Genetic Consequences of Multiple Translocations of the Banded Hare-Wallaby in Western Australia

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Tissue Samples
2.2. DNA Extraction and Nuclear Microsatellite Amplification
2.3. Statistical Analyses of Genetic Data
2.4. Population Modelling
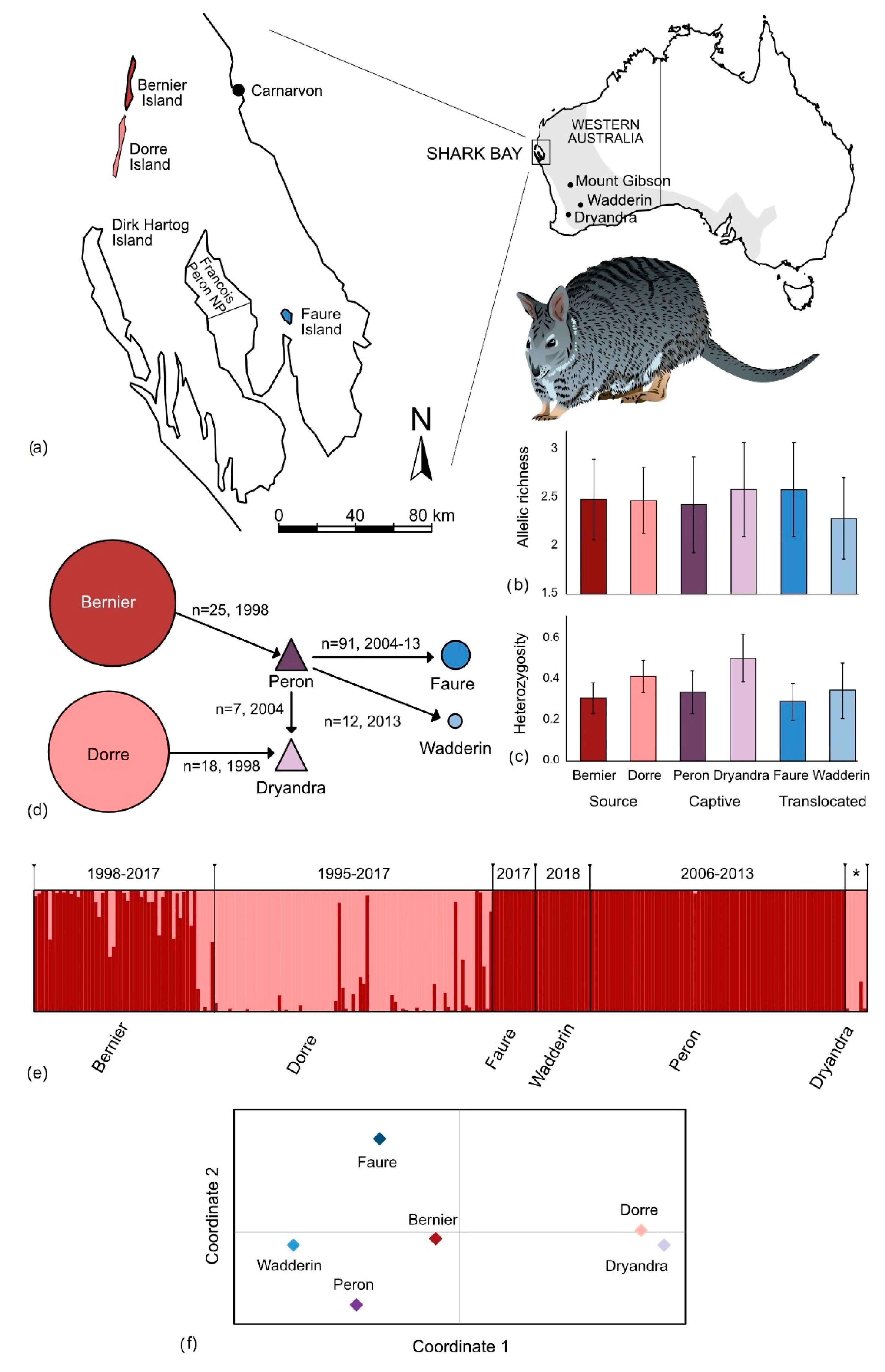
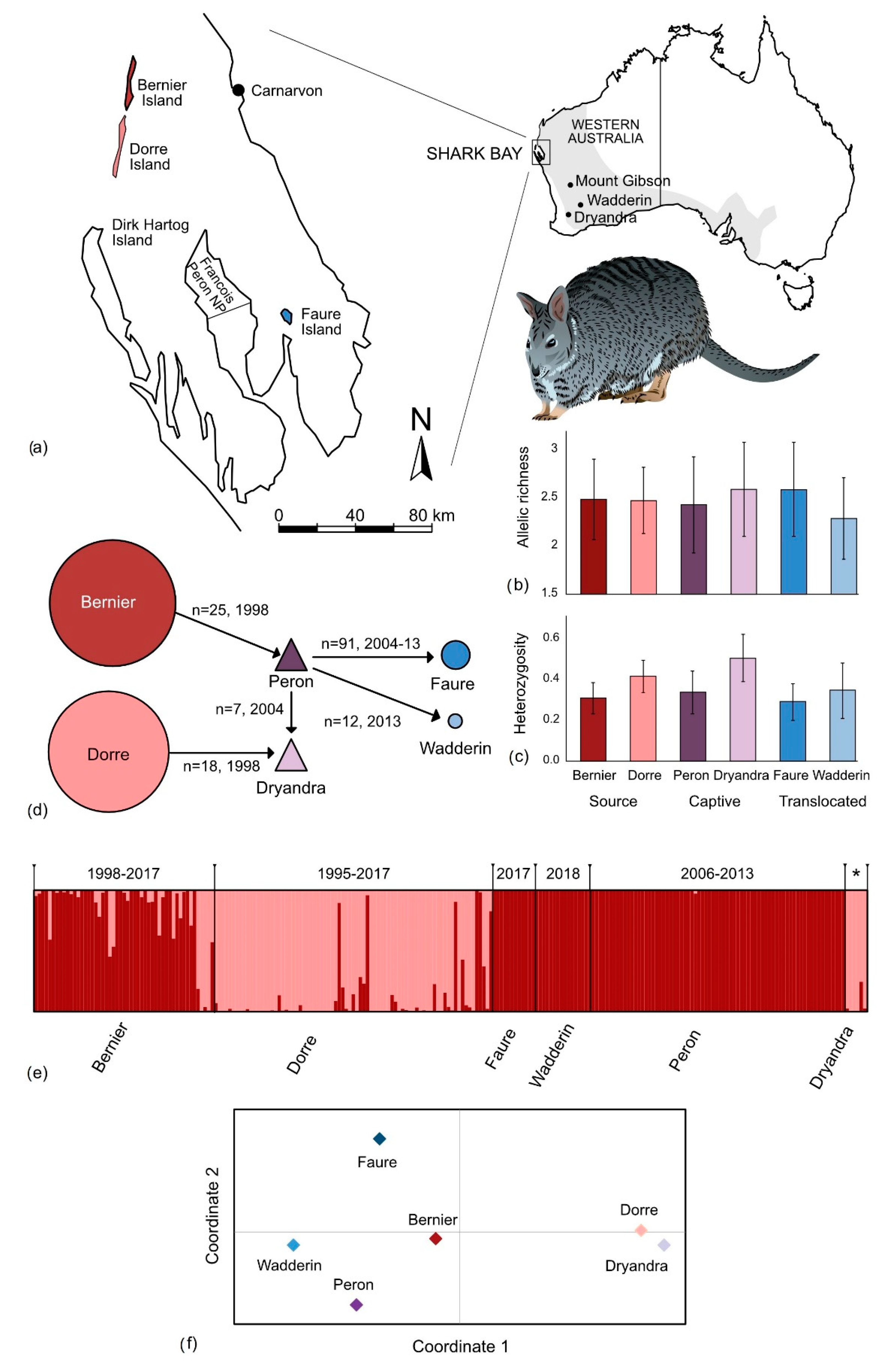
3. Results
3.1. Marker Performance
3.2. Genetic Diversity
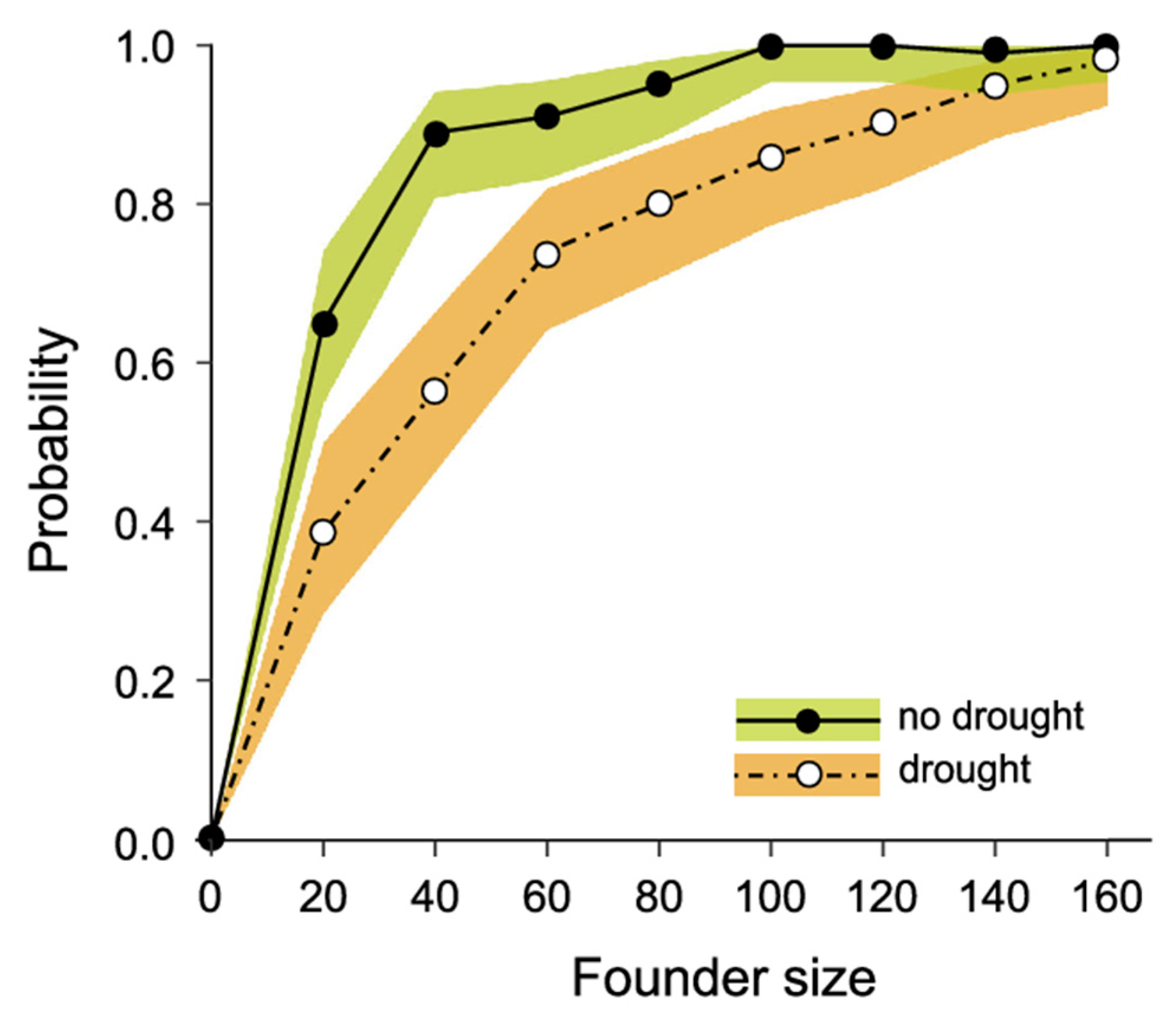
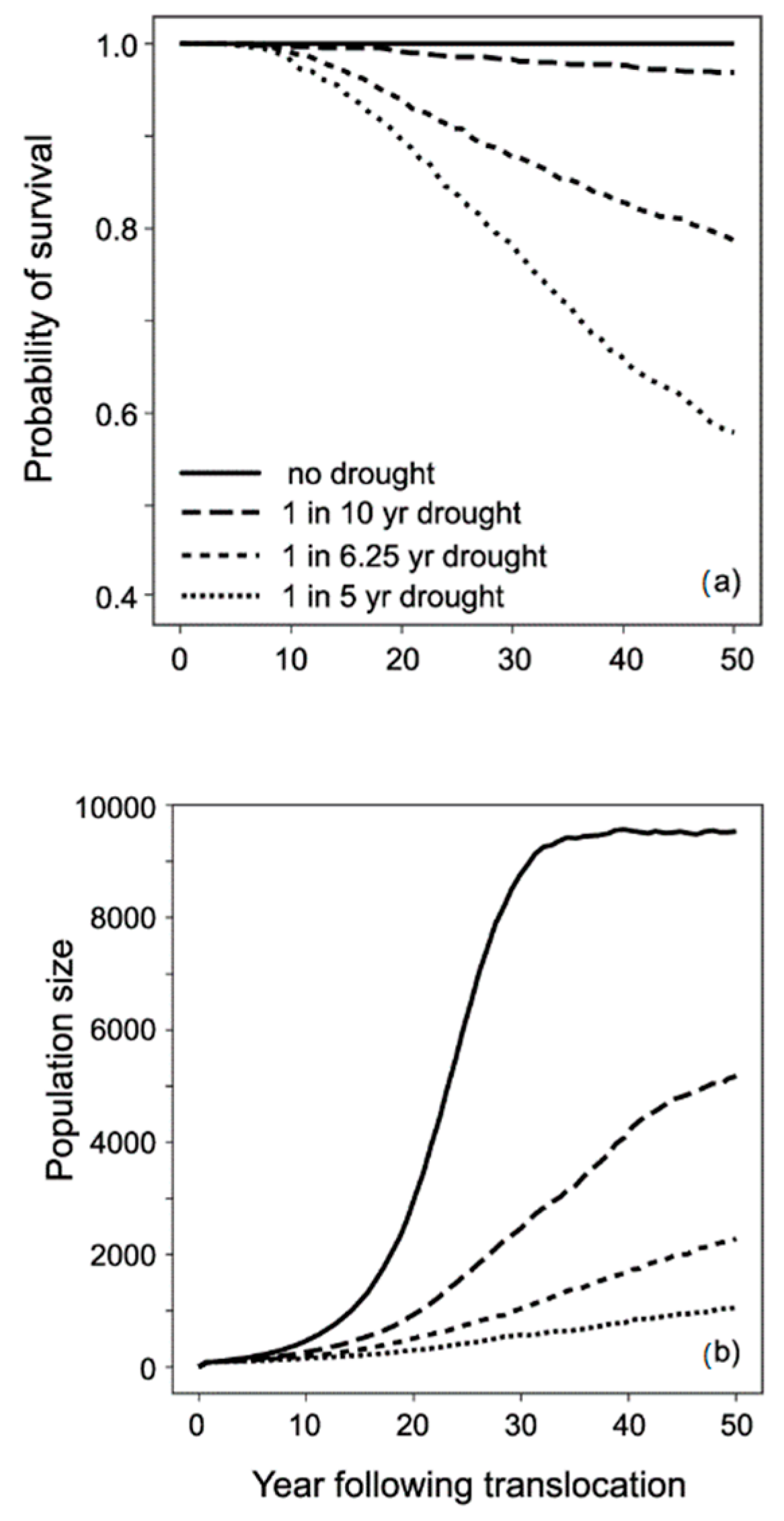
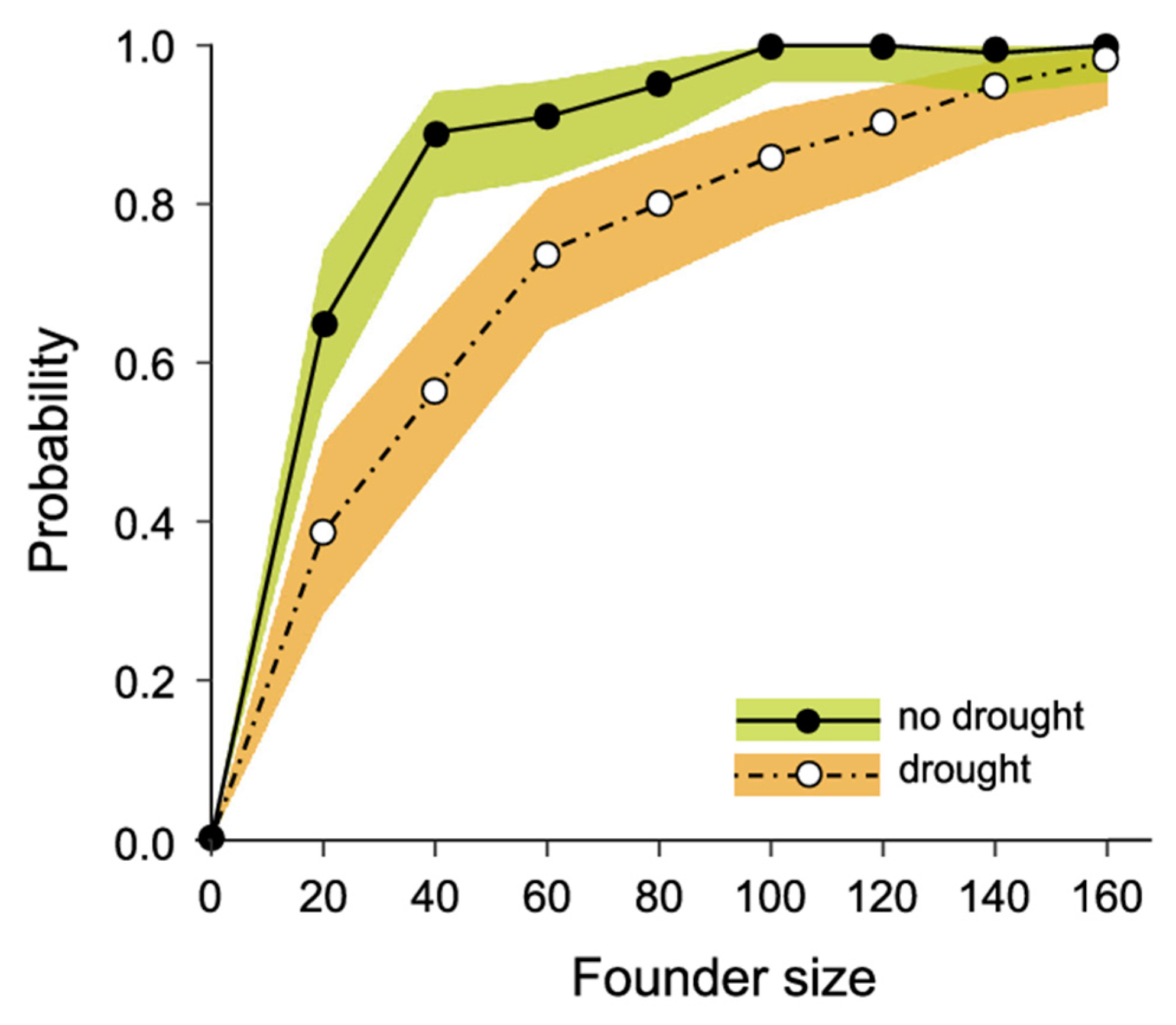
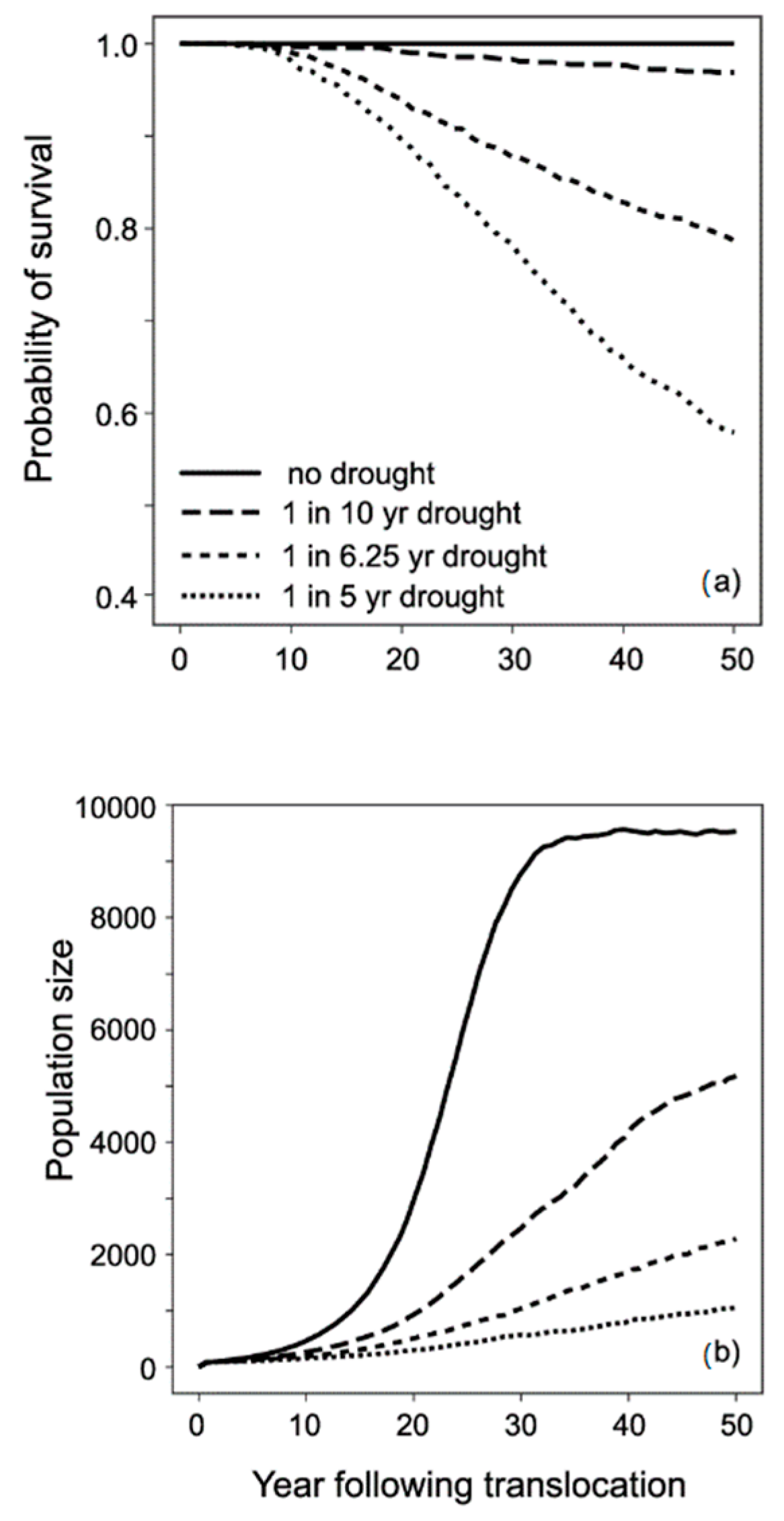
3.3. Modelling Conservation Translocations
4. Discussion
4.1. Genetic Diversity in Remnant Populations
4.2. Impact of Past Translocations on Genetic Diversity
