Nuclear Orthologs Derived from Whole Genome Sequencing Indicate Cryptic Diversity in the Bemisia tabaci (Insecta: Aleyrodidae) Complex of Whiteflies

 , and
, and
Abstract
1. Introduction:
2. Materials and Methods
2.1. Sample Preparation and Sequencing
2.2. Ortholog Prediction and Phylogenomic Analyses
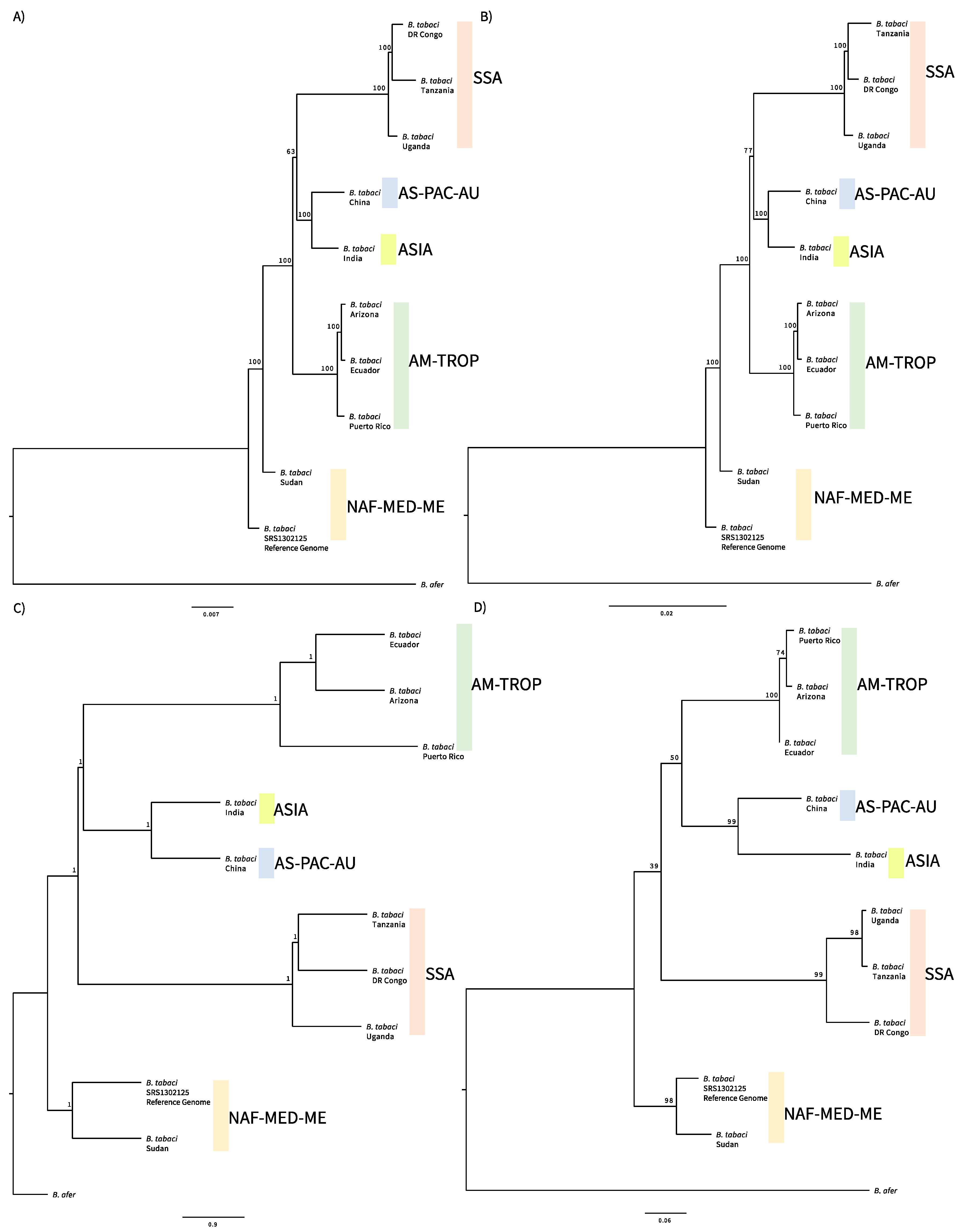
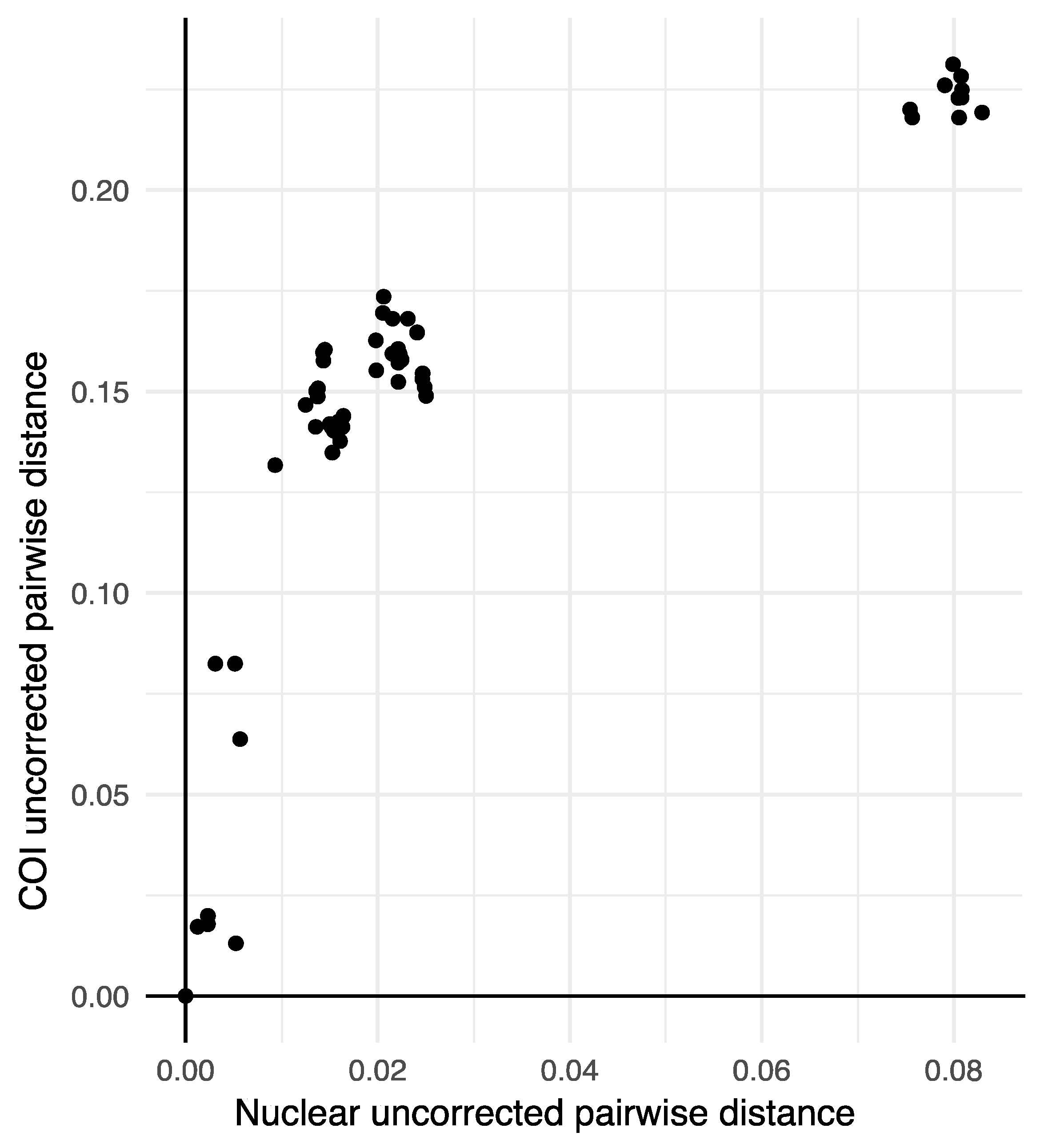
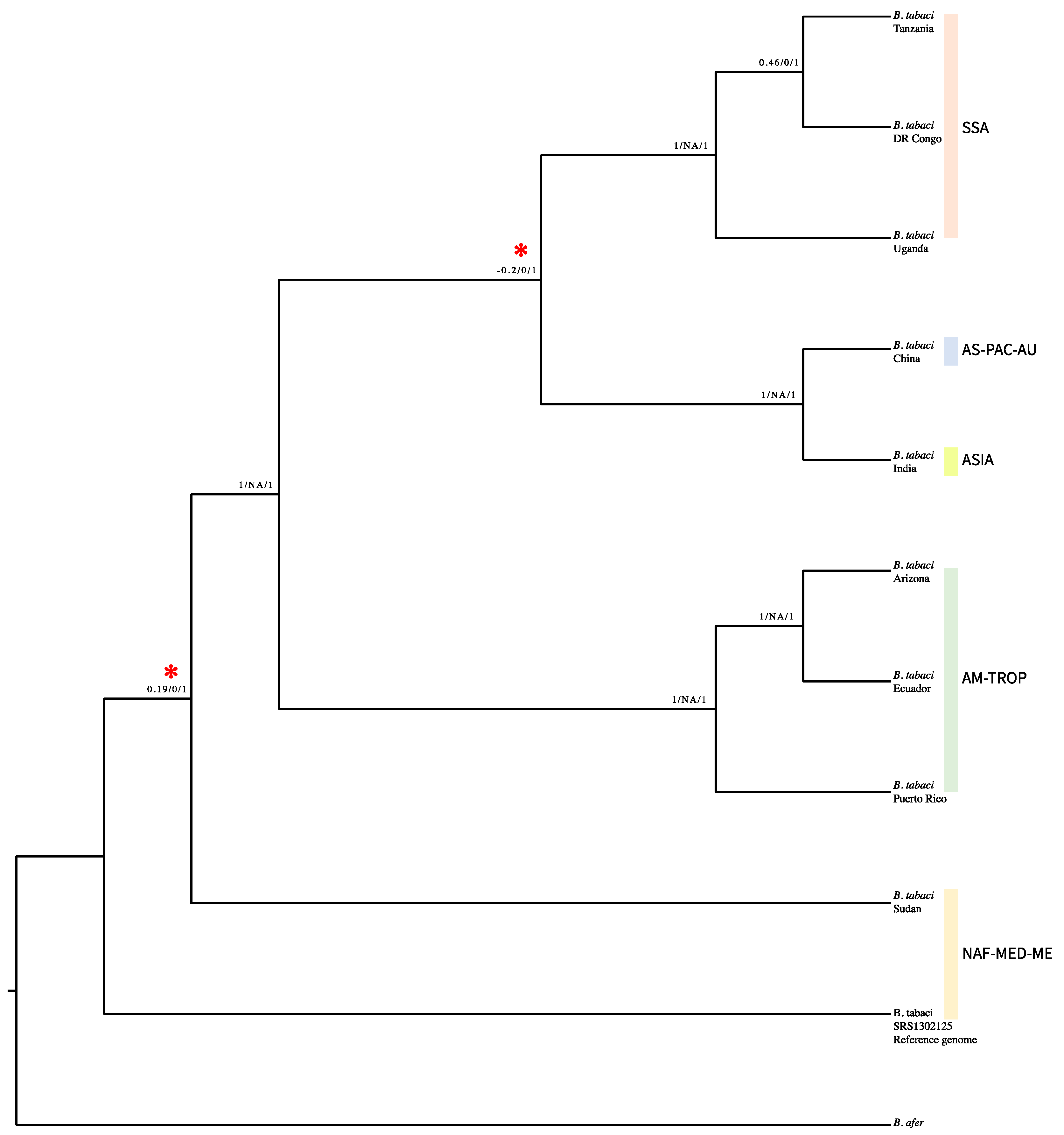
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mound, L.A.; Halsey, S.H. Whitefly of the World. A Systematic Catalogue of the Aleyrodidae (Homoptera) with Host Plant and Natural Enemy Data; John Wiley and Sons: Hoboken, NJ, USA, 1978. [Google Scholar]
- Greathead, A.H. Bemisia tabaci: A Literature Survey on the Cotton Whitefly with an Annotated Bibliography. In Host Plants; Cock, M.J.W., Ed.; CAB International Institutes, Biological Control: Silwood Park, UK, 1986; pp. 17–26. [Google Scholar]
- Bird, J. A whitefly-transmitted mosaic of Jatropha Gossypifolia. Tech. Paper 1957, 22, 1–35. [Google Scholar]
- Costa, H.S.; Brown, J.K. Variation in biological characteristics and esterase patterns among populations of Bemisia tabaci, and the association of one population with silverleaf symptom induction. Entomol. Exp. Appl. 1991, 61, 211–219. [Google Scholar] [CrossRef]
- Bedford, I.D.; Briddon, R.W.; Brown, J.K.; Rosell, R.C.; Markham, P.G. Geminivirus transmission and biological characterisation of Bemisia tabaci (Gennadius) biotypes from different geographic regions. Ann. Appl. Biol. 1994, 125, 311–325. [Google Scholar] [CrossRef]
- De Barro, P.J.; Trueman, J.W.H.; Frohlich, D.R. Bemisia argentifolii is a race of B. tabaci (Hemiptera: Aleyrodidae): The molecular genetic differentiation of B. tabaci populations around the world. Bull. Entomol. Res. 2005, 95, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.K. Phylogenetic Biology of the Bemisia tabaci Sibling Species Group. In Bemisia: Bionomics and Management of a Global Pest; Stansly, P.A., Naranjo, S.E., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 31–67. ISBN 978-90-481-2460-2. [Google Scholar]
- Gill, R.J.; Brown, J.K. Systematics of Bemisia and Bemisia Relatives: Can Molecular Techniques Solve the Bemisia tabaci Complex Conundrum–A Taxonomist’s Viewpoint. In Bemisia: Bionomics and Management of a Global Pest; Stansly, P.A., Naranjo, S.E., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 5–29. ISBN 978-90-481-2460-2. [Google Scholar]
- Oliveira, M.R.V.; Henneberry, T.J.; Anderson, P. History, current status, and collaborative research projects for Bemisia tabaci. Crop Prot. 2001, 20, 709–723. [Google Scholar] [CrossRef]
- Sseruwagi, P.; Legg, J.P.; Maruthi, M.N.; Colvin, J.; Rey, M.E.C.; Brown, J.K. Genetic diversity of Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) populations and presence of the B biotype and a non-B biotype that can induce silverleaf symptoms in squash, in Uganda. Ann. Appl. Biol. 2005, 147, 253–265. [Google Scholar] [CrossRef]
- Mugerwa, H.; Rey, M.E.C.; Alicai, T.; Ateka, E.; Atuncha, H.; Ndunguru, J.; Sseruwagi, P. Genetic diversity and geographic distribution of Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) genotypes associated with cassava in East Africa. Ecol. Evol. 2012, 2, 2749–2762. [Google Scholar] [CrossRef]
- Barbosa, L.D.F.; Marubayashi, J.M.; Marchi, B.R.D.; Yuki, V.A.; Pavan, M.A.; Moriones, E.; Navas-Castillo, J.; Krause-Sakate, R. Indigenous American species of the Bemisia tabaci complex are still widespread in the Americas. Pest Manag. Sci. 2014, 70, 1440–1445. [Google Scholar] [CrossRef]
