A Phylogenomic Supertree of Birds

 , , , ,
, , , ,  ,
, 
Abstract

1. Introduction
2. Materials and Methods
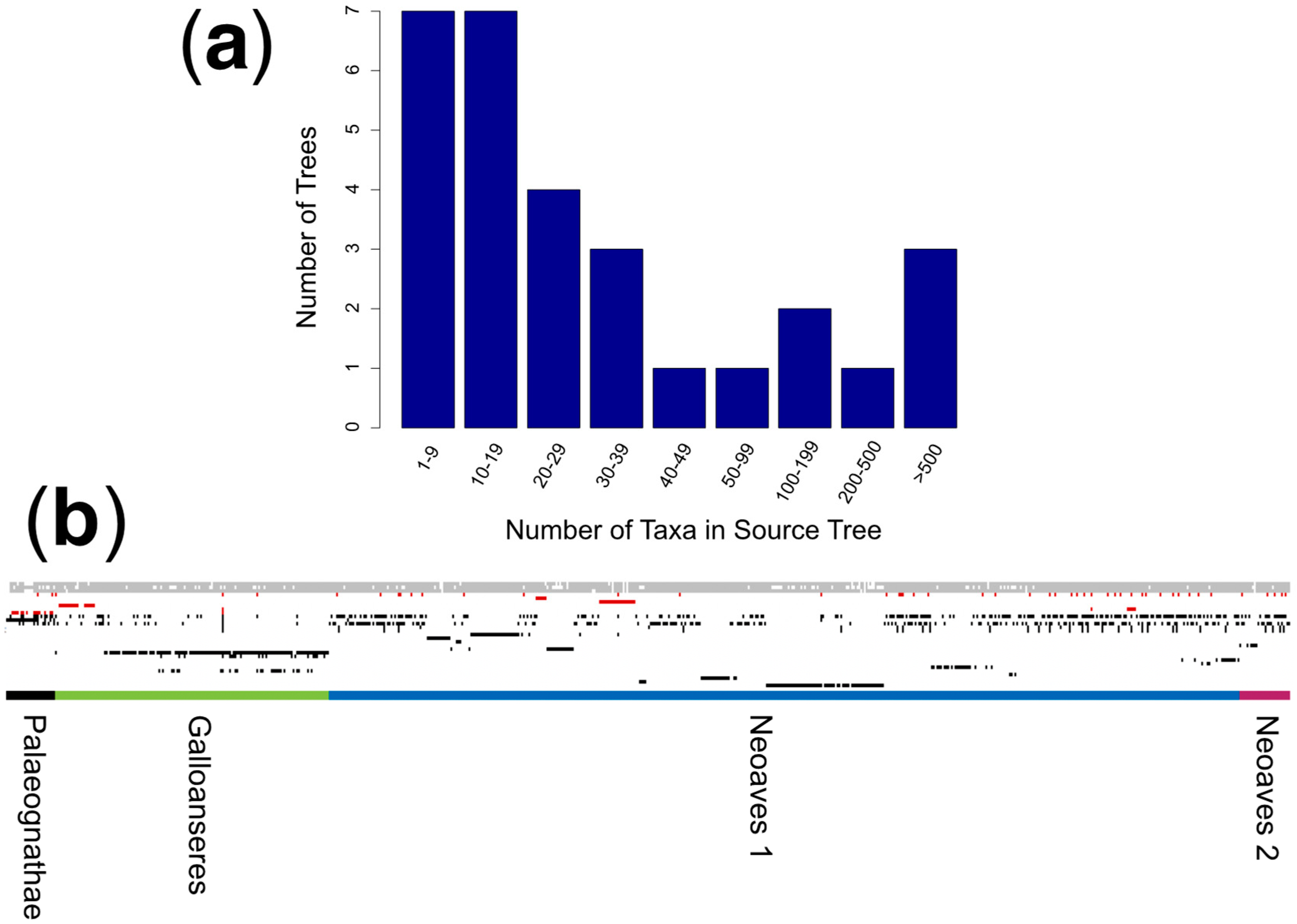
2.1. Source Tree Selection and Taxonomic Reconciliation
2.2. MRP and MRL Supertree Searches
2.3. Estimating Branch Lengths and A Calibrated Time Tree
3. Results
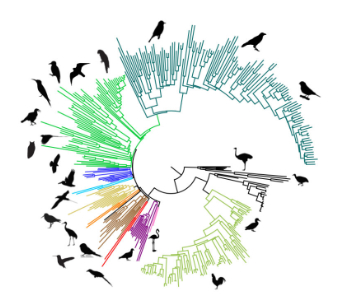