4.3. Towards an Optimal Translocation Protocol for L. fasciatus
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frankham, R.; Ballou, J.D.; Ralls, K.; Eldridge, M.D.B.; Dudash, M.R.; Fenster, C.B.; Lacy, R.C.; Sunnucks, P. Genetic Management of Fragmented Animal and Plant Populations; Oxford University Press: Oxford, UK, 2017; p. 432. [Google Scholar]
- Fischer, J.; Lindenmayer, D.B. An assessment of the published results of animal relocations. Biol. Conserv. 2000, 96, 1–11. [Google Scholar] [CrossRef]
- Germano, J.M.; Bishop, P.J. Suitability of Amphibians and Reptiles for Translocation. Conserv. Biol. 2009, 23, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Ottewell, K.; Dunlop, J.; Thomas, N.; Morris, K.; Coates, D.; Byrne, M. Evaluating success of translocations in maintaining genetic diversity in a threatened mammal. Biol. Conserv. 2014, 171, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Perez, I.; Anadon, J.D.; Diaz, M.; Nicola, G.G.; Tella, J.L.; Gimenez, A. What is wrong with current translocations? A review and a decision-making proposal. Front. Ecol. Environ. 2012, 10, 494–501. [Google Scholar] [CrossRef] [Green Version]
- Sheean, V.A.; Manning, A.D.; Lindenmayer, D.B. An assessment of scientific approaches towards species relocations in Australia. Austral Ecol. 2012, 37, 204–215. [Google Scholar] [CrossRef]
- Weeks, A.R.; Sgro, C.M.; Young, A.G.; Frankham, R.; Mitchell, N.J.; Miller, K.A.; Byrne, M.; Coates, D.J.; Eldridge, M.D.B.; Sunnucks, P.; et al. Assessing the benefits and risks of translocations in changing environments: A genetic perspective. Evol. Appl. 2011, 4, 709–725. [Google Scholar] [CrossRef] [Green Version]
- Short, J. Predation by feral cats key to the failure of a long-term reintroduction of the western barred bandicoot (Perameles bougainville). Wildl. Res. 2016, 43, 38–50. [Google Scholar] [CrossRef]
- Brashares, J.S.; Werner, J.R.; Sinclair, A.R.E. Social ‘meltdown’ in the demise of an island endemic: Allee effects and the Vancouver Island marmot. J. Anim. Ecol. 2010, 79, 965–973. [Google Scholar] [CrossRef] [PubMed]
- IUCN/SSC. Guidelines for Reintroductions and Other Conservation Translocations; Version 1.0; IUCN Special Survival Commission: Gland, Switzerland; p. viiii + 57.
