the Tuber brumale aggr. Mycorrhiza 2014, 24, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Molinier, V.; Murat, C.; Peter, M.; Gollotte, A.; De la Varga, H.; Meier, B.; Egli, S.; Belfiori, B.; Paolocci, F.; Wipf, D. SSR-based identification of genetic groups within European populations of Tuber aestivum Vittad. Mycorrhiza 2016, 26, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Qiao, P.; Tian, W.; Liu, P.; Yu, F.; Chen, J.; Deng, X.; Wan, S.; Wang, R.; Wang, Y.; Guo, H. Phylogeography and population genetic analyses reveal the speciation of the Tuber indicum complex. Fungal Genet. Biol. 2018, 113, 14–23. [Google Scholar] [CrossRef]
- Riccioni, C.; Rubini, A.; Türkoğlu, A.; Belfiori, B.; Paolocci, F. Ribosomal DNA polymorphisms reveal genetic structure and a phylogeographic pattern in the Burgundy truffle Tuber aestivum Vittad. Mycologia 2019, 111, 26–39. [Google Scholar] [CrossRef]
- Gandeboeuf, D.; Dupré, C.; Roeckel-Drévet, P.; Nicolas, P.; Chevalier, G. Grouping and identification of Tuber species using RAPD markers. Can. J. Bot. 1997, 75, 36–45. [Google Scholar] [CrossRef]
- Mello, A.; Fontana, A.; Meotto, F.; Comandini, O.; Bonfante, P. Molecular and morphological characterization of T. magnatum mycorrhizas in a long-term survey. Microbiol. Res. 2001, 155, 279–284. [Google Scholar] [CrossRef]
- Mello, A.; Murat, C.; Vizzini, A.; Gavazza, V.; Bonfante, P. Tuber magnatum Pico, a species of limited geographical distribution: Its genetic diversity inside and outside a truffle ground. Environ. Microbiol. 2005, 7, 55–65. [Google Scholar] [CrossRef]
- Riccioni, C.; Rubini, A.; Belfiori, B.; Gregori, G.; Paolocci, F. Tuber magnatum: The Special One. What Makes It so Different from the Other Tuber spp. In True Truffle (Tuber spp.) in the World: Soil Ecology, Systematics and Biochemistry; Zambonelli, A., Iotti, M., Murat, C., Eds.; Springer: Cham, Switzerland, 2016; pp. 87–103. [Google Scholar] [CrossRef]
- Frizzi, G.; Lalli, G.; Miranda, M.; Pacioni, G. Intraspecific isozyme variability in Italian populations of the white truffle Tuber magnatum. Mycol. Res. 2001, 105, 365–369. [Google Scholar] [CrossRef]
- Paolocci, F.; Rubini, A.; Riccioni, C.; Arcioni, S. Reevaluation of the life cycle of Tuber magnatum. Appl. Environ. Microbiol. 2006, 72, 2390–2393. [Google Scholar] [CrossRef] [PubMed]
- Marjanović, Ž.; Grebenc, T.; Marković, M.; Glišić, A.; Milenković, M. Ecological specificities and molecular diversity of truffles (genus Tuber) originating from mid-west of the Balkan Peninsula. Sydowia 2010, 62, 67–87. [Google Scholar]
- Tabouret, P. Exclusif! Description d’un site français producteur de truffe blanche d’Italie. Le Trufficulteur 2011, 78, 18. [Google Scholar]
- Tabouret, P. Constitution d’une fédération Suisse de la truffe et de la trufficulture. Le Trufficulteur 2012, 81, 25. [Google Scholar]
- Paolocci, F.; Rubini, A.; Granetti, B.; Arcioni, S. Rapid molecular approach for a reliable identification of Tuber spp. ectomycorrhizae. FEMS Microbiol. Ecol. 1999, 28, 23–30. [Google Scholar] [CrossRef]
- Rubini, A.; Topini, F.; Riccioni, C.; Paolocci, F.; Arcioni, S. Isolation and characterization of polymorphic microsatellite loci in white truffle (Tuber magnatum). Mol. Ecol. Notes 2004, 4, 116–118. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Szpiech, Z.A.; Jakobsson, M.; Rosenberg, N.A. ADZE: A rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 2008, 24, 2498–2504. [Google Scholar] [CrossRef]
