Genetic Patterns and Climate Modelling Reveal Challenges for Conserving Sclerolaena napiformis (Amaranthaceae s.l.) an Endemic Chenopod of Southeast Australia


Abstract
1. Introduction
2. Materials and Methods
2.1. The Study Species
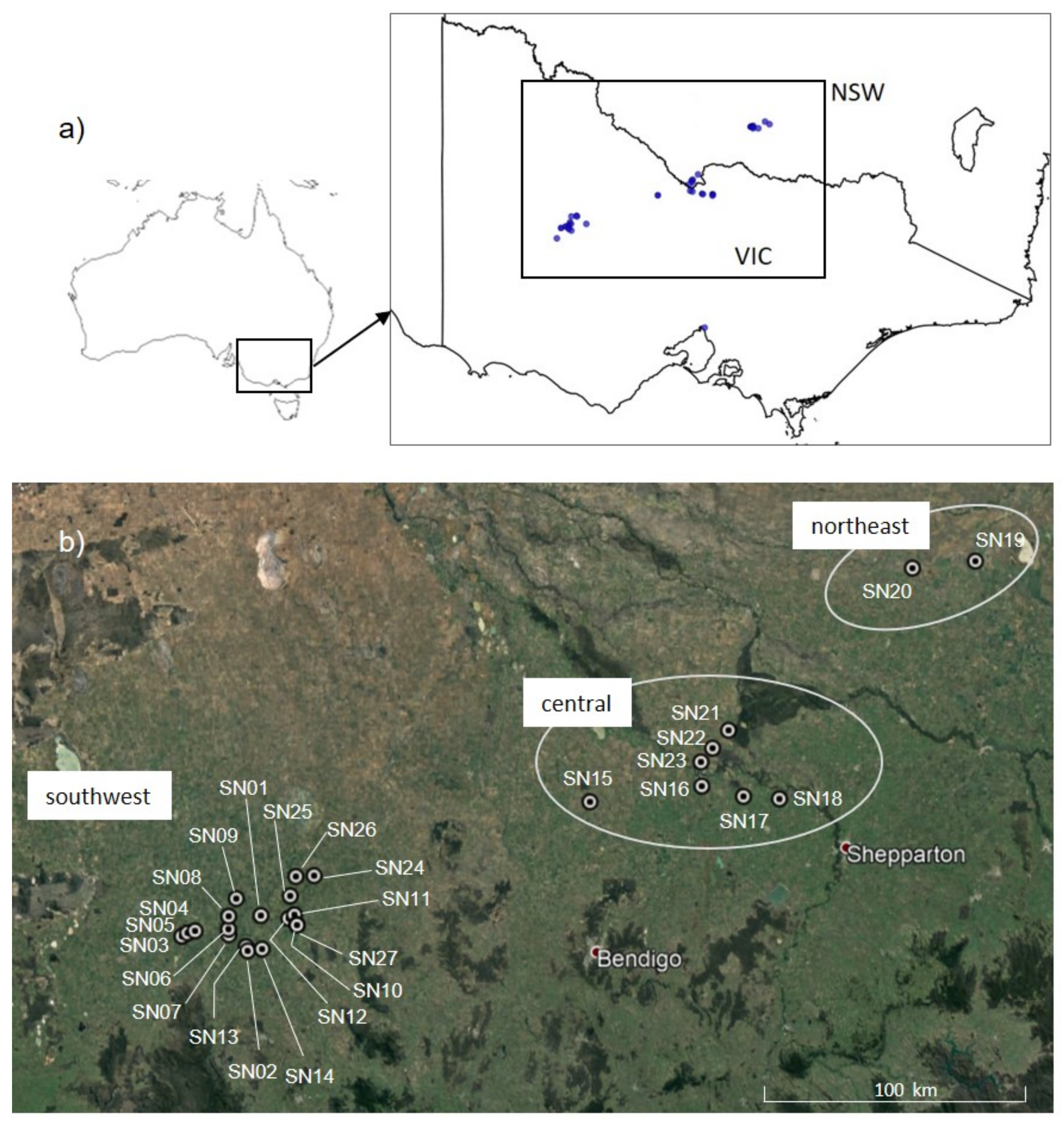
2.2. Sampling
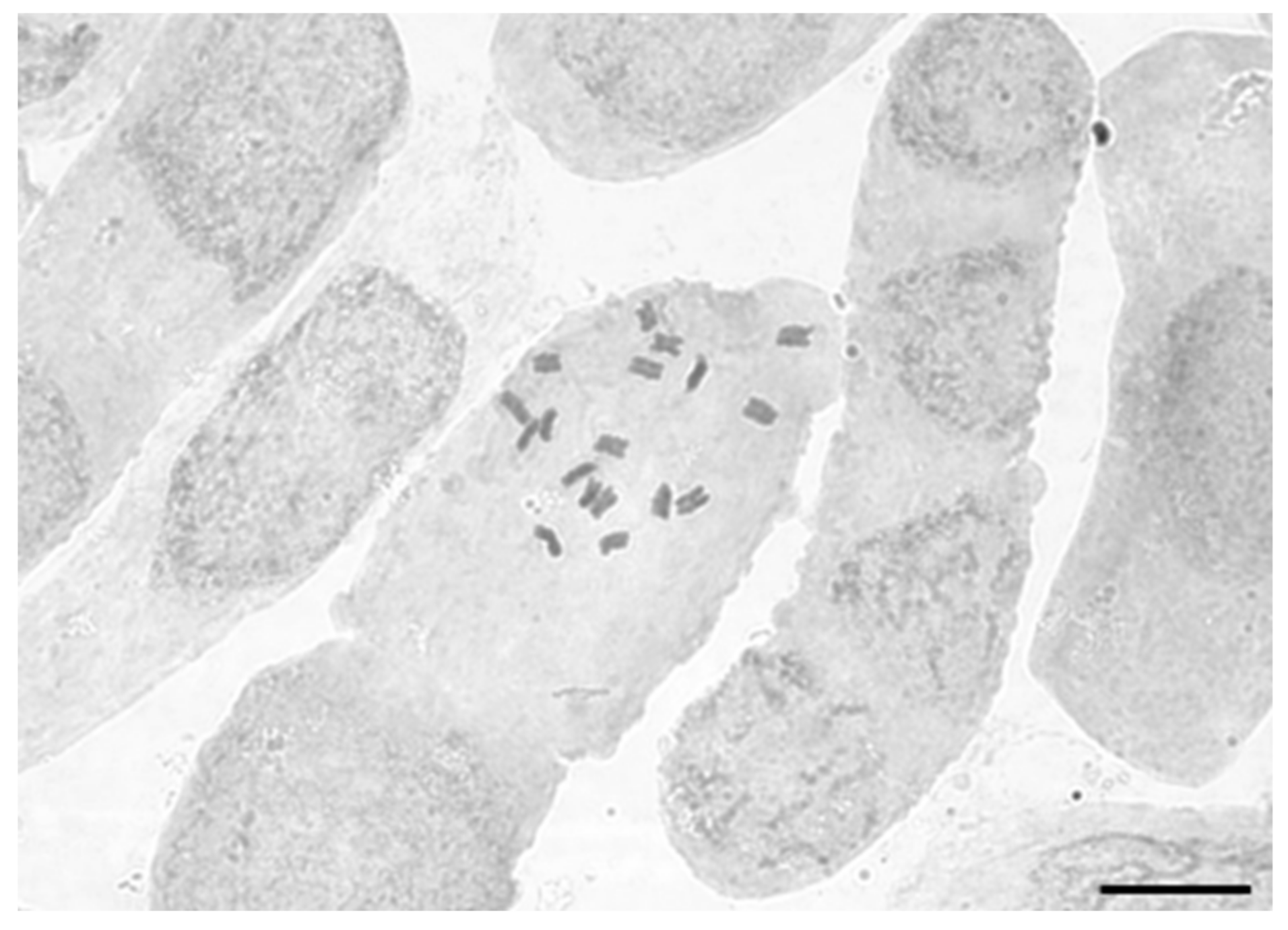
2.3. Chromosome Counts
2.4. DNA Isolation and Sequencing
2.4.1. Whole Genome Sequencing
2.4.2. ddRADseq