- Saltz, D. A long-term systematic approach to planning reintroductions: The Persian fallow deer and the Arabian oryx in Israel. Anim. Conserv. 1998, 1, 245–252. [Google Scholar] [CrossRef]
- Holzapfel, S.A.; Robertson, H.A.; McLennan, J.A.; Sporle, W.; Hackwell, K.; Impey, M. Kiwi (Apteryx spp.) recovery plan 2008–2018. In Threatened Species Recovery Plan; Department of Conservation: Wellington, New Zealand, 2008; Volume 60. [Google Scholar]
- Lloyd, B.D.; Powlesland, R.G. The decline of kakapo (Strigops habroptilus) and attempts at conservation by translocation. Biol. Conserv. 1994, 69, 75–85. [Google Scholar] [CrossRef]
- Weeks, A.R.; Heinze, D.; Perrin, L.; Stoklosa, J.; Hoffmann, A.A.; van Rooyen, A.; Kelly, T.; Mansergh, I. Genetic rescue increases fitness and aids rapid recovery of an endangered marsupial population. Nat. Comm. 2017, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Frankham, R. Genetics and extinction. Biol. Conserv. 2005, 126, 131–140. [Google Scholar] [CrossRef]
- Allendorf, F.W. Genetic Drift and the Loss of Alleles Versus Heterozygosity. Zoo Biol. 1986, 5, 181–190. [Google Scholar] [CrossRef]
- Gilpin, M.E.; Soule, M.E. Minimum viable populations: Processes of species extinction. In Conservation Biology: The Science of Scarcity and Diversity; Gilpin, M.E., Soule, M.E., Eds.; Sinauer: Sunderland, MA, USA, 1986; pp. 19–24. [Google Scholar]
- Lacy, R.C. Lessons from 30 years of population viability analysis of wildlife populations. Zoo Biol. 2019, 38, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Tracy, L.N.; Wallis, G.P.; Efford, M.G.; Jamieson, I.G. Preserving genetic diversity in threatened species reintroductions: How many individuals should be released? Anim. Conserv. 2011, 14, 439–446. [Google Scholar] [CrossRef]
- Pacioni, C.; Wayne, A.F.; Page, M. Guidelines for genetic management in mammal translocation programs. Biol. Conserv. 2019, 237, 105–113. [Google Scholar] [CrossRef]
- Grueber, C.E.; Fox, S.; McLennan, E.A.; Gooley, R.M.; Pemberton, D.; Hogg, C.J.; Belov, K. Complex problems need detailed solutions: Harnessing multiple data types to inform genetic management in the wild. Evol. Appl. 2019, 12, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Kelly, E.; Phillips, B. How many and when? Optimising targeted gene flow for a step change in the environment. Ecol. Lett. 2019, 22, 447–457. [Google Scholar] [CrossRef]
- Ramalho, C.E.; Ottewell, K.M.; Chambers, B.K.; Yates, C.J.; Wilson, B.A.; Bencini, R.; Barrett, G. Demographic and genetic viability of a medium-sized ground-dwelling mammal in a fire prone, rapidly urbanizing landscape. PLoS ONE 2018, 13, e0191190. [Google Scholar] [CrossRef] [Green Version]
- Woinarski, J.C.Z.; Burbidge, A.A.; Harrison, P.L. Ongoing unraveling of a continental fauna: Decline and extinction of Australian mammals since European settlement. Proc. Natl. Acad. Sci. USA 2015, 112, 4531–4540. [Google Scholar] [CrossRef] [Green Version]
- Burbidge, A.A.; McKenzie, N.L. Patterns in the Modern Decline of Western-Australia Vertebrate Fauna—Causes and Conservation Implications. Biol. Conserv. 1989, 50, 143–198. [Google Scholar] [CrossRef]
- Cardillo, M.; Bromham, L. Body size and risk of extinction in Australian mammals. Conserv. Biol. 2001, 15, 1435–1440. [Google Scholar] [CrossRef]
- Chisholm, R.; Taylor, R. Null-hypothesis significance testing and the critical weight range for Australian mammals. Conserv. Biol. 2007, 21, 1641–1645. [Google Scholar] [CrossRef] [PubMed]
- Woinarski, J.C.Z. Critical-weight-range marsupials in northern Australia are declining: A commentary on Fisher et al. (2014) ’The current decline of tropical marsupials in Australia: Is history repeating?’. Glob. Ecol. Biogeogr. 2015, 24, 118–122. [Google Scholar] [CrossRef]
- Molloy, S.W.; Davis, R.A.; Van Etten, E.J.B. Species distribution modelling using bioclimatic variables to determine the impacts of a changing climate on the western ringtail possum (Pseudocheirus occidentals; Pseudocheiridae). Environ. Conserv. 2014, 41, 176–186. [Google Scholar] [CrossRef]
- Reckless, H.J.; Murray, M.; Crowther, M.S. A review of climatic change as a determinant of the viability of koala populations. Wildl. Res. 2017, 44, 458–470. [Google Scholar] [CrossRef]
- Shortridge, G.C. An account of the geographical distribution of the Marsuprals and Monotremes of South-West Australia, having special reference to the specimens collected during the Balston Expedition of 1904-1907. Proc. Zool. Soc. Lond. 1909, 1909, 803–848. [Google Scholar]
- Thomas, N.; (Department of Biodiversity, Conservation and Attractions, Perth, WA, Australia). Personal communication, 2018.
- Chapman, T.F.; Sims, C.; Thomas, N.D.; Reinhold, L. Assessment of mammal populations on Bernier and Dorre Island 2006–2013. In Report for the Department of Parks and Wildlife; Department of Parks and Wildlife: Kensington, Australia, 2015; p. 56. [Google Scholar]
- Short, J.; Turner, B.; Majors, C.; Leone, J. The fluctuating abundance of endangered mammals on Bernier and Dorre Islands, Western Australia—conservation implications. Aust. Mammal. 1997, 20, 53–61. [Google Scholar]
- Hardman, B.; Moro, D.; Calver, M. Direct evidence implicates feral cat predation as the primary cause of failure of a mammal reintroduction programme. Ecol. Manag. Restor. 2016, 17, 152–158. [Google Scholar] [CrossRef]
- Morris, K.; Sims, C.; Himbeck, K.; Christensen, P.; Sercombe, N.; Ward, B.; Noakes, N. Project Eden—fauna recovery on Peron Peninsula, Shark Bay: Western Shield review—February 2003. Conserv. Sci. West. Aust. 2004, 5, 202–234. [Google Scholar]
- Short, J.; Turner, B. The Distribution and Abundance of the Banded and Rufous Hare-Wallabies, Lagostrophus fasciatus and Lagorchestes hirsutus. Biol. Conserv. 1992, 60, 157–166. [Google Scholar] [CrossRef]
- Ruykys, L.; Smith, M.; Kanowski, J. Translocation of Banded Hare-wallabies (Lagostrophus fasciatus) to Mt Gibson Sanctuary and Faure Island, WA; Department of Biodiversity, Conservation and Attractions: Kensington, Australia, 2017; p. 44. [Google Scholar]
- Short, J.; Hide, A. Successful reintroduction of the brushtail possum to Wadderin Sanctuary in the eastern wheatbelt of Western Australia. Aust. Mammal. 2014, 36, 229–241. [Google Scholar] [CrossRef]
- Short, J.; (Wildlife Research and Management Pty Ltd., Perth, WA, Australia). Personal communication, 2020.