- El Mousadik, A.; Petit, R.J. High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L.) Skeels] endemic to Morocco. Theor. Appl. Genet. 1996, 92, 832–839. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Agapow, P.M.; Burt, A. Indices of multilocus linkage disequilibrium. Mol. Ecol. Notes 2011, 1, 101–102. [Google Scholar] [CrossRef]
- Stenberg, P.; Lundmark, M.; Saura, A. MLGsim: A program for detecting clones using a simulation approach. Mol. Ecol. Resour. 2003, 3, 329–331. [Google Scholar] [CrossRef]
- Rousset, F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 1997, 145, 1219–1228. [Google Scholar] [PubMed]
- Hardy, O.J.; Vekemans, X. SPAGEDI: A versatile computer program to analyze spatial genetic structure at the individual or population level. Mol. Ecol. Notes 2002, 2, 618–620. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Chen, C.; Durand, E.; Forbes, F.; Francois, O. Bayesian clustering algorithms ascertaining spatial population structure: A new computer program and a comparison study. Mol. Ecol. Notes 2007, 7, 747–756. [Google Scholar] [CrossRef]
- Durand, E.; Jay, F.; Gaggiotti, O.E.; François, O. Spatial inference of admixture proportions and secondary contact zones. Mol. Biol. Evol. 2009, 26, 1963–1973. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Dent, A.E.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, N.A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Jay, F.; Manel, S.; Alvarez, N.; Durand, E.Y.; Thuiller, W.; Holderegger, R.; Taberlet, P.; François, O. Forecasting changes in population genetic structure of alpine plants in response to global warming. Mol. Ecol. 2012, 21, 2354–2368. [Google Scholar] [CrossRef] [PubMed]
- How to Display Ancestry Coefficients (Q-matrix) on a Geographic Map. Available online: http://membres-timc.imag.fr/Olivier.Francois/TESS_Plot.html (accessed on 4 November 2019).
- Bertault, G.; Raymond, M.; Berthomieu, A.; Callot, G.; Fernandez, D. Trifling variation in truffles. Nature 1998, 394, 734. [Google Scholar] [CrossRef]
- Murat, C.; Riccioni, C.; Belfiori, B.; Cichocki, N.; Labbé, J.; Morin, E.; Tisserant, E.; Paolocci, F.; Rubini, A.; Martin, F. Distribution and localization of microsatellites in the Perigord black truffle genome and identification of new molecular markers. Fungal Genet. Biol. 2011, 48, 592–601. [Google Scholar] [CrossRef]
- Molinier, V.; Murat, C.; Morin, E.; Gollotte, A.; Wipf, D.; Martin, F. First identification of polymorphic microsatellite markers in the Burgundy truffle, Tuber aestivum (Tuberaceae). Appl. Plant. Sci. 2013, 1, 1200220. [Google Scholar] [CrossRef]
- Pomarico, M.; Figliuolo, G.; Rana, G.L. Tuber spp. Biodiversity in one of the southernmost European distribution areas. Biodivers. Conserv. 2007, 16, 3447–3461. [Google Scholar] [CrossRef]
- Petit, R.J.; Brewer, S.; Bordács, S.; Burg, K.; Cheddadi, R.; Coart, E.; Cottrel, J.; Csaikl, U.M.; Van Dam, B.; Deans, J.D.; et al. Identification of refugia and post-glacial colonisation routes of European white oaks based on chloroplast DNA and fossil pollen evidence. For Ecol. Manag. 2002, 156, 49–74. [Google Scholar] [CrossRef]