2.5. Quality Filtering and Bioinformatics
2.5.1. Nuclear DNA Assembly
2.5.2. Population Samples (ddRADseq)
2.5.3. Population Samples (cp Haplotyping)
2.6. Analysis of Genetic Diversity and Structure
2.7. Distribution Modelling
3. Results
3.1. Chromosome Counts
3.2. Data assembly Statistics
3.3. Regional Genetic Diversity and Structure
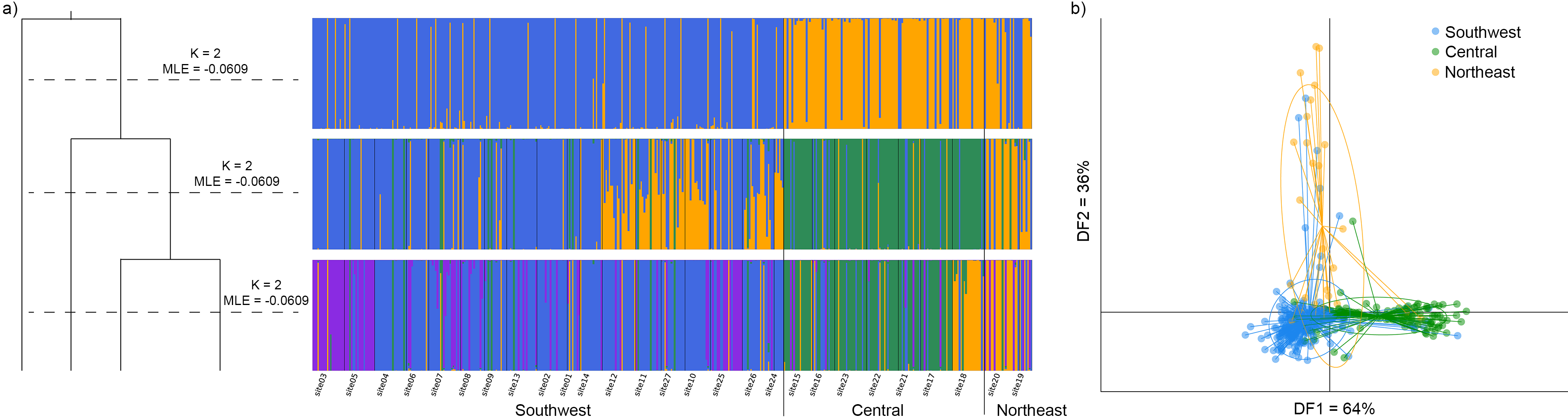
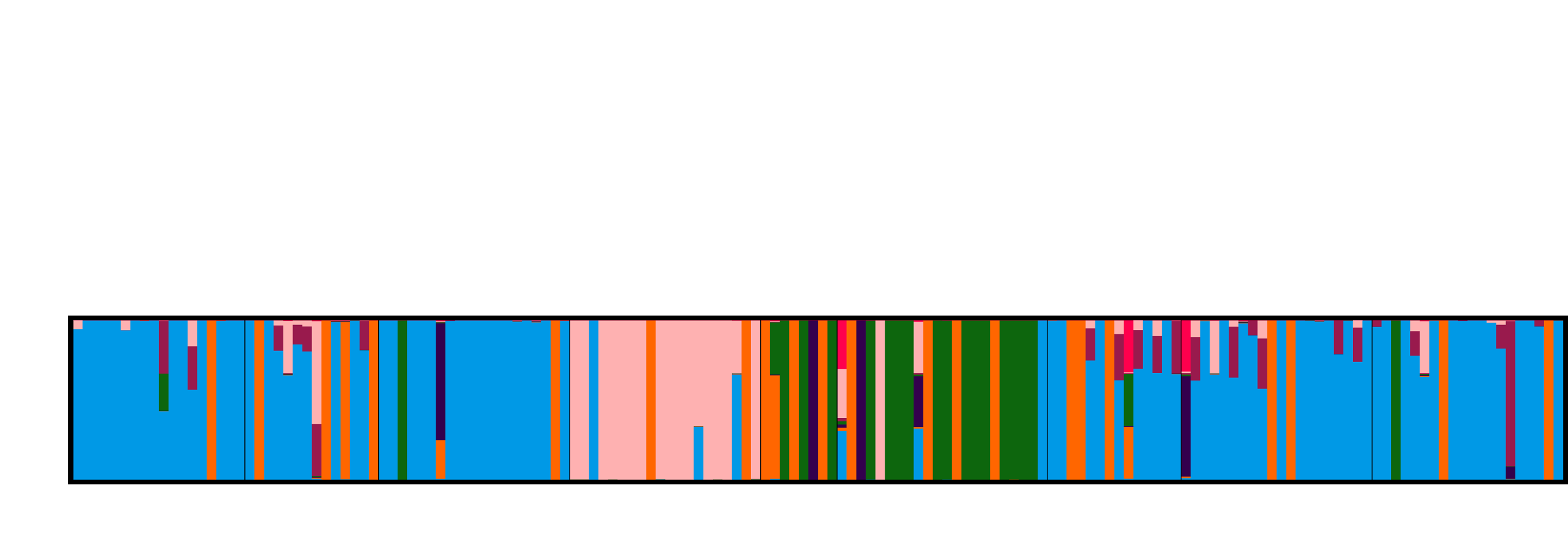