- Morris, K.; Page, M.; Thomas, N.; Ottewell, K. A strategic framework for the reconstruction and conservation of the vertebrate fauna of Dirk Hartog Island 2016–2030; Department of Parks and Wildlife: Perth, Australia, 2017; p. 26. [Google Scholar]
- Harris, R.M.B.; Beaumont, L.J.; Vance, T.R.; Tozer, C.R.; Remenyi, T.A.; Perkins-Kirkpatrick, S.E.; Mitchell, P.J.; Nicotra, A.B.; McGregor, S.; Andrew, N.R.; et al. Biological responses to the press and pulse of climate trends and extreme events. Nat. Clim. Chang. 2018, 8, 579–587. [Google Scholar] [CrossRef]
- Sunnucks, P.; Hales, D.F. Numerous transposed sequences of mitochondrial cytochrome oxidase I-II in aphids of the genus Sitobion (Hemiptera: Aphididae). Mol. Biol. Evol. 1996, 13, 510–524. [Google Scholar] [CrossRef]
- Taylor, A.C.; Cooper, D.W. A set of tammar wallaby (Macropus eugenii) microsatellites tested for genetic linkage. Mol. Ecol. 1998, 7, 925–926. [Google Scholar]
- Pope, L.C.; Sharp, A.; Moritz, C. Population structure of the yellow-footed rock-wallaby Petrogale xanthopus (Gray, 1854) inferred from mtDNA sequences and microsatellite loci. Mol. Ecol. 1996, 5, 629–640. [Google Scholar] [CrossRef]
- Spencer, P.B.S.; Odorico, D.M.; Jones, S.J.; Marsh, H.D.; Miller, D.J. Highly Variable Microsatellites in Isolated Colonies of the Rock-Wallaby (Petrogale assimilis). Mol. Ecol. 1995, 4, 523–525. [Google Scholar] [CrossRef]
- Rousset, F. GENEPOP ‘ 007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef]
- Guo, S.W.; Thompson, E.A. Performing the exact test of hardy-weinberg proportion for multiple alleles. Biometrics 1992, 48, 361–372. [Google Scholar] [CrossRef]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Res. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, N.A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Kalinowski, S.T. HP-RARE 1.0: A computer program for performing rarefaction on measures of allelic richness. Mol. Ecol. Notes 2005, 5, 187–189. [Google Scholar] [CrossRef]
- Hill, W.G. Estimation of Effective Population-Size from Data on Linkage Disequilibrium. Genet. Res. 1981, 38, 209–216. [Google Scholar] [CrossRef] [Green Version]
- Waples, R.S. A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conserv. Genet. 2006, 7, 167–184. [Google Scholar] [CrossRef] [Green Version]
- Waples, R.S.; Do, C. Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: A largely untapped resource for applied conservation and evolution. Evol. Appl. 2010, 3, 244–262. [Google Scholar] [CrossRef]
- Do, C.; Waples, R.S.; Peel, D.; Macbeth, G.M.; Tillett, B.J.; Ovenden, J.R. NEESTIMATOR v2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Res. 2014, 14, 209–214. [Google Scholar] [CrossRef]
- Piry, S.; Luikart, G.; Cornuet, J.M. BOTTLENECK: A computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Cornuet, J.M.; Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 1996, 144, 2001–2014. [Google Scholar]
- Luikart, G.; Sherwin, W.B.; Steele, B.M.; Allendorf, F.W. Usefulness of molecular markers for detecting population bottlenecks via monitoring genetic change. Mol. Ecol. 1998, 7, 963–974. [Google Scholar] [CrossRef]
- Luikart, G.; Allendorf, F.W.; Cornuet, J.M.; Sherwin, W.B. Distortion of allele frequency distributions provides a test for recent population bottlenecks. J. Hered. 1998, 89, 238–247. [Google Scholar] [CrossRef]
- Queller, D.C.; Goodnight, K.F. Estimating Relatedness Using Genetic-Markers. Evolution 1989, 43, 258–275. [Google Scholar] [CrossRef]
- Weiser, E.L.; Grueber, C.E.; Jamieson, I.G. AlleleRetain: A program to assess management options for conserving allelic diversity in small, isolated populations. Mol. Ecol. Res. 2012, 12, 1161–1167. [Google Scholar] [CrossRef]
- Weiser, E.L.; Grueber, C.E.; Jamieson, I.G. Simulating Retention of Rare Alleles in Small Populations to Assess Management Options for Species with Different Life Histories. Conserv. Biol. 2013, 27, 335–344. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.R-project.org/ (accessed on 11 May 2019).
- Sims, C.; (Department of Biodiversity, Conservation and Attractions, Perth, WA, Australia). Personal communication, 2018.
- Richards, J.D.; Short, J.; Prince, R.I.T.; Friend, J.A.; Courtenay, J.M. The biology of banded (Lagostrophus fasciatus) and rufous (Lagorchestes hirsutus) hare-wallabies (Diprotodontia: Macropodidae) on Dorre and Bernier Islands, Western Australia. Wildl. Res. 2001, 28, 311–322. [Google Scholar] [CrossRef]
- Lacy, R.C. VORTEX—A computer-simulation model for population viability analysis. Wildl. Res. 1993, 20, 45–65. [Google Scholar] [CrossRef]
- Lacy, R.C.; Pollak, J.P. Vortex: A Stochastic Simulation of the Extinction Process; Version 10.2.9; Chicago Zoological Society: Brookfield, IL, USA, 1993; Available online: http://www.vortex10.org/Vortex10.aspx (accessed on 11 May 2019).
- Eldridge, M.D.B.; King, J.M.; Loupis, A.K.; Spencer, P.B.S.; Taylor, A.C.; Pope, L.C.; Hall, G.P. Unprecedented low levels of genetic variation and inbreeding depression in an island population of the black-footed rock-wallaby. Conserv. Biol. 1999, 13, 531–541. [Google Scholar] [CrossRef]
- Chapuis, M.P.; Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 2007, 24, 621–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankham, R. Do island populations have less genetic variation than mainland populations? Heredity 1997, 78, 311–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, S. Evolution in Mendelian populations. Genetics 1931, 16, 0097–0159. [Google Scholar]
- Pacioni, C.; Wayne, A.F.; Spencer, P.B.S. Genetic outcomes from the translocations of the critically endangered woylie. Curr. Zool. 2013, 59, 294–310. [Google Scholar] [CrossRef] [Green Version]
- Thavornkanlapachai, R.; Mills, H.R.; Ottewell, K.; Dunlop, J.; Sims, C.; Morris, K.; Donaldson, F.; Kennington, W.J. Mixing Genetically and Morphologically Distinct Populations in Translocations: Asymmetrical Introgression in A Newly Established Population of the Boodie (Bettongia lesueur). Genes 2019, 10, 729. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.; Hughes, J. Microsatellite and mitochondrial DNA variation defines island genetic reservoirs for reintroductions of an endangered Australian marsupial, Perameles bougainville. Conserv. Genet. 2008, 9, 547–557. [Google Scholar] [CrossRef]
- Eldridge, M.D.B.; Neaves, L.E.; Spencer, P.B.S. Genetic analysis of three remnant populations of the rufous hare-wallaby (Lagorchestes hirsutus) in arid Australia. Aust. Mammal. 2019, 41, 123–131. [Google Scholar] [CrossRef]
- Frankham, R. Effective Population Size Adult Population Size Ratios in Wildlife—A Review. Genet. Res. 1995, 66, 95–107. [Google Scholar] [CrossRef]
- Nunney, L. The influence of variation in female fecundity on effective population size. Biol. J. Linn. Soc. 1996, 59, 411–425. [Google Scholar] [CrossRef]
- Waples, R.S.; Luikart, G.; Faulkner, J.R.; Tallmon, D.A. Simple life-history traits explain key effective population size ratios across diverse taxa. Proc. R. Soc. B Biol. Sci. 2013, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churchill, D.M. Late Quaternary eustatic changes in the Swan River district. J. R. Soc. West. Aust. 1959, 42, 53–55. [Google Scholar]
- Short, J.; Turner, B.; Majors, C. The Distribution, Relative Abundance, and Habitat Preferences of Rare Macropods and Bandicoots on Barrow, Boodie, Bernier and Dorre Islands; CSIRO Division of Wildlife and Ecology: Midland, Australia, 1989; p. 64. [Google Scholar]
- Goldstein, D.B.; Pollock, D.D. Launching microsatellites: A review of mutation processes and methods of phylogenetic inference. J. Hered. 1997, 88, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; (Australian Wildlife Conservancy, Perth, WA, Australia). Personal communication, 2020.