- Cock, M.J.W. Bemisia Tabaci, an Update 1986–1992 on the Cotton Whitefly with an Annotated Bibliography; CAB International Institutes, Biological Control: Silwood Park, UK, 1993. [Google Scholar]
- Brown, J.K.; Frohlich, D.R.; Rosell, R.C. The Sweetpotato or Silverleaf Whiteflies: Biotypes of Bemisia tabaci or a Species Complex? Annu. Rev. Entomol. 1995, 40, 511–534. [Google Scholar] [CrossRef]
- Costa, A.S.; Russell, R.C. Failure of Bemisia tabaci to breed on cassava plants in Brazil (Homoptera: Aleyrodidae). Cienc. Cult. Saõ Paulo 1975, 2, 388–390. [Google Scholar]
- Brown, J.K.; Bird, J. Whitefly-transmitted geminiviruses in the Americas and the Caribbean Basin: Past and present. Plant Dis. 1992, 76, 220–225. [Google Scholar] [CrossRef]
- Perring, T.M.; Cooper, A.D.; Rodriguez, R.J.; Farrar, C.A.; Bellows, T.S. Identification of a whitefly species by genomic and behavioral studies. Science 1993, 259, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Chowda-Reddy, R.; Kirankumar, M.; Seal, S.E.; Muniyappa, V.; Valand, G.B.; Govindappa, M.; Colvin, J. Bemisia tabaci Phylogenetic Groups in India and the Relative Transmission Efficacy of Tomato leaf curl Bangalore virus by an Indigenous and an Exotic Population. J. Integr. Agric. 2012, 11, 235–248. [Google Scholar] [CrossRef]
- Bellows, T.S.; Perring, T.M.; Gill, R.J.; Headrick, D.H. Description of a Species of Bemisia (Homoptera: Aleyrodidae). Ann. Entomol. Soc. Am. 1994, 87, 195–206. [Google Scholar] [CrossRef]
- Rosell, R.C.; Bedford, I.D.; Frohlich, D.R.; Gill, R.J.; Brown, J.K.; Markham, P.G. Analysis of Morphological Variation in Distinct Populations of Bemisia tabaci (Homoptera: Aleyrodidae). Ann. Entomol. Soc. Am. 1997, 90, 575–589. [Google Scholar] [CrossRef]
- Dinsdale, A.; Cook, L.; Riginos, C.; Buckley, Y.M.; Barro, P.D. Refined Global Analysis of Bemisia tabaci (Hemiptera: Sternorrhyncha: Aleyrodoidea: Aleyrodidae) Mitochondrial Cytochrome Oxidase 1 to Identify Species Level Genetic Boundaries. Ann. Entomol. Soc. Am. 2010, 103, 196–208. [Google Scholar] [CrossRef]
- Hadjistylli, M.; Roderick, G.K.; Brown, J.K. Global Population Structure of a Worldwide Pest and Virus Vector: Genetic Diversity and Population History of the Bemisia tabaci Sibling Species Group. PLoS ONE 2016, 11, e0165105. [Google Scholar] [CrossRef] [PubMed]
- De Barro, P.J.; Liu, S.-S.; Boykin, L.M.; Dinsdale, A.B. Bemisia tabaci: A Statement of Species Status. Annu. Rev. Entomol. 2011, 56, 1–19. [Google Scholar] [CrossRef]
- Brown, J.K.; Idris, A.M. Genetic Differentiation of Whitefly Bemisia tabaci Mitochondrial Cytochrome Oxidase I, and Phylogeographic Concordance with the Coat Protein of the Plant Virus Genus Begomovirus. Ann. Entomol. Soc. Am. 2005, 98, 827–837. [Google Scholar] [CrossRef]
- Frohlich, D.R.; Torres-Jerez, I.; Bedford, I.D.; Markham, P.G.; Brown, J.K. A phylogeographical analysis of the Bemisia tabaci species complex based on mitochondrial DNA markers. Mol. Ecol. 1999, 8, 1683–1691. [Google Scholar] [CrossRef]
- Boykin, L.M.; Bell, C.D.; Evans, G.; Small, I.; De Barro, P.J. Is agriculture driving the diversification of the Bemisia tabaci species complex (Hemiptera: Sternorrhyncha: Aleyrodidae)? Dating, diversification and biogeographic evidence revealed. BMC Evol. Biol. 2013, 13, 228. [Google Scholar] [CrossRef] [PubMed]