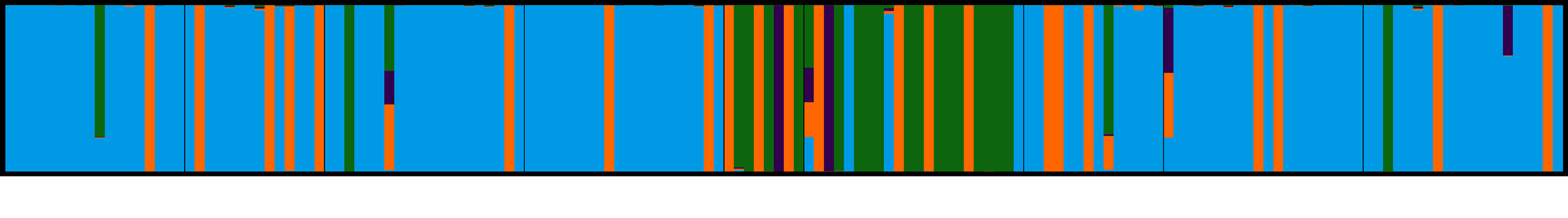
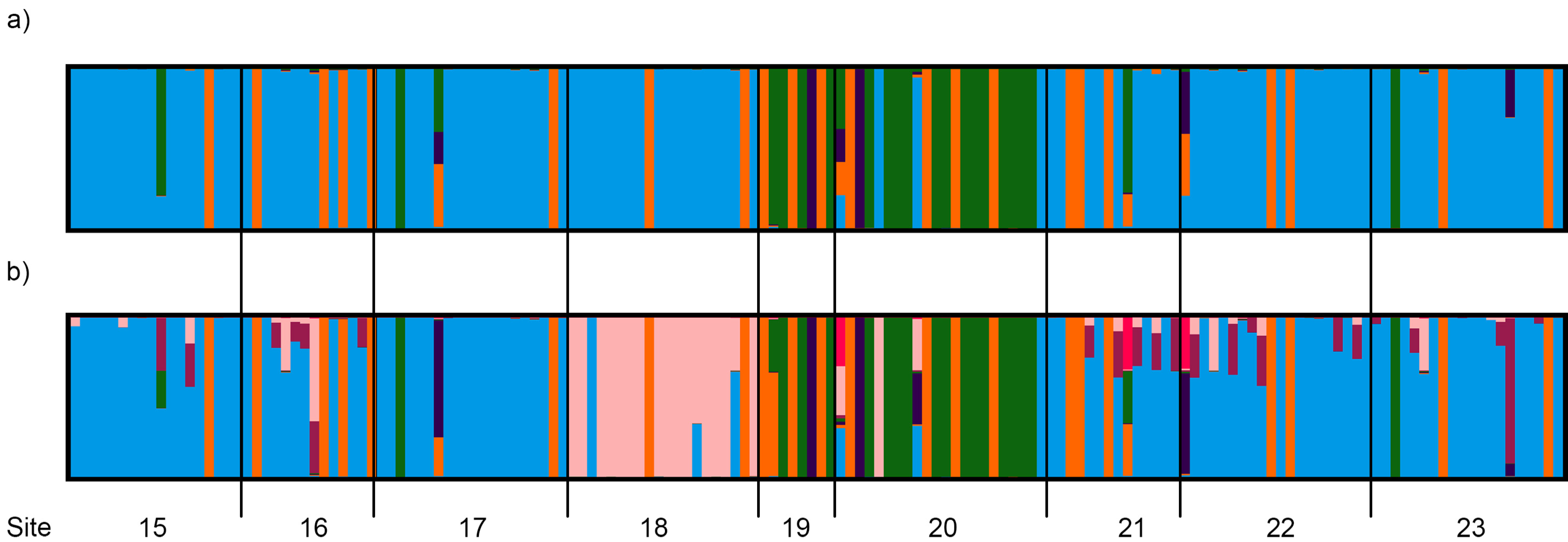
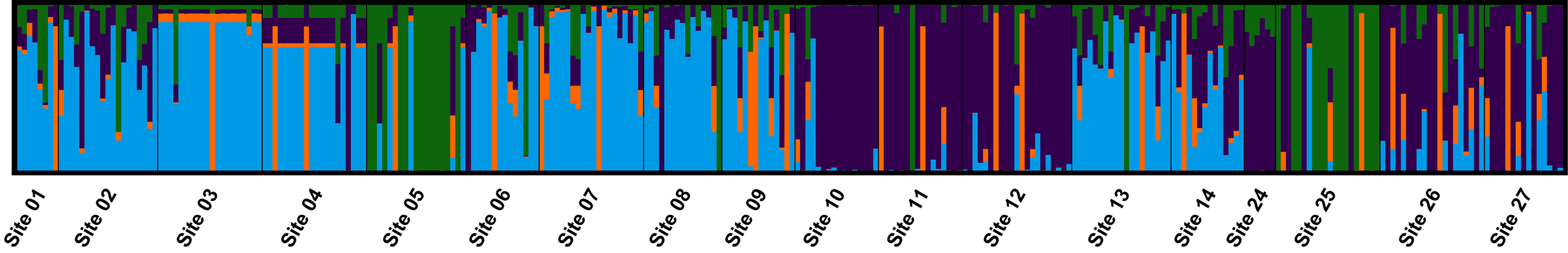
3.4. Population Genetic Structure
3.4.1. Overview of All Regions
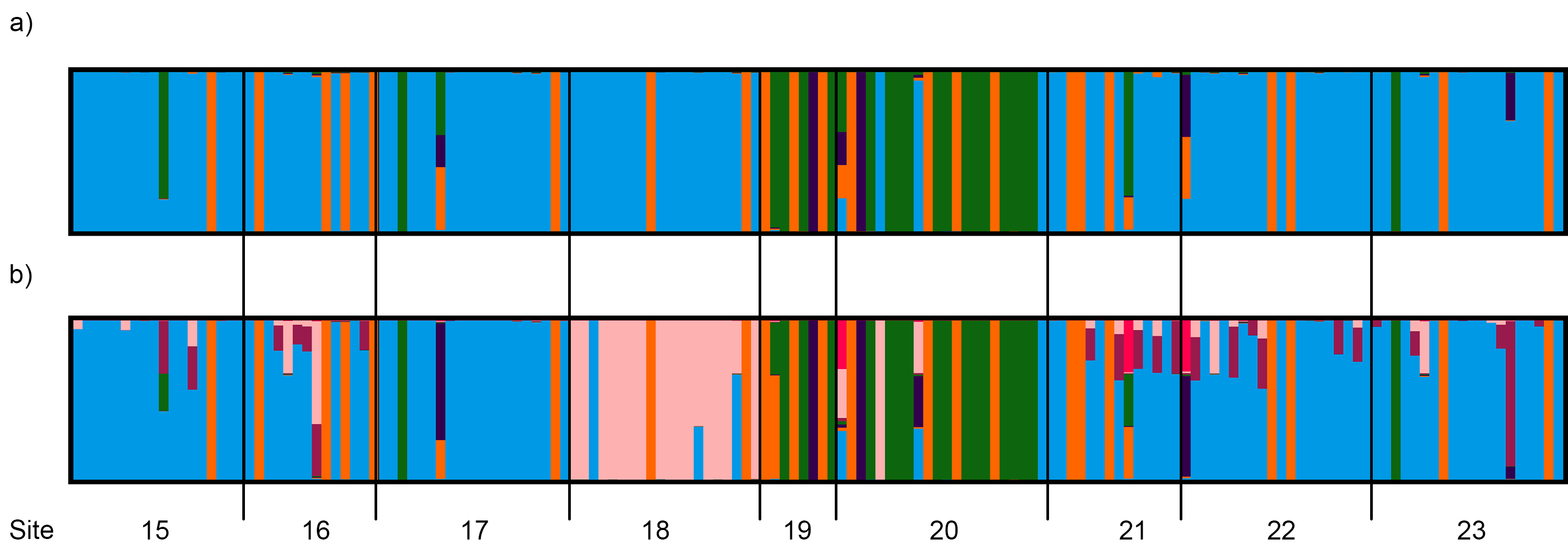
3.4.2. Central and Northeast Regions
3.4.3. Southwest Region
3.5. Chloroplast Genomes and Haplotyping
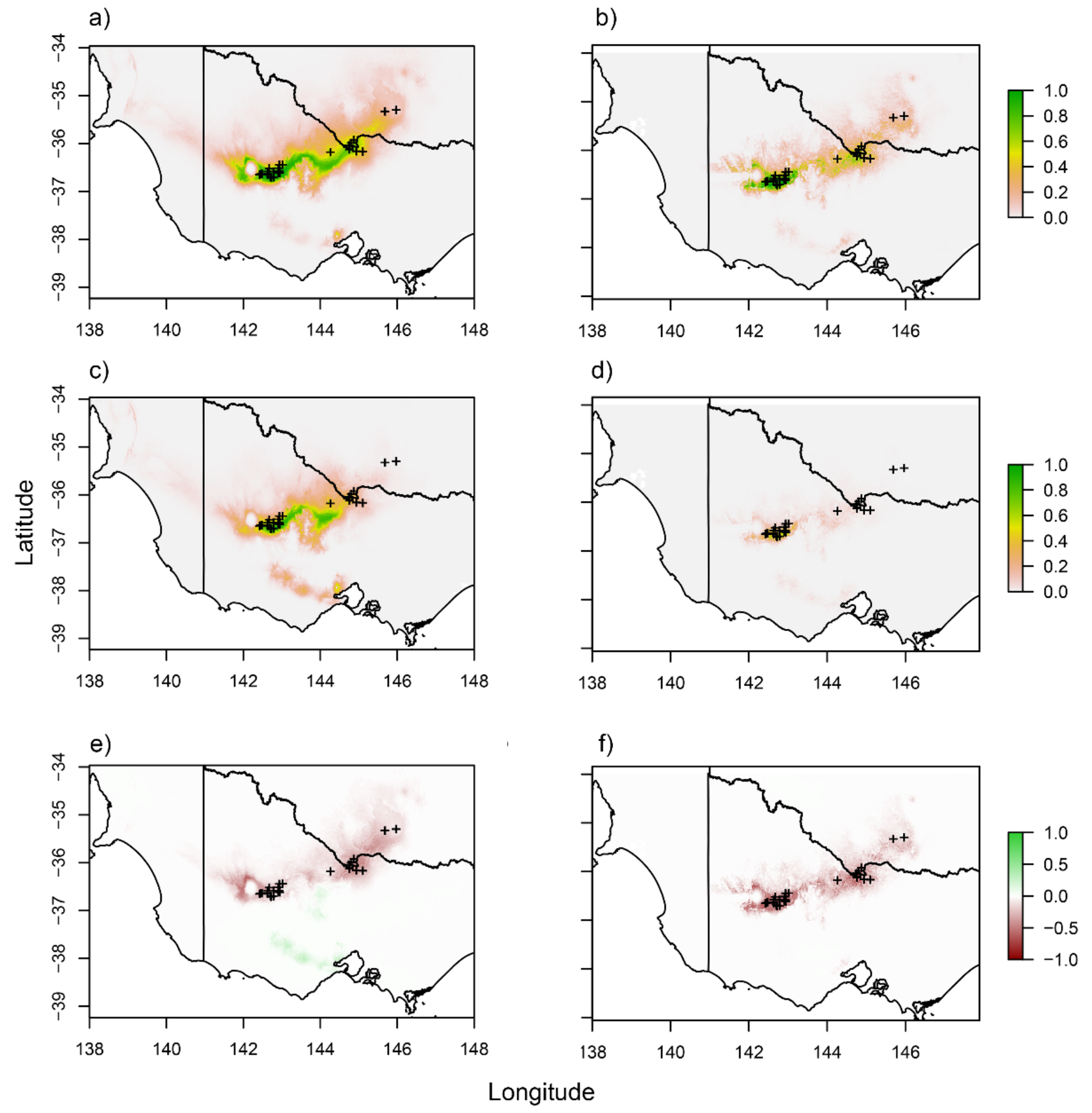
3.6. Distribution Modelling
4. Discussion
4.1. Fragmentation, Genetic Structure, and Gene-Flow
4.2. Genetic Diversity and Inbreeding
4.3. Considerations for Persistence of S. napiformis
5. Conclusions and Implications for Conservation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Accessibility
References
- International Union for Conservation of Nature (IUCN). The IUCN Red List of Threatened Species. 2020. Available online: https://www.iucnredlist.org (accessed on 21 May 2020).