- Bertault, G.; Rousset, F.; Fernandez, D.; Berthomieu, A.; Hochberg, M.E.; Callot, G.; Raymond, M. Population genetics and dynamics of the black truffle in a man-made truffle field. Heredity 2001, 86, 451–458. [Google Scholar] [CrossRef]
- García-Cunchillos, I.; Sánchez, S.; Barriuso, J.J.; Pérez-Collazos, E. Population genetics of the westernmost distribution of the glaciations-surviving black truffle Tuber melanosporum. Mycorrhiza 2014, 24, 89–100. [Google Scholar] [CrossRef]
- Merényi, Z.; Varga, T.; Hubai, A.G.; Pitlik, P.; Erős, Á.; Trappe, J.M.; Bratek, Z. Challenges in the delimitation of morphologically similar species: A case study of Tuber brumale agg. (Ascomycota, Pezizales). Mycol. Prog. 2017, 16, 613. [Google Scholar] [CrossRef]
- Widmer, A.; Lexer, C. Glacial refugia: Sanctuaries for allelic richness, but not for gene diversity. Trends Ecol. Evol. 2001, 16, 267–269. [Google Scholar] [CrossRef]
- Taberlet, P.; Fumagalli, L.; Wust-Saucy, A.G.; Cosson, J.F. Comparative phylogeography and postglacial colonisation routes in Europe. Mol. Ecol. 1998, 7, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Grivet, D.; Petit, R.J. Chloroplast DNA phylogeography of the hornbeam in Europe: Evidence for a bottleneck at the outset of postglacial colonization. Conserv. Genet. 2003, 4, 47–56. [Google Scholar] [CrossRef]
- Magri, D. Patterns of post-glacial spread and the extent of glacial refugia of European beech (Fagus sylvatica). J. Biogeogr. 2008, 35, 450–463. [Google Scholar] [CrossRef]
- Sanmartín, I. Dispersal vs vicariance in the mediterranean: Historical biogeography of the Paleartic Pachydeminae (Coleoptera, Scarabeodidea). J. Biogeog. 2003, 30, 1883–1897. [Google Scholar] [CrossRef]
- De Giuli, C.; Masini, F.; Torre, D. Island endemism in the Eastern Mediterranean mammalian paleofaunas patterns in the Gargano paleo-arcipelago. Atti Accad. Naz. Lin. 1990, 85, 247–262. [Google Scholar]
- Kaiser, K. The climate of Europe during the Quaternary Ice Age. In Quaternary Geology and Climate; Wright, H.E., Jr., Ed.; National Academy of Sciences: Washington, DC, USA, 1969. [Google Scholar]
- Gridelli, E. IL problema delle specie a diffusione attuale transadriatica con particolare riguardo ai Coleotteri. Ital. J. Zool. 1950, 17, 421–441. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population No. | Population Name (Sampling Location) | Locality | Sample Size* | Mean No. of Alleles** | Expected Heterozygosity** | Allelic Richness** |
|---|---|---|---|---|---|---|
| 1 | Valle del Crati | Italy (Calabria) | 3 | 1.5 ± 0.19 | 0.22 ± 0.08 | ND++ |
| 2 | Potenza | Italy (Basilicata) | 14 (12) | 2.9 ± 0.35 | 0.37 ± 0.06 | 1.81 ± 0.15 |
| 3 | Benevento, Avellino | Italy (Campania) | (5) | 2.3 ± 0.37 | 0.39 ± 0.09 | 2.05 ± 0.29 |
| 4 | Isernia, Campobasso | Italy (Molise) | (16) | 3.4 ± 0.53 | 0.56 ± 0.06 | 2.27 ± 0.18 |
| 5 | Agnone | Italy (Molise) | (10) | 3.4 ± 0.73 | 0.46 ± 0.11 | 2.21 ± 0.33 |
| 6 | Castel del Giudice | Italy (Molise) | (16) | 2.9 ± 0.48 | 0.42 ± 0.09 | 1.95 ± 0.23 |
| 7 | Ateleta, SP. Avellana | Italy (Abruzzo-Molise) | 36 (18) | 4.6 ± 0.89 | 0.50 ± 0.10 | 2.17 ± 0.26 |
| 8 | Roio del Sangro | Italy (Abruzzo-Molise) | (10) | 2.9 ± 0.48 | 0.41 ± 0.09 | 1.98 ± 0.23 |
| 9 | Quadri, Rosello | Italy (Abruzzo) | (14) | 2.9 ± 0.58 | 0.44 ± 0.11 | 2.04 ± 0.27 |
| 10 | Valle Roveto | Italy (Abruzzo) | (7) | 2.3 ± 0.41 | 0.34 ± 0.09 | 1.83 ± 0.25 |