- Tay, W.T.; Elfekih, S.; Court, L.N.; Gordon, K.H.J.; Delatte, H.; De Barro, P.J. The Trouble with MEAM2: Implications of Pseudogenes on Species Delimitation in the Globally Invasive Bemisia tabaci (Hemiptera: Aleyrodidae) Cryptic Species Complex. Genome Biol. Evol. 2017, 9, 2732–2738. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gawel, N.J.; Bartlett, A.C. Characterization of differences between whiteflies using RAPD-PCR. Insect Mol. Biol. 1993, 2, 33–38. [Google Scholar] [CrossRef] [PubMed]
- De Barro, P.J.; Driver, F.; Trueman, J.W.H.; Curran, J. Phylogenetic Relationships of World Populations of Bemisia tabaci (Gennadius) Using Ribosomal ITS1. Mol. Phylogenet. Evol. 2000, 16, 29–36. [Google Scholar] [CrossRef] [PubMed]
- De la Rúa, P.; Simón, B.; Cifuentes, D.; Martinez-Mora, C.; Cenis, J.L. New insights into the mitochondrial phylogeny of the whitefly Bemisia tabaci (Hemiptera: Aleyrodidae) in the Mediterranean Basin. J. Zool. Syst. Evol. Res. 2006, 44, 25–33. [Google Scholar] [CrossRef]
- Baoli, Q.; Coats, S.A.; Shunxiang, R.; Idris, A.M.; Caixia, X.; Brown, J.K. Phylogenetic relationship of native and introduced Bemisia tabaci (Homoptera: Aleyrodidae) from China and India based on mtCOI DNA sequencing and host plant comparisons. Prog. Nat. Sci. 2007, 17, 645–654. [Google Scholar] [CrossRef]
- Boykin, L.M.; Shatters, R.G.; Rosell, R.C.; McKenzie, C.L.; Bagnall, R.A.; De Barro, P.; Frohlich, D.R. Global relationships of Bemisia tabaci (Hemiptera: Aleyrodidae) revealed using Bayesian analysis of mitochondrial COI DNA sequences. Mol. Phylogenet. Evol. 2007, 44, 1306–1319. [Google Scholar] [CrossRef]
- Ahmed, M.Z.; Ren, S.-X.; Mandour, N.S.; Maruthi, M.N.; Naveed, M.; Qiu, B.-L. Phylogenetic analysis of Bemisia tabaci (Hemiptera: Aleyrodidae) populations from cotton plants in Pakistan, China, and Egypt. J. Pest Sci. 2010, 83, 135–141. [Google Scholar] [CrossRef]
- Brown, W.M.; George, M.; Wilson, A.C. Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1979, 76, 1967–1971. [Google Scholar] [CrossRef]
- Ballard, J.W.O.; Whitlock, M.C. The incomplete natural history of mitochondria. Mol. Ecol. 2004, 13, 729–744. [Google Scholar] [CrossRef]
- Latorre, A.; Hernández, C.; Martínez, D.; Castro, J.A.; Ramón, M.; Moya, A. Population structure and mitochondrial DNA gene flow in Old World populations of Drosophila subobscura. Heredity 1992, 68, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Besansky, N.J.; Lehmann, T.; Fahey, G.T.; Fontenille, D.; Braack, L.E.O.; Hawley, W.A.; Collins, F.H. Patterns of Mitochondrial Variation within and Between African Malaria Vectors, Anopheles gambiae and An. arabiensis, Suggest Extensive Gene Flow. Genetics 1997, 147, 1817–1828. [Google Scholar] [PubMed]
- Pearce, R.L.; Wood, J.J.; Artukhin, Y.; Birt, T.P.; Damus, M.; Friesen, V.L. Mitochondrial dna suggests high gene flow in ancient murrelets. Condor 2002, 104, 84–91. [Google Scholar] [CrossRef]
- Harpending, H.C. Signature of Ancient Population Growth in a Low-Resolution Mitochondrial DNA Mismatch Distribution. Hum. Biol. 1994, 66, 591–600. [Google Scholar] [PubMed]
- Schneider, S.; Excoffier, L. Estimation of Past Demographic Parameters from the Distribution of Pairwise Differences When the Mutation Rates Vary Among Sites: Application to Human Mitochondrial DNA. Genetics 1999, 152, 1079–1089. [Google Scholar] [PubMed]
- Toews, D.P.L.; Brelsford, A. The biogeography of mitochondrial and nuclear discordance in animals. Mol. Ecol. 2012, 21, 3907–3930. [Google Scholar] [CrossRef]
- Elfekih, S.; Etter, P.; Tay, W.T.; Fumagalli, M.; Gordon, K.; Johnson, E.; Barro, P.D. Genome-wide analyses of the Bemisia tabaci species complex reveal contrasting patterns of admixture and complex demographic histories. PLoS ONE 2018, 13, e0190555. [Google Scholar] [CrossRef]