- Garner, B.A.; Hoban, S.; Luikart, G. IUCN red list and the value of integrating genetics. Conserv. Genet. 2020, 21, 795–801. [Google Scholar] [CrossRef]
- McGuigan, K.; Sgrò, C.M. Evolutionary consequences of cryptic genetic variation. Trends Ecol. Evol. 2009, 24, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Sgrò, C.M.; Lowe, A.J.; Hoffmann, A.A. Building evolutionary resilience for conserving biodiversity under climate change. Evol. Appl. 2011, 4, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Griffin, P.; Dillon, S.; Catullo, R.; Rane, R.; Byrne, M.; Jordan, R.; Oakeshott, J.; Weeks, A.; Joseph, L.; et al. A framework for incorporating evolutionary genomics into biodiversity conservation and management. Clim. Chang. Responses 2015, 2, 1–23. [Google Scholar] [CrossRef]
- Reed, D.H.; Frankham, R. Correlation between fitness and genetic diversity. Conserv. Biol. 2003, 17, 230–237. [Google Scholar] [CrossRef]
- Kramer, A.T.; Havens, K. Plant conservation genetics in a changing world. Trends Plant Sci. 2009, 14, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Ouborg, N.J.; Vergeer, P.; Mix, C. The rough edges of the conservation genetics paradigm for plants. J. Ecol. 2006, 94, 1233–1248. [Google Scholar] [CrossRef]
- Dunlop, M.; Hilbert, D.; Ferrier, S.; House, A.; Liedloff, A.; Prober, S.M.; Smyth, A.; Martin, T.G.; Harwood, T.; Williams, K.J.; et al. A report prepared for the Department of Sustainability, Environment, Water, Population and Communities and the Department of Climate Change and Energy Efficiency. In The Implications of Climate Change for Biodiversity Conservation and the National Reserve System: Final Synthesis; CSIRO Climate Adaptation Flagship: Canberra, Australia, 2012. [Google Scholar]
- Mable, B.K. Conservation of adaptive potential and functional diversity: Integrating old and new approaches. Conserv. Genet. 2019, 20, 89–100. [Google Scholar] [CrossRef]
- Pina-Martins, F.; Baptista, J.; Pappas, G., Jr.; Paulo, O.S. New insights into adaptation and population structure of cork oak using genotyping by sequencing. Glob. Chang. Biol. 2019, 25, 337–350. [Google Scholar] [CrossRef]
- Willi, Y.; Hoffmann, A.A. Demographic factors and genetic variation influence population persistence under environmental change. J. Evol. Biol. 2009, 22, 124–133. [Google Scholar] [CrossRef]
- Mimura, M.; Yahara, T.; Faith, D.P.; Vazquez-Dominguez, E.; Colautti, R.I.; Araki, H.; Javadi, F.; Nunez-Farfan, J.; Mori, A.S.; Zhou, S.; et al. Understanding and monitoring the consequences of human impacts on intraspecific variation. Evol. Appl. 2017, 10, 121–139. [Google Scholar] [CrossRef] [PubMed]
- Willi, Y.; Van Buskirk, J.; Hoffmann, A.A. Limits to the adaptive potential of small populations. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 433–458. [Google Scholar] [CrossRef]
- Vilas, A.; Perez-Figueroa, A.; Quesada, H.; Caballero, A. Allelic diversity for neutral markers retains a higher adaptive potential for quantitative traits than expected heterozygosity. Mol. Ecol. 2015, 24, 4419–4432. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.A.; Sgrò, C.M. Climate change and evolutionary adaptation. Nature 2011, 470, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Cunze, S.; Heydel, F.; Tackenberg, O. Are plant species able to keep pace with the rapidly changing climate. PLoS ONE 2013, 8, e67909. [Google Scholar] [CrossRef]
- Vitt, P.; Havens, K.; Kramer, A.T.; Sollenberger, D.; Yates, E. Assisted migration of plants: Changes in latitudes, changes in attitudes. Biol. Conserv. 2010, 143, 18–27. [Google Scholar] [CrossRef]
- Franklin, I.R.; Frankham, R. How large must populations be to retain evolutionary potential? Anim. Conserv. 1998, 1, 69–70. [Google Scholar] [CrossRef]
- Nunney, L.; Campbell, K.A. Assessing minimum viable population size–Demography meets population genetics. Trends Ecol. Evol. 1993, 8, 234–239. [Google Scholar] [CrossRef]
- Commonwealth of Australia. Interim Biogeographical Regionalisation of Australia, Version 7. 2020. Available online: https://www.environment.gov.au/land/nrs/science/ibra (accessed on 17 July 2020).
- EPBC. Environment Protection and Biodiversity Conservation Act 1999. 2016. Available online: https://www.legislation.gov.au/Details/C2016C00777 (accessed on 17 July 2020).
- Mavromihalis, J. National Recovery Plan for the Turnip Copperburr Sclerolaena napiformis; Victorian Government Department of Sustainability and Environment (DSE): Melbourne, Australia, 2010.