| 11 | Chieti | Italy (Abruzzo) | (5) | 2.3 ± 0.41 | 0.37 ± 0.12 | 2.08 ± 0.35 |
| 12 | Ascoli Piceno | Italy (Marche) | (10) | 2.8 ± 0.56 | 0.37 ± 0.11 | 1.92 ± 0.30 |
| 13 | Montecastrilli | Italy (Umbria) | (9) | 2.8 ± 0.59 | 0.42 ± 0.10 | 2.06 ± 0.29 |
| 14 | Fabro | Italy (Umbria) | (11) | 2.8 ± 0.41 | 0.42 ± 0.07 | 1.96 ± 0.21 |
| 15 | Appennino umbro | Italy (Umbria) | (9) | 2.4 ± 0.50 | 0.36 ± 0.10 | 1.85 ± 0.27 |
| 16 | Gubbio | Italy (Umbria) | (25) | 3.8 ± 0.75 | 0.45 ± 0.09 | 1.97 ± 0.22 |
| 17 | Città di Castello | Italy (Umbria) | (12) | 2.9 ± 0.69 | 0.39 ± 0.10 | 1.93 ± 0.28 |
| 18 | Montemaggiore | Italy (Umbria) | (12) | 2.3 ± 0.53 | 0.29 ± 0.11 | 1.68 ± 0.27 |
| 19 | Pietralunga | Italy (Umbria) | (7) | 2.4 ± 0.42 | 0.36 ± 0.10 | 1.92 ± 0.28 |
| 20 | Firenze | Italy (Toscana) | (32) | 4.1 ± 0.90 | 0.47 ± 0.08 | 2.04 ± 0.21 |
| 21 | Val di Zena | Italy (Emilia-Romagna) | (10) | 2.9 ± 0.58 | 0.43 ± 0.10 | 2.02 ± 0.27 |
| 22 | Pianoro | Italy (Emilia-Romagna) | (9) | 2.5 ± 0.53 | 0.38 ± 0.11 | 1.94 ± 0.20 |
| 23 | Alba | Italy (Piemonte) | (7) | 2.8 ± 0.49 | 0.42 ± 0.09 | 2.12 ± 0.29 |
| 24 | Asti | Italy (Piemonte) | (9) | 2.5 ± 0.42 | 0.40 ± 0.10 | 1.98 ± 0.27 |
| 25 | Langhe | Italy (Piemonte) | (15) | 3.0 ± 0.76 | 0.42 ± 0.11 | 2.01 ± 0.30 |
| 26 | Pavia | Italy (Lombardia) | (11) | 2.4 ± 0.42 | 0.36 ± 0.10 | 1.85 ± 0.26 |
| 27 | Istria | Croatia-Slovenia | (17) | 3.0 ± 0.60 | 0.41 ± 0.10 | 1.93 ± 0.25 |
| 28 | Southern Hungary | Hungary | 14 | 3.1 ± 0.74 | 0.37 ± 0.11 | 1.91 ± 0.30 |
| 29 | Šabac | Serbia | 6 | 2.1 ± 0.44 | 0.28 ± 0.11 | 1.78 ± 0.31 |
| 30 | Valjevo | Serbia | 7 | 2.0 ± 0.33 | 0.33 ± 0.10 | 1.66 ± 0.27 |
| 31 | Anina | Romania | 3 | 1.4 ± 0.18 | 0.17 ± 0.08 | ND++ |
| 32 | Craiova | Romania | 22 | 3.6 ± 0.84 | 0.44 ± 0.12 | 2.04 ± 0.31 |
| 33 | Oryahovo | Bulgaria | 6 | 2.0 ± 0.27 | 0.39 ± 0.09 | 1.89 ± 0.22 |
| 34 | Razgrad | Bulgaria | 6 | 1.9 ± 0.40 | 0.29 ± 0.11 | 1.75 ± 0.32 |
| 35 | Kastoria | Greece | 18 | 3.6 ± 0.71 | 0.47 ± 0.11 | 2.13 ± 0.28 |
| 36 | Elatochori | Greece | 6 | 2.5 ± 0.46 | 0.40 ± 0.10 | 2.15 ± 0.33 |
| Mean | Italy | - | 12.46 ± 1.52 | 2.84 ± 0.12 | 0.40 ± 0.01 | 1.99 ± 0.03 |
| Mean | Pannonia/Balkans | - | 10.50 ± 2.09 | 2.52 ± 0.24 | 0.36 ± 0.03 | 1.92 ± 0.05 |
| Mean | Total | - | 11.92 ± 1.23 | 2.75 ± 0.11 | 0.39 ± 0.01 | 1.97 ± 0.02 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belfiori, B.; D’Angelo, V.; Riccioni, C.; Leonardi, M.; Paolocci, F.; Pacioni, G.; Rubini, A. Genetic Structure and Phylogeography of Tuber magnatum Populations. Diversity 2020, 12, 44. https://doi.org/10.3390/d12020044
Belfiori B, D’Angelo V, Riccioni C, Leonardi M, Paolocci F, Pacioni G, Rubini A. Genetic Structure and Phylogeography of Tuber magnatum Populations. Diversity. 2020; 12(2):44. https://doi.org/10.3390/d12020044
Chicago/Turabian StyleBelfiori, Beatrice, Valentina D’Angelo, Claudia Riccioni, Marco Leonardi, Francesco Paolocci, Giovanni Pacioni, and Andrea Rubini. 2020. "Genetic Structure and Phylogeography of Tuber magnatum Populations" Diversity 12, no. 2: 44. https://doi.org/10.3390/d12020044
APA StyleBelfiori, B., D’Angelo, V., Riccioni, C., Leonardi, M., Paolocci, F., Pacioni, G., & Rubini, A. (2020). Genetic Structure and Phylogeography of Tuber magnatum Populations. Diversity, 12(2), 44. https://doi.org/10.3390/d12020044