- Hsieh, C.-H.; Ko, C.-C.; Chung, C.-H.; Wang, H.-Y. Multilocus approach to clarify species status and the divergence history of the Bemisia tabaci (Hemiptera: Aleyrodidae) species complex. Mol. phylogenetics Evol. 2014, 76, 172–180. [Google Scholar] [CrossRef]
- McKenzie, C.L.; Bethke, J.A.; Byrne, F.J.; Chamberlin, J.R.; Dennehy, T.J.; Dickey, A.M.; Gilrein, D.; Hall, P.M.; Ludwig, S.; Oetting, R.D.; et al. Distribution of Bemisia tabaci (Hemiptera: Aleyrodidae) Biotypes in North America After the Q Invasion. J. Econ. Entomol. 2012, 105, 753–766. [Google Scholar] [CrossRef]
- Wosula, E.N.; Chen, W.; Fei, Z.; Legg, J.P. Unravelling the Genetic Diversity among Cassava Bemisia tabaci Whiteflies Using NextRAD Sequencing. Genome Biol. Evol. 2017, 9, 2958–2973. [Google Scholar] [CrossRef]
- Chen, W.; Wosula, E.N.; Hasegawa, D.K.; Casinga, C.; Shirima, R.R.; Fiaboe, K.K.M.; Hanna, R.; Fosto, A.; Goergen, G.; Tamò, M.; et al. Genome of the African cassava whitefly Bemisia tabaci and distribution and genetic diversity of cassava-colonizing whiteflies in Africa. Insect Biochem. Mol. Biol. 2019, 110, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hasegawa, D.K.; Arumuganathan, K.; Simmons, A.M.; Wintermantel, W.M.; Fei, Z.; Ling, K.-S. Estimation of the Whitefly Bemisia tabaci Genome Size Based on k-mer and Flow Cytometric Analyses. Insects 2015, 6, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hasegawa, D.K.; Kaur, N.; Kliot, A.; Pinheiro, P.V.; Luan, J.; Stensmyr, M.C.; Zheng, Y.; Liu, W.; Sun, H.; et al. The draft genome of whitefly Bemisia tabaci MEAM1, a global crop pest, provides novel insights into virus transmission, host adaptation, and insecticide resistance. BMC Biol. 2016, 14, 110. [Google Scholar] [CrossRef] [PubMed]
- Strimmer, K.; Haeseler, A. von Likelihood-mapping: A simple method to visualize phylogenetic content of a sequence alignment. Proc. Natl. Acad. Sci. USA 1997, 94, 6815–6819. [Google Scholar] [CrossRef] [PubMed]
- Pease, J.B.; Brown, J.W.; Walker, J.F.; Hinchliff, C.E.; Smith, S.A. Quartet Sampling distinguishes lack of support from conflicting support in the green plant tree of life. Am. J. Bot. 2018, 105, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony (and Other Methods) 4.0. B5. 2001. Available online: http://citeseerx.ist.psu.edu/viewdoc/summary?doi=10.1.1.458.6867 (accessed on 26 August 2019).
- Puillandre, N.; Lambert, A.; Brouillet, S.; Achaz, G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol. Ecol. 2012, 21, 1864–1877. [Google Scholar] [CrossRef] [PubMed]
- Sweet, A.D.; Boyd, B.M.; Allen, J.M.; Villa, S.M.; Valim, M.P.; Rivera-Parra, J.L.; Wilson, R.E.; Johnson, K.P. Integrating phylogenomic and population genomic patterns in avian lice provides a more complete picture of parasite evolution. Evolution 2018, 72, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.K.; Coats, S.A.; Bedford, I.D.; Markham, P.G.; Bird, J.; Frohlich, D.R. Characterization and distribution of esterase electromorphs in the whitefly, Bemisia tabaci (Genn.) (Homoptera: Aleyrodidae). Biochem. Genet. 1995, 33, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Costa, H.S.; Brown, J.K.; Sivasupramaniam, S.; Bird, J. Regional distribution, insecticide resistance, and reciprocal crosses between the A and B biotypes of Bemisia tabaci. Int. J. Trop. Insect Sci. 1993, 14, 255–266. [Google Scholar] [CrossRef]
- Perring, T.M. The Bemisia tabaci species complex. Crop Prot. 2001, 20, 725–737. [Google Scholar] [CrossRef]