- Cunningham, G.M.; Mulham, W.E.; Milthorpe, P.L.; Leigh, J.H. Plants of Western New South Wales; CSIRO Publishing: Melbourne, Australia, 1992. [Google Scholar]
- Conn, B.J. Natural regions and vegetation of Victoria. In Flora of Victoria; Foreman, D.B., Walsh, N.G., Eds.; Inkata Press: Melbourne, Australia, 1993; Volume 1, pp. 79–158. [Google Scholar]
- Van Rossum, F.; Martin, H.; Le Cadre, S.; Brachi, B.; Christenhusz, M.J.M.; Touzet, P. Phylogeography of a widely distributed species reveals a cryptic assemblage of distinct genetic lineages needing separate conservation strategies. Perspect. Plant. Ecol. Evol. Syst. 2018, 35, 44–51. [Google Scholar] [CrossRef]
- Weeks, A.R.; Sgrò, C.M.; Young, A.G.; Frankham, R.; Mitchell, N.J.; Miller, K.A.; Byrne, M.; Coates, D.J.; Eldridge, M.D.; Sunnucks, P.; et al. Assessing the benefits and risks of translocations in changing environments: A genetic perspective. Evol. Appl. 2011, 4, 709–725. [Google Scholar] [CrossRef]
- Prober, S.M.; Byrne, M.; McLean, E.H.; Steane, D.A.; Potts, B.M.; Vaillancourt, R.E.; Stock, W.D. Climate-adjusted provenancing: A strategy for climate-resilient ecological restoration. Front. Ecol. Evol. 2015, 3, 65. [Google Scholar] [CrossRef]
- Walsh, N.G. Maireana obrienii (Chenopodiaceae), a new species from eastern Australia. Muelleria 2013, 31, 61–64. [Google Scholar]
- Walsh, N.G.; Sluiter, I.R.K. Lectotypification of Atriplex stipitata Benth. (Chenopodiaceae) and recognition of a new subspecies. Muelleria 2020, 38, 101–109. [Google Scholar]
- Frankham, R. Genetic rescue of small inbred populations: Meta-analysis reveals large and consistent benefits of gene flow. Mol. Ecol. 2015, 24, 2610–2618. [Google Scholar] [CrossRef]
- Frankham, R.; Ballou, J.D.; Eldridge, M.D.; Lacy, R.C.; Ralls, K.; Dudash, M.R.; Fenster, C.B. Predicting the probability of outbreeding depression. Conserv. Biol. 2011, 25, 465–475. [Google Scholar] [CrossRef]
- Wilson, P.G. Sclerolaena napiformis Paul G. Wilson sp. nov. Flora Aust. 1984, 4, 330. [Google Scholar]
- Angiosperm Phylogeny Group (APG). An update of the classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef]
- Kadereit, G.; Newton, R.J.; Vandelook, F. Evolutionary ecology of fast seed germination—A case study in Amaranthaceae/Chenopodiaceae. Perspect. Plant. Ecol. Evol. Syst. 2017, 29, 1–11. [Google Scholar] [CrossRef]
- Woinarski, J.C.Z.; Braby, M.F.; Burbidge, A.A.; Coates, D.; Garnett, S.T.; Fensham, R.J.; Legge, S.M.; McKenzie, N.L.; Silcock, J.L.; Murphy, B.P. Reading the black book: The number, timing, distribution and causes of listed extinctions in Australia. Biol. Conserv. 2019, 239, 108261. [Google Scholar] [CrossRef]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 2012, 7, 1–11. [Google Scholar] [CrossRef]
- Amor, M.D.; Jackson, C.J.; Holmes, G.D.; James, E.A. Characterization of the complete chloroplast genome of Sclerolaena napiformis Wilson, an endangered Australian chenopod. Mitochondrial DNA B 2020, 5, 1332–1333. [Google Scholar] [CrossRef]
- Murray, B.; Young, A.G. Widespread chromosome variation in the endangered grassland forb Rutidosis leptorrhynchoides F. Muell. (Asteraceae: Gnaphalieae). Ann. Bot. 2001, 87, 83–90. [Google Scholar] [CrossRef]
- Jackson, R.C. Chromosomal evolution in Haplopappus gracilis: A centric transposition race. Evolution 1973, 27, 243–256. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.E.A. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. In Gene Prediction; Methods in Molecular Biology Series; Kollmar, M., Ed.; Humana: New York, NY, USA, 2019; Volume 1962, pp. 227–245. [Google Scholar]
- Wood, D.E.; Salzburg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 1415, R46. [Google Scholar] [CrossRef]
- Catchen, J.; Hohenlohe, P.; Bassham, S.; Amores, A.; Cresko, W. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef]
- Eaton, D.A.R.; Overcast, I. Ipyrad: Interactive assembly and analysis of RADseq datasets. Bioinformatics 2020, 36, 2592–2594. [Google Scholar] [CrossRef] [PubMed]
- Foll, M.; Gaggiotti, O.E. A genome scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [PubMed]
- Luu, K.; Blum, M.; Privé, F. Pcadapt: Fast Principal Component Analysis for Outlier Detection. R package Version 4.1.0. 2019. Available online: https://CRAN.R-project.org/package=pcadapt (accessed on 4 July 2020).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: http://www.r-project.org (accessed on 1 June 2020).
- Paradis, E. Pegas: An R package for population genetics with an integrated–modular approach. Bioinformatics 2010, 26, 419–420. [Google Scholar] [CrossRef]
- Raj, A.; Stephens, M.; Pritchard, J.K. FastSTRUCTURE: Variational inference of population structure in large SNP data sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of spatially structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R package Version 2.5-4. 2019. Available online: https://cran.r-project.org./package=vegan (accessed on 4 July 2020).
- Kamvar, Z.N.; Tabima, J.F.; Grunwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef]
- Goudet, J. Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef]
- Winter, D.J. Mmod: An R library for the calculation of population differentiation statistics. Mol. Ecol. Resour. 2012, 12, 1158–1160. [Google Scholar] [CrossRef]
- Hijmans, R.J.; Phillips, S.; Leathwick, J.; Elith, J. Package ‘Dismo’. 2011. Available online: http://cran.r-project.org/web/packages/dismo/index.html (accessed on 4 July 2020).
- Hijmans, R.J.; van Etten, J. Raster: Geographic Analysis and Modeling with Raster Data. R Package Version 2.0-12. 2012. Available online: http://CRAN.R-project.org/package=raster (accessed on 4 July 2020).