- Sonsthagen, S.A.; Wilson, R.E.; Underwood, J.G. Genetic implications of bottleneck effects of differing severities on genetic diversity in naturally recovering populations: An example from Hawaiian coot and Hawaiian gallinule. Ecol. Evol. 2017, 7, 9925–9934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heber, S.; Briskie, J.V. Population Bottlenecks and Increased Hatching Failure in Endangered Birds. Conserv. Biol. 2010, 24, 1674–1678. [Google Scholar] [CrossRef]
- Easton, L.J.; Bishop, P.J.; Whigham, P.A. Balancing act: Modelling sustainable release numbers for translocations. Anim. Conserv. 2019. [Google Scholar] [CrossRef]
- Allendorf, F.W.; Ryman, N. The role of genetics in population viability analysis. In Population Viability Analysis; Beissinger, S.R., McCullough, D.R., Eds.; University of Chicago Press: Chicago, IL, USA, 2002; pp. 50–85. [Google Scholar]
- Pacioni, C.; Rafferty, C.; Morley, K.; Stevenson, S.; Chapman, A.; Wickins, M.; Verney, T.; Deegan, G.; Trocini, S.; Spencer, P.B.S. Augmenting the conservation value of rehabilitated wildlife by integrating genetics and population modeling in the post-rehabilitation decision process. Curr. Zool. 2018, 64, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Allendorf, F.W.; Leary, R.F.; Spruell, P.; Wenburg, J.K. The problems with hybrids: Setting conservation guidelines. Trends Ecol. Evol. 2001, 16, 613–622. [Google Scholar] [CrossRef]
- Edmands, S. Between a rock and a hard place: Evaluating the relative risks of inbreeding and outbreeding for conservation and management. Mol. Ecol. 2007, 16, 463–475. [Google Scholar] [CrossRef]
- Frankham, R.; Ballou, J.D.; Eldridge, M.D.B.; Lacy, R.C.; Ralls, K.; Dudash, M.R.; Fenster, C.B. Predicting the Probability of Outbreeding Depression. Conserv. Biol. 2011, 25, 465–475. [Google Scholar] [CrossRef]
- Armbruster, P.; Bradshaw, W.E.; Steiner, A.L.; Holzapfel, C.M. Evolutionary responses to environmental stress by the pitcher-plant mosquito, Wyeomyia smithii. Heredity 1999, 83, 509–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmands, S. Heterosis and outbreeding depression in interpopulation crosses spanning a wide range of divergence. Evolution 1999, 53, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Marr, A.B.; Keller, L.F.; Arcese, P. Heterosis and outbreeding depression in descendants of natural immigrants to an inbred population of song sparrows (Melospiza melodia). Evolution 2002, 56, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Tymchuk, W.E.; Sundstrom, L.F.; Devlin, R.H. Growth and survival trade-offs and outbreeding depression in rainbow trout (Oncorhynchus mykiss). Evolution 2007, 61, 1225–1237. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Life History Parameter | Value | Used in |
|---|---|---|
| Species description | ||
| Inbreeding depression | V | |
| Lethal equivalents | 3.14 | V |
| % due to recessive lethal | 50 | V |
| EV concordance of reproduction and survival | 0.5 | V |
| EV correlation among populations | 0.5–0.8 | V |
| Reproductive system | ||
| Reproductive system | polygynous | V/A |
| Duration of breeding cycle in days | 274 | V/A |
| Age of first offspring for females/males | 9 months/18 months | V/A |
| Maximum age of reproduction | 8 years | V/A |
| Maximum lifespan | 10 years | V/A |
| Maximum number of broods per breeding cycle | 1 | V/A |
| Maximum number of progeny per brood | 1 | V/A |
| Mean female number of progeny per breeding cycle | 1 | A |
| Mean male lifetime reproductive success (±SD) | 7 ± 3 | A |
| Sex ratio at birth | 50 | V/A |
| Reproductive rates | ||
| % adult females breeding | 90% with DDR | V |
| EV in % breeding | 18 | V |
| Distribution of broods per breeding cycle | ||
| 0 broods | 0 | V |
| 1 brood | 100% | V |
| Number of offspring per female brood | ||
| 1 offspring | 100% | V |
| Mate monopolisation | ||
| % males in breeding pool | 85 | V |
| % males successfully siring offspring | 63 | V |
| Mortality rates | ||
| Females | ||
| Mortality age 0 to 1 (±SD) | 40 (±10) | V/A |
| Annual mortality after age 1 (±SD) | 10 (±3) | V/A |
| Males | ||
| Mortality age 0 to 1 (±SD) | 40 (±10) | V/A |
| Annual mortality after age 1 (±SD) | 10 (±3) | V/A |
| Catastrophes | ||
| Number of types of catastrophes | 1 (drought) | V |
| Frequency | 1 in 6.25 calendar yearsa | V |
| Severity | 50% reduction in survival and reproduction | V |
| Initial population size | ||
| Bernier | 2000 | V/A |
| Dorre | 2000 | V/A |
| Faure | 300 | V/A |
| Carrying capacity, K (SD due to EV) | ||
| Bernier | 3000 (300) | V/A |
| Dorre | 3000 (300) | V/A |
| Faure | 1000 (100) or 3000 (300) | V/A |
| DHI | (1000) | V/A |
| Mt. Gibson | 5000 (500) | V/A |
| Genetic management | ||
| Number of neutral loci to be modelled | 7 empirical, 1 simulated | V |
| Initial minor allele frequency | 0.05 | A |
| Scenario settings | ||
| No. replicates | 1000/100 | V/A |
| No. years | 50 calendar years | V/A |
| Target Population | Scenario | Description |
|---|---|---|
| Dirk Hartog Island | 1 | One translocation from Bernier Island in year 1 |
| 2 | Individuals translocated from Bernier Island only, half in each of first two years | |