- Banks, G.K.; Bedford, I.D.; Beitia, F.J.; Rodriguez-Cerezo, E.; Markham, P.G. A Novel Geminivirus of Ipomoea indica (Convolvulacae) from Southern Spain. Plant Dis. 1999, 83, 486. [Google Scholar] [CrossRef] [PubMed]
- Simón, B.; Cenis, J.L.; Demichelis, S.; Rapisarda, C.; Caciagli, P.; Bosco, D. Survey of Bemisia tabaci (Hemiptera: Aleyrodidae) biotypes in Italy with the description of a new biotype (T) from Euphorbia characias. Bull. Entomol. Res. 2003, 93, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Demichelis, S.; Arnò, C.; Bosco, D.; Marian, D.; Caciagli, P. Characterization of biotype T of Bemisia tabaci associated with Euphorbia characias in Sicily. Phytoparasitica 2005, 33, 196–208. [Google Scholar] [CrossRef]
- Delatte, H.; Reynaud, B.; Granier, M.; Thornary, L.; Lett, J.M.; Goldbach, R.; Peterschmitt, M. A new silverleaf-inducing biotype Ms of Bemisia tabaci (Hemiptera: Aleyrodidae) indigenous to the islands of the south-west Indian Ocean. Bull. Entomol. Res. 2005, 95, 29–35. [Google Scholar] [CrossRef] [PubMed]
- De Barro, P.J.; Liebregts, W.; Carver, M. Distribution and identity of biotypes of Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) in member countries of the Secretariat of the Pacific Community. Aust. J. Entomol. 1998, 37, 214–218. [Google Scholar] [CrossRef]
- Qiu, B.-L.; Ren, S.X.; Wen, S.Y.; Mandour, N.S. Population differentiation of three biotypes of Bemisia tabaci (Hemiptera: Aleyrodidae) in China by DNA polymorphism. J. South China Agric. Univ. 2006, 27, 29–33. [Google Scholar]
- Qiu, B.-L.; Chen, Y.; Liu, L.; Peng, W.; Li, X.; Ahmed, M.Z.; Mathur, V.; Du, Y.; Ren, S. Identification of three major Bemisia tabaci biotypes in China based on morphological and DNA polymorphisms. Prog. Nat. Sci. 2009, 19, 713–718. [Google Scholar] [CrossRef]
- Qiu, B.-L.; Dang, F.; Li, S.-J.; Ahmed, M.Z.; Jin, F.-L.; Ren, S.-X.; Cuthbertson, A.G.S. Comparison of biological parameters between the invasive B biotype and a new defined Cv biotype of Bemisia tabaci (Hemiptera: Aleyradidae) in China. J. Pest Sci 2011, 84, 419–427. [Google Scholar] [CrossRef]
- Zang, L.-S.; Liu, S.S.; Liu, Y.Q.; Ruan, Y.M.; Wan, F.H. Competition between the B biotype and a non-B biotype of the whitefly, Bemisia tabaci (Homoptera: Aleyrodidae) in Zhejang, China. Biodivers. Sci. 2005, 13, 181–187. [Google Scholar] [CrossRef][Green Version]
- Zang, L.-S.; Chen, W.-Q.; Liu, S.-S. Comparison of performance on different host plants between the B biotype and a non-B biotype of Bemisia tabaci from Zhejiang, China. Entomol. Exp. Appl. 2006, 121, 221–227. [Google Scholar] [CrossRef]
- Zang, L.-S.; Tong, J.; Jing, X.; Shu-Sheng, L.; You-Jun, Z. SCAR molecular markers of the B biotype and two non-B populations of the whitefly, Bemisia tabaci (Hemiptera: Aleyrodidae). Chin. J. Agric. Biotechnol. 2006, 3, 189–194. [Google Scholar]
- Li, J.; Tang, Q.; Bai, R.; Li, X.; Jiang, J.; Zhai, Q.; Yan, F. Comparative Morphology and Morphometry of Six Biotypes of Bemisia tabaci (Hemiptera: Aleyrodidae) from China. J. Integr. Agric. 2013, 12, 846–852. [Google Scholar] [CrossRef]
- Andrews, S. FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 12 July 2018).