- Auld, B.A.; Martin, P.M. Morphology and distribution of Bassia birchii (F. Muell.) F. Muell. [native sheep forage shrub, semi-arid eastern Australia]. Proc. Linn. Soc. N. S. W. 1976, 100, 167–178. [Google Scholar]
- Novaes, R.M.; De Lemos Filho, J.P.; Ribeiro, R.A.; Lovato, M.B. Phylogeography of Plathymenia reticulata (Leguminosae) reveals patterns of recent range expansion towards northeastern Brazil and southern Cerrados in Eastern Tropical South America. Mol. Ecol. 2010, 19, 985–998. [Google Scholar] [CrossRef]
- Peter, B.M.; Slatkin, M. Detecting range expansions from genetic data. Evolution 2013, 67, 3274–3289. [Google Scholar] [CrossRef]
- Amor, M.D.; Johnson, J.C.; James, E.A. Identification of clonemates and genetic lineages using next-generation sequencing (ddRADseq) guides conservation of a rare species, Bossiaea vombata (Fabaceae). Perspect. Plant Ecol. Evol. Syst. 2020, 45, 125544. [Google Scholar] [CrossRef]
- Harper, J.L. Population Biology of Plants; Academic Press: London, UK, 1977. [Google Scholar]
- Silvertown, J.W.; Lovett-Doust, J. Introduction to Plant Population Biology; Blackwell: Oxford, UK, 1993. [Google Scholar]
- Ouborg, N.J.; Piquot, Y.; Groenendael, V. Population genetics, molecular markers and the study of dispersal in plants. J. Ecol. 1999, 87, 551–568. [Google Scholar] [CrossRef]
- James, E.A.; Jordan, R. Limited structure and widespread diversity suggest potential buffers to genetic erosion in a threatened grassland shrub Pimelea spinescens (Thymelaeaceae). Conserv. Genet. 2014, 15, 305–317. [Google Scholar] [CrossRef]
- Llorens, T.M.; Ayre, D.J.; Whelan, R. Anthropogenic fragmentation may not alter pre-existing patterns of genetic diversity and differentiation in perennial shrubs. Mol. Ecol. 2018, 27, 1541–1555. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, R.; Quesada, M.; Ashworth, L.; Herrerias-Diego, Y.; Lobo, J. Genetic consequences of habitat fragmentation in plant populations: Susceptible signals in plant traits and methodological approaches. Mol. Ecol. 2008, 17, 5177–5188. [Google Scholar] [CrossRef]
- McDougall, K.; Kirkpatrick, J.B. Conservation of Lowland Native Grassland in South-Eastern Australia; World Wide Fund for Nature: Sydney, Australia, 1994.
- Commonwealth of Australia. The National Recovery Plan for the Plains-Wanderer (Pedionomus torquatus); Department of the Environment: Canberra, Australia, 2016.
- Peakall, R.; Oliver, I.; Turnbull, C.L.; Beattie, A.J. Genetic diversity in an ant-dispersed chenopod Sclerolaena diacantha. Aust. J. Ecol. 1993, 18, 171–179. [Google Scholar] [CrossRef]
- Peakall, R.; Beattie, A.J. Does ant dispersal of seeds in Sclerolaena diacantha (Chenopodiaceae) generate local spatial genetic structure? Heredity 1995, 75, 351. [Google Scholar] [CrossRef]
- Goodwillie, C.; Kalisz, S.; Eckert, C.G. The evolutionary enigma of mixed mating systems in plants: Occurrence, theoretical explanations, and empirical evidence. Ann. Rev. Ecol. Evol. Syst. 2005, 36, 47–79. [Google Scholar] [CrossRef]
- Laenen, B.; Tedder, A.; Nowak, M.D.; Toräng, P.; Wunder, J.; Wötzel, S.; Steige, K.A.; Kourmpetis, Y.; Odong, T.; Drouzas, A.D.; et al. Demography and mating system shape the genomewide impact of purifying selection in Arabis alpina. Proc. Natl. Acad. Sci. USA 2018, 115, 816–821. [Google Scholar] [CrossRef]
- Eckert, C.G.; Kalisz, S.; Geber, M.A.; Sargent, R.; Elle, E.; Cheptou, P.O.; Goodwillie, C.; Johnston, M.O.; Kelly, J.K.; Moeller, D.A.; et al. Plant mating systems in a changing world. Trends Ecol. Evol. 2010, 25, 35–43. [Google Scholar] [CrossRef]
- Jordan, C.Y.; Lohse, K.; Turner, F.; Thomson, M.; Gharbi, K.; Ennos, R.A. Maintaining their genetic distance: Little evidence for introgression between widely hybridizing species of Geum with contrasting mating systems. Mol. Ecol. 2018, 27, 1214–1228. [Google Scholar] [CrossRef]
- Mustin, K.; Benton, T.G.; Dytham, C.; Travis, J.M.J. The dynamics of climate-induced range shifting; Perspectives from simulation modelling. Oikos 2009, 118, 131–137. [Google Scholar] [CrossRef]
- Scott, J.K.; Webber, B.L.; Murphy, H.; Kriticos, D.J.; Ota, N.; Loechel, B. Weeds and Climate Change: Supporting Weed Management Adaptation. 2014. Available online: www.AdaptNRM.org (accessed on 18 September 2020).