| 3 | Individuals translocated from Bernier and Dorre Islands, half from one island in year 1 and half from other island in year 2 | |
| 4 | Individuals translocated from Bernier and Dorre Islands, half from each island in year 1 | |
| 5–7 | Best performing scenario from 1 to 4, with drought frequencies of no drought, 1 in 10 years, and 1 in 5 years | |
| Dirk Hartog Islandand Mount Gibson Wildlife Sanctuary | 8 | To Dirk Hartog Island: six individuals from Bernier and six from Dorre in BHW year 1; 50 from Bernier in BHW year 2; 50 from Dorre in BHW year 3To Mount Gibson: 23 individuals from Bernier, 39 from Dorre, and 10 from Faure in BHW year 1; 37 from Bernier, one from Dorre, and 20 from Faure in BHW year 3 |
| Population | N a | HWE b | NA | PA | AR (±s.e.) | HE (±s.e.) | HO (±s.e.) | FIS (±s.e.) | Ne (95% CI) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Bernier Island (all) | 51 | 1/6 | 20 | 0 | 2.47 (0.15) | 0.36 (0.09) | 0.30 (0.08) | 0.14 (0.05) | ||
| Bernier (1998) | 6 | n/a | 0.38 (0.11) | 0.43 (0.15) | −0.06 (0.17) | n/a | ||||
| Bernier (2010–2011) | 9 | n/a | 0.34 (0.10) | 0.23 (0.07) | 0.31 (0.10) | n/a | ||||
| Bernier (2016–2017) | 33 | 2.40 (0.15) | 0.36 (0.08) | 0.31 (0.07) | 0.11 (0.04) | 82 (12, ∞) | ||||
| Dorre Island (all) | 79 | 2/6 | 23 | 1 | 2.46 (0.10) | 0.42 (0.08) | 0.41 (0.08) | 0.03 (0.07) | ||
| Dorre (1995–1996) | 7 | n/a | 0.44 (0.08) | 0.61 (0.11) | −0.39 (0.07) | n/a | ||||
| Dorre (1999–2000) | 8 | n/a | 0.45 (0.09) | 0.45 (0.09) | 0.00 (0.07) | n/a | ||||
| Dorre (2013) | 11 | 2.41 (0.29) | 0.42 (0.08) | 0.43 (0.09) | 0.01 (0.08) | n/a | ||||
| Dorre (2016–2017) | 52 | 2.40 (0.11) | 0.40 (0.07) | 0.38 (0.08) | 0.07 (0.10) | 140 (29, ∞) c | ||||
| Faure Island (2017) | 10 | 0/6 | 18 | 2 | 2.57 (0.40) | 0.39 (0.08) | 0.29 (0.09) | 0.36 (0.14) | n/a | |
| Wadderin (2018) | 17 | 1/5 | 16 | 0 | 2.27 (0.27) | 0.36 (0.10) | 0.34 (0.13) | 0.09 (0.20) | n/a | |
| Peron CBC (2006–2013) | 73 | 1/5 | 19 | 1 | 2.41 (0.15) | 0.36 (0.10) | 0.33 (0.10) | 0.08 (0.08) | 20 (7, 57) | |
| Dryandra (1999–2002) | 6 | 0/6 | 18 | 0 | n/a | 0.40 (0.08) | 0.50 (0.12) | −0.28 (0.15) | n/a | |
| 236 | µ (±s.d.) | 2.34 (0.13) | 0.39 (0.02) | 0.37 (0.08) | 0.07 (0.21) |
| Population (Time Period) | n | Bottleneck Test | |
|---|---|---|---|
| Wilcoxon (One-Tailed, H Excess) | Mode Shift | ||
| Bernier Island (all) | 51 | 0.281 | No |
| Bernier Island (2016/2017) | 33 | 0.500 | No |
| Bernier Island (2010/2011) | 9 | 0.109 | Yes |
| Bernier Island (1998) | 6 | 0.016 | Yes |
| Dorre Island (all) | 79 | 0.039 | No |
| Dorre Island (2016/2017) | 52 | 0.422 | No |
| Dorre Island (2013) | 11 | 0.008 | Yes |
| Dorre Island (1999/2000) | 8 | 0.008 | Yes |
| Dorre Island (1995/1996) | 7 | 0.016 | Yes |
| Peron CBC (from Bernier; t = 25, y = 1998) | 73 | 0.219 | Yes |
| Dryandra (from Dorre and Peron CBC; t = 25, y = 1998) | 6 | 0.578 | No |
| Faure Island (from Peron CBC; t = 91, y = 2004 to 2013) | 10 | 0.281 | No |
| Wadderin (from Peron CBC; t = 12, y = 2013) | 17 | 0.031 | Yes |
| Scenario | Description | P (surv) | HE | N |
|---|---|---|---|---|
| 1 | 100 in year 1 from Bernier Island | 0.76 | 0.311 | 2094 |
| 2 | 50 from Bernier Island in year 1 and year 2 | 0.79 | 0.330 | 2489 |
| 3 | 50 from Bernier Island in year 1, 50 from Dorre Island in year 2 | 0.80 | 0.368 | 2251 |
| 4 | 50 from Bernier Island, 50 from Dorre Island in year 1 | 0.79 | 0.362 | 2277 |
| Conservative Current Census Sizes of Source Populations | Census Sizes Following Drought | |
|---|---|---|
| NBernier = 2000, NDorre = 2000, NFaure = 300 | NBernier = 500, NDorre = 500, NFaure = 75 | |
| (a) Target Populations | ||
| Dirk Hartog Island | ||
| P (surv) | 0.83 | 0.81 |
| N | 2363 | 2436 |
| HE | 0.367 | 0.367 |
| Mount Gibson | ||
| P (surv) | 0.84 | 0.84 |
| N | 1600 | 1565 |
| HE | 0.372 | 0.372 |
| (b) Source Populations | ||
| Bernier Island | ||
| P (surv) | 0.99 | 0.94 |
| N | 1640 | 1474 |
| HE | 0.359 | 0.353 |
| Dorre Island | ||
| P (surv) | 0.98 | 0.95 |
| N | 1647 | 1465 |
| HE | 0.417 | 0.408 |
| Faure Island * | ||
| P (surv) | 0.93/0.93 | 0.60/0.63 |
| N | 495/1401 | 386/901 |
| HE | 0.369/0.374 | 0.344/0.343 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
White, D.J.; Ottewell, K.; Spencer, P.B.S.; Smith, M.; Short, J.; Sims, C.; Mitchell, N.J. Genetic Consequences of Multiple Translocations of the Banded Hare-Wallaby in Western Australia. Diversity 2020, 12, 448. https://doi.org/10.3390/d12120448
White DJ, Ottewell K, Spencer PBS, Smith M, Short J, Sims C, Mitchell NJ. Genetic Consequences of Multiple Translocations of the Banded Hare-Wallaby in Western Australia. Diversity. 2020; 12(12):448. https://doi.org/10.3390/d12120448
Chicago/Turabian StyleWhite, Daniel J., Kym Ottewell, Peter B. S. Spencer, Michael Smith, Jeff Short, Colleen Sims, and Nicola J. Mitchell. 2020. "Genetic Consequences of Multiple Translocations of the Banded Hare-Wallaby in Western Australia" Diversity 12, no. 12: 448. https://doi.org/10.3390/d12120448
APA StyleWhite, D. J., Ottewell, K., Spencer, P. B. S., Smith, M., Short, J., Sims, C., & Mitchell, N. J. (2020). Genetic Consequences of Multiple Translocations of the Banded Hare-Wallaby in Western Australia. Diversity, 12(12), 448. https://doi.org/10.3390/d12120448