- Kriventseva, E.V.; Rahman, N.; Espinosa, O.; Zdobnov, E.M. OrthoDB: The hierarchical catalog of eukaryotic orthologs. Nucleic Acids Res. 2008, 36, D271–D275. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.P.; Dietrich, C.H.; Friedrich, F.; Beutel, R.G.; Wipfler, B.; Peters, R.S.; Allen, J.M.; Petersen, M.; Donath, A.; Walden, K.K.O.; et al. Phylogenomics and the evolution of hemipteroid insects. Proc. Natl. Acad. Sci. USA 2018, 115, 12775–12780. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup the Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Mirarab, S.; Nguyen, N.; Guo, S.; Wang, L.-S.; Kim, J.; Warnow, T. PASTA: Ultra-Large Multiple Sequence Alignment for Nucleotide and Amino-Acid Sequences. J. Comput. Biol. 2014, 22, 377–386. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Vaidya, G.; Lohman, D.J.; Meier, R. SequenceMatrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Mirarab, S.; Reaz, R.; Bayzid, M.S.; Zimmermann, T.; Swenson, M.S.; Warnow, T. ASTRAL: Genome-scale coalescent-based species tree estimation. Bioinformatics 2014, 30, i541–i548. [Google Scholar] [CrossRef] [PubMed]
- Vachaspati, P.; Warnow, T. ASTRID: Accurate Species TRees from Internode Distances. BMC Genom. 2015, 16, S3. [Google Scholar] [CrossRef] [PubMed]
- Rabiee, M.; Sayyari, E.; Mirarab, S. Multi-allele species reconstruction using ASTRAL. Mol. Phylogenet. Evol. 2019, 130, 286–296. [Google Scholar] [CrossRef]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A Greedy Algorithm for Aligning DNA Sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef]
- Morgulis, A.; Coulouris, G.; Raytselis, Y.; Madden, T.L.; Agarwala, R.; Schäffer, A.A. Database indexing for production MegaBLAST searches. Bioinformatics 2008, 24, 1757–1764. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Boykin, L.M.; Savill, A.; De Barro, P. Updated mtCOI reference dataset for the Bemisia tabaci species complex. F1000Research 2017, 6, 1835. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-F.; Jiang, L.-Y.; Qiao, G.-X. Use of a mitochondrial COI sequence to identify species of the subtribe Aphidina (Hemiptera, Aphididae). Zookeys 2011, 122, 1–17. [Google Scholar]
- Lee, H.-C.; Yang, M.-M.; Yeh, W.-B. Identification of Two Invasive Cacopsylla chinensis (Hemiptera: Psyllidae) Lineages Based on Two Mitochondrial Sequences and Restriction Fragment Length Polymorphism of Cytochrome Oxidase I Amplicon. J. Econ. Entomol. 2008, 101, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Percy, D.M. Radiation, Diversity, and Host-Plant Interactions Among Island and Continental Legume-Feeding Psyllids. Evolution 2003, 57, 2540–2556. [Google Scholar] [CrossRef] [PubMed]
- Bess, E.C.; Catanach, T.A.; Johnson, K.P. The importance of molecular dating analyses for inferring Hawaiian biogeographical history: A case study with bark lice (Psocidae: Ptycta). J. Biogeogr. 2014, 41, 158–167. [Google Scholar] [CrossRef]
- Byrne, F.J.; Devonshire, A.L. Biochemical evidence of haplodiploidy in the whitefly Bemisia tabaci. Biochem. Genet. 1996, 34, 93–107. [Google Scholar] [CrossRef] [PubMed]
- McMeniman, C.J.; Barker, S.C. Transmission ratio distortion in the human body louse, Pediculus humanus (Insecta: Phthiraptera). Heredity 2006, 96, 63–68. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Andersen, J.C.; Wu, J.; Gruwell, M.E.; Gwiazdowski, R.; Santana, S.E.; Feliciano, N.M.; Morse, G.E.; Normark, B.B. A phylogenetic analysis of armored scale insects (Hemiptera: Diaspididae), based upon nuclear, mitochondrial, and endosymbiont gene sequences. Mol. Phylogenet. Evol. 2010, 57, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Hodson, C.N.; Hamilton, P.T.; Dilworth, D.; Nelson, C.J.; Curtis, C.I.; Perlman, S.J. Paternal Genome Elimination in Liposcelis Booklice (Insecta: Psocodea). Genetics 2017, 206, 1091–1100. [Google Scholar] [CrossRef]