- Jurado, E.; Westoby, M. Germination biology of selected central Australian plants. Aust. J. Ecol. 1992, 17, 341–348. [Google Scholar] [CrossRef]
- Navie, S.C.; Cowley, R.A.; Rogers, R.W. The relationship between distance from water and the soil seed bank in a grazed semi-arid subtropical rangeland. Aust. J. Bot. 1996, 44, 421–431. [Google Scholar] [CrossRef]
- Auld, B.A. Aspects of the population ecology of galvanised burr (Sclerolaena birchii). Aust. Range. J. 1981, 3, 142–148. [Google Scholar] [CrossRef]
- Carta, F.E.; Parsons, R.F. Notes on the germination of the endangered species Sclerolaena napiformis (Chenopodiaceae). Cunninghamiana 2005, 9, 215–218. [Google Scholar]
- Plummer, J.A.; Bell, D.T. The effect of temperature, light and gibberellic acid (GA3) on the germination of Australian everlasting daisies (Asteraceae, tribe Inuleae). Aust. J. Bot. 1995, 43, 93–102. [Google Scholar] [CrossRef]
- Bykova, O.; Chuine, I.; Morin, X. Highlighting the importance of water availability in reproductive processes to understand climate change impacts on plant biodiversity. Perspect. Plant. Ecol. Evol. Syst. 2019, 37, 20–25. [Google Scholar] [CrossRef]
- Kinloch, J.E.; Friedel, M.H. Soil seed reserves in arid grazing lands of central Australia. Part 1: Seed bank and vegetation dynamics. J. Arid Environ. 2005, 60, 133–161. [Google Scholar] [CrossRef]
- Ralls, K.; Ballou, J.D.; Dudash, M.R.; Eldridge, M.D.B.; Fenster, C.B.; Lacy, R.C.; Sunnucks, P.; Frankham, R. Call for a paradigm shift in the genetic management of fragmented populations. Conserv. Lett. 2018, 11, e12412. [Google Scholar] [CrossRef]
- Krauss, S.L.; Dixon, B.; Dixon, K.W. Rapid genetic decline in a translocated population of the endangered plant Grevillea scapigera. Conserv. Biol. 2002, 16, 986–994. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
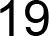{kind=link}
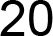{kind=link}
{kind=link}
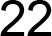{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Region | Number of Samples Collected | Estimated Area (ha) | Estimated Pop Size |
|---|---|---|---|---|
| SN01 | Southwest | 9 | 0.6 | 50 |
| SN02 | Southwest | 25 | 1.0 | 380 |
| SN03 | Southwest | 20 | 0.3 | 65 |
| SN04 | Southwest | 21 | 0.06 | 40 |
| SN05 | Southwest | 20 | 0.15 | 340 |
| SN06 | Southwest | 20 | 1.25 | 200 |
| SN07 | Southwest | 20 | 1.8 | 80 |
| SN08 | Southwest | 20 | 0.56 | 150 |
| SN09 | Southwest | 20 | 1.0 | 150 |
| SN10 | Southwest | 20 | 0.8 | 170 |
| SN11 | Southwest | 20 | 0.45 | 210 |
| SN12 | Southwest | 20 | 3.0 | 120 |
| SN13 | Southwest | 20 | 1.125 | 150 |
| SN14 | Southwest | 20 | 0.8 | 130 |
| SN15 | Central | 20 | 0.7 | 400 |
| SN16 | Central | 20 | 1.2 | 250 |
| SN17 | Central | 23 | 0.11 | 120 |
| SN18 | Central | 19 | 1.14 | 350 |
| SN19 | Northeast | 12 | 0.44 | 70 |
| SN20 | Northeast | 23 | 0.24 | 500 |
| SN21 | Central | 20 | 1.05 | 500 |
| SN22 | Central | 20 | 1.5 | 100 |
| SN23 | Central | 20 | 5.0 | 80 |
| SN24 | Southwest | 6 | 0.05 | 40 |
| SN25 | Southwest | 23 | 1.25 | 120 |
| SN26 | Southwest | 21 | 1.8 | 120 |
| SN27 | Southwest | 19 | 0.9 | 180 |
| All Regions | Southwest | Central | Northeast | |
|---|---|---|---|---|
| No. individuals in assembly | 452 | 296 | 126 | 30 |
| Total loci | 3827 | 4043 | 5169 | 5011 |
| Informative sites | 5518 | 4971 | 3659 | 4082 |
| Percent reads mapped to reference | 31.2 | 31.3 | 31.3 | 30.0 |
| Mean error rate of base calls (± SD) | 0.003 (0.001) | 0.003 (0.001) | 0.003 (0.001) | 0.003 (0.001) |
| Unlinked SNPs | 3093 | 3045 | 2837 | 3367 |
| Mean locus depth (± SD) | 15.31 (8.83) | 15.21 (17.24) | 15.65 (7.20) | 15.37 (9.93) |
| Mean individuals per locus (± SD) | 163.38 (43.45) | 108.11 (28.63) | 46.64 (12.39) | 12.17 (3.59) |
| N | n | Ho | He | FST | GST | FIS | Mantel’s R (p-Value) | |
|---|---|---|---|---|---|---|---|---|
| All regions | 27 | 452 | 0.017 | 0.056 | 0.156 | 0.209 | 0.617 | 0.088 (0.001) * |
| Southwest | 18 | 296 | 0.027 | 0.058 | 0.205 | 0.304 | 0.517 | −0.009 (0.646) |
| Central | 7 | 126 | 0.040 | 0.112 | 0.225 | 0.288 | 0.652 | 0.103 (0.038) * |
| Northeast | 2 | 30 | 0.049 | 0.258 | 0.200 | 0.369 | 0.830 | 0.106 (0.153) |
| Region | Northeast | Central | Southwest |
|---|---|---|---|
| Northeast | - | 0.017 | 0.014 |
| Central | 0.228 | - | 0.010 |
| Southwest | 0.180 | 0.189 | - |
| Haplotype | Variables Sites | |||||||
|---|---|---|---|---|---|---|---|---|
| 125,670 - | 125,801 - | 134,268 rpoC1 Gene | 134,367 rpoC1 Gene | 134,376 rpoC1 Gene | 115,143 matK CDS | 115,263 matK CDS | ||
| n | ||||||||
| H1 | 351 | A | C | G | C | G | T | A |
| H2 | 21 | T | C | G | C | G | T | A |
| H3 | 5 | A | T | A | C | G | T | C |
| H4 | 1 | A | T | G | C | G | T | C |
| 1 H4A | 8 | A | T | G | C | N | T | C |
| 1 H5A * | 1 | A | C | G | C | A | N | N |
| 1 H5B * | 2 | A | C | N | C | N | C | C |
| H6 | 1 | A | C | G | T | G | T | A |
| Reference: | ||||||||
| SN20 | 1 | A | C | G | C | G | T | A |
| TR01 | 1 | A | T | G | C | G | T | C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amor, M.D.; Walsh, N.G.; James, E.A. Genetic Patterns and Climate Modelling Reveal Challenges for Conserving Sclerolaena napiformis (Amaranthaceae s.l.) an Endemic Chenopod of Southeast Australia. Diversity 2020, 12, 417. https://doi.org/10.3390/d12110417
Amor MD, Walsh NG, James EA. Genetic Patterns and Climate Modelling Reveal Challenges for Conserving Sclerolaena napiformis (Amaranthaceae s.l.) an Endemic Chenopod of Southeast Australia. Diversity. 2020; 12(11):417. https://doi.org/10.3390/d12110417
Chicago/Turabian StyleAmor, Michael D., Neville G. Walsh, and Elizabeth A. James. 2020. "Genetic Patterns and Climate Modelling Reveal Challenges for Conserving Sclerolaena napiformis (Amaranthaceae s.l.) an Endemic Chenopod of Southeast Australia" Diversity 12, no. 11: 417. https://doi.org/10.3390/d12110417
APA StyleAmor, M. D., Walsh, N. G., & James, E. A. (2020). Genetic Patterns and Climate Modelling Reveal Challenges for Conserving Sclerolaena napiformis (Amaranthaceae s.l.) an Endemic Chenopod of Southeast Australia. Diversity, 12(11), 417. https://doi.org/10.3390/d12110417