- De la Filia, A.G.; Andrewes, S.; Clark, J.M.; Ross, L. The unusual reproductive system of head and body lice (Pediculus humanus). Med. Vet. Entomol. 2018, 32, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.C.; Schwarz, M.P.; Crespi, B.J.; Cooper, S.J.B. Phylogenetics of gall-inducing thrips on Australian Acacia. Biol. J. Linn. Soc. 2001, 74, 73–86. [Google Scholar] [CrossRef][Green Version]
- Park, D.-S.; Suh, S.-J.; Hebert, P.D.N.; Oh, H.-W.; Hong, K.-J. DNA barcodes for two scale insect families, mealybugs (Hemiptera: Pseudococcidae) and armored scales (Hemiptera: Diaspididae). Bull. Entomol. Res. 2011, 101, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Yang, Q.; Li, H.; Song, F.; Stejskal, V.; Opit, G.P.; Cai, W.; Li, Z.; Shao, R. The Highly Divergent Mitochondrial Genomes Indicate That the Booklouse, Liposcelis bostrychophila (Psocoptera: Liposcelididae) Is a Cryptic Species. G3-Genes Genomes Genet. 2018, 8, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.P.; Cruickshank, R.H.; Adams, R.J.; Smith, V.S.; Page, R.D.M.; Clayton, D.H. Dramatically elevated rate of mitochondrial substitution in lice (Insecta: Phthiraptera). Mol. PHYLOGENET. Evol. 2003, 26, 231–242. [Google Scholar] [CrossRef]
- Horowitz, A.R.; Gerling, D. Seasonal Variation of Sex Ratio in Bemisia tabaci on Cotton in Israel. Environ. Entomol. 1992, 21, 556–559. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nuclear Lineages (Current) | Mitochondrial Major Clades a | Biotypes d | |||
|---|---|---|---|---|---|
| Name | Acronym | Number | Name | ||
| Sub-Saharan Africa | SSA | I | Sub-Saharan Africa—East and South | S | |
| II | Sub-Saharan Africa—West | ||||
| North Africa–Mediterranean–Middle East | III | North Africa–Mediterranean–Middle East | B, B1, B2 | ||
| NAF–MED–ME | J, L, Q | ||||
| Non-Ug/MS | |||||
| Asia 2 | ASIA | IV | Asia 2 | K, P, ZHJ2 | |
| ZHJ1 | |||||
| G | |||||
| Cv, I | |||||
| Asia–Pacific–Australia | V | Asia–Pacific Islands–Australia | H, M, Na (Nauru), | ||
| AS–PAC–AU | An | ||||
| ZHJ3 | |||||
| American Tropics: North-Central America and Caribbean | AM-TROP | VI | American Tropics: North and Central/ Caribbean | A, A1, A2, C, D, F, O, N, R | |
| VII | American Tropics: South America b | - | |||
| Unassigned groups c: | |||||
| Uganda | - | ||||
| Italy | T | ||||
| Benin | E | ||||
| Geographical Acronym | Country | State/Province | City | Plant Host | Reads | Gbp | Coverage (X) | Mapped Genes | Aligned Genes | Aligned bp | Best Blast Match | % Pairwise Identity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AM-TROP | Puerto Rico | - | - | Jatropha gossypifolia | 106,701,526 | 320 | 52 | 2192 | 2183 | 3,662,977 | KX397317 | 99.7 |
| AM-TROP | Ecuador | Guayas | Isla Puná | Datura stramonium | 80,297,211 | 241 | 39 | 2192 | 2183 | 3,669,262 | EU427728 | 98.6 |
| AM-TROP | United States | Arizona | Tucson | Gossypium hirsutum | 86,382,401 | 259 | 42 | 2192 | 2183 | 3,672,677 | AY521259 | 100 |
| NAF–MED–ME | Sudan | Al Jazirah | Wad Mani | Phaseolus vulgaris | 94,837,626 | 285 | 46 | 2193 | 2184 | 3,672,242 | FJ188567 | 99.0 |
| SSA | DR Congo | - | Lulimba | Manihot esculenta | 92,877,327 | 279 | 45 | 2193 | 2184 | 3,668,485 | AF344246 | 99.7 |
| SSA | Uganda | Kajera | Bukoba | Manihot esculenta | 133,240,668 | 400 | 65 | 2193 | 2184 | 3,668,433 | AM040604 | 99.7 |
| SSA | Tanzania | Dar es-Salam | Dar es-Salam | Manihot esculenta | 103,951,478 | 312 | 51 | 2193 | 2184 | 3,671,055 | JQ286457 | 99.7 |
| AS–PAC–AU | China | Hainan | Hainan | Gossypium hirsutum | 97,765,484 | 293 | 48 | 2193 | 2184 | 3,672,435 | HG918196 | 99.8 |
| ASIA | India | Ahmedabad | New Delhi | Nicotiana tabacum | 103,114,416 | 309 | 50 | 2193 | 2184 | 3,671,735 | KF751570 | 100 |
| B. afer | Australia | New South Wales | Sydney | Hardenbergia sp. | 86,662,872 | 277 | 45 | 1938 | 1929 | 3,002,554 | AJ784260 | 87.9 |
| (A) | Geographical Acronym | Specimen | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| 1 | B. afer | - | ||||||||||
| 2 | NAF-MED-ME | SRS1302125 | 0.0755 | - | ||||||||
| 3 | NAF-MED-ME | Sudan | 0.0757 | 0.0057 | - | |||||||
| 4 | AM-TROP | Arizona | 0.0805 | 0.0164 | 0.0138 | - | ||||||
| 5 | AM-TROP | Ecuador | 0.0806 | 0.0163 | 0.0138 | 0.0012 | - | |||||
| 6 | AM-TROP | Puerto Rico | 0.0808 | 0.0159 | 0.0136 | 0.0023 | 0.0023 | - | ||||
| 7 | SSA | DR Congo | 0.0808 | 0.0223 | 0.0199 | 0.0221 | 0.0221 | 0.0222 | - | |||
| 8 | SSA | Tanzania | 0.0830 | 0.0250 | 0.0225 | 0.0246 | 0.0247 | 0.0249 | 0.0051 | - | ||
| 9 | SSA | Uganda | 0.0808 | 0.0221 | 0.0198 | 0.0221 | 0.0221 | 0.0221 | 0.0031 | 0.0052 | - | |
| 10 | AS–PAC–AU | China | 0.0799 | 0.0161 | 0.0135 | 0.0152 | 0.0153 | 0.0154 | 0.0215 | 0.0241 | 0.0216 | - |
| 11 | ASIA | India | 0.0791 | 0.0150 | 0.0125 | 0.0143 | 0.0143 | 0.0145 | 0.0205 | 0.0231 | 0.0206 | 0.0093 |
| (B) | Geographical Acronym | Specimen | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| 1 | B. afer | - | ||||||||||
| 2 | NAF–MED–ME | SRS1302125 | 0.2200 | - | ||||||||
| 3 | NAF–MED–ME | Sudan | 0.2180 | 0.0637 | - | |||||||
| 4 | AM-TROP | Arizona | 0.2229 | 0.1439 | 0.1508 | - | ||||||
| 5 | AM-TROP | Ecuador | 0.2180 | 0.1412 | 0.1487 | 0.0171 | - | |||||
| 6 | AM-TROP | Puerto Rico | 0.2229 | 0.1426 | 0.1501 | 0.0178 | 0.0199 | - | ||||
| 7 | SSA | DR Congo | 0.2282 | 0.1594 | 0.1552 | 0.1588 | 0.1602 | 0.1581 | - | |||
| 8 | SSA | Tanzania | 0.2193 | 0.1489 | 0.1579 | 0.1531 | 0.1545 | 0.1511 | 0.0825 | - | ||
| 9 | SSA | Uganda | 0.2249 | 0.1524 | 0.1627 | 0.1592 | 0.1606 | 0.1572 | 0.0824 | 0.0131 | - | |
| 10 | AS–PAC–AU | China | 0.2312 | 0.1377 | 0.1412 | 0.1410 | 0.1348 | 0.1403 | 0.1594 | 0.1646 | 0.1681 | - |
| 11 | ASIA | India | 0.2260 | 0.1419 | 0.1467 | 0.1597 | 0.1576 | 0.1604 | 0.1695 | 0.1681 | 0.1736 | 0.1317 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Moya, R.S.; Brown, J.K.; Sweet, A.D.; Walden, K.K.O.; Paredes-Montero, J.R.; Waterhouse, R.M.; Johnson, K.P. Nuclear Orthologs Derived from Whole Genome Sequencing Indicate Cryptic Diversity in the Bemisia tabaci (Insecta: Aleyrodidae) Complex of Whiteflies. Diversity 2019, 11, 151. https://doi.org/10.3390/d11090151
de Moya RS, Brown JK, Sweet AD, Walden KKO, Paredes-Montero JR, Waterhouse RM, Johnson KP. Nuclear Orthologs Derived from Whole Genome Sequencing Indicate Cryptic Diversity in the Bemisia tabaci (Insecta: Aleyrodidae) Complex of Whiteflies. Diversity. 2019; 11(9):151. https://doi.org/10.3390/d11090151
Chicago/Turabian Stylede Moya, Robert S., Judith K. Brown, Andrew D. Sweet, Kimberly K. O. Walden, Jorge R. Paredes-Montero, Robert M. Waterhouse, and Kevin P. Johnson. 2019. "Nuclear Orthologs Derived from Whole Genome Sequencing Indicate Cryptic Diversity in the Bemisia tabaci (Insecta: Aleyrodidae) Complex of Whiteflies" Diversity 11, no. 9: 151. https://doi.org/10.3390/d11090151
APA Stylede Moya, R. S., Brown, J. K., Sweet, A. D., Walden, K. K. O., Paredes-Montero, J. R., Waterhouse, R. M., & Johnson, K. P. (2019). Nuclear Orthologs Derived from Whole Genome Sequencing Indicate Cryptic Diversity in the Bemisia tabaci (Insecta: Aleyrodidae) Complex of Whiteflies. Diversity, 11(9), 151. https://doi.org/10.3390/d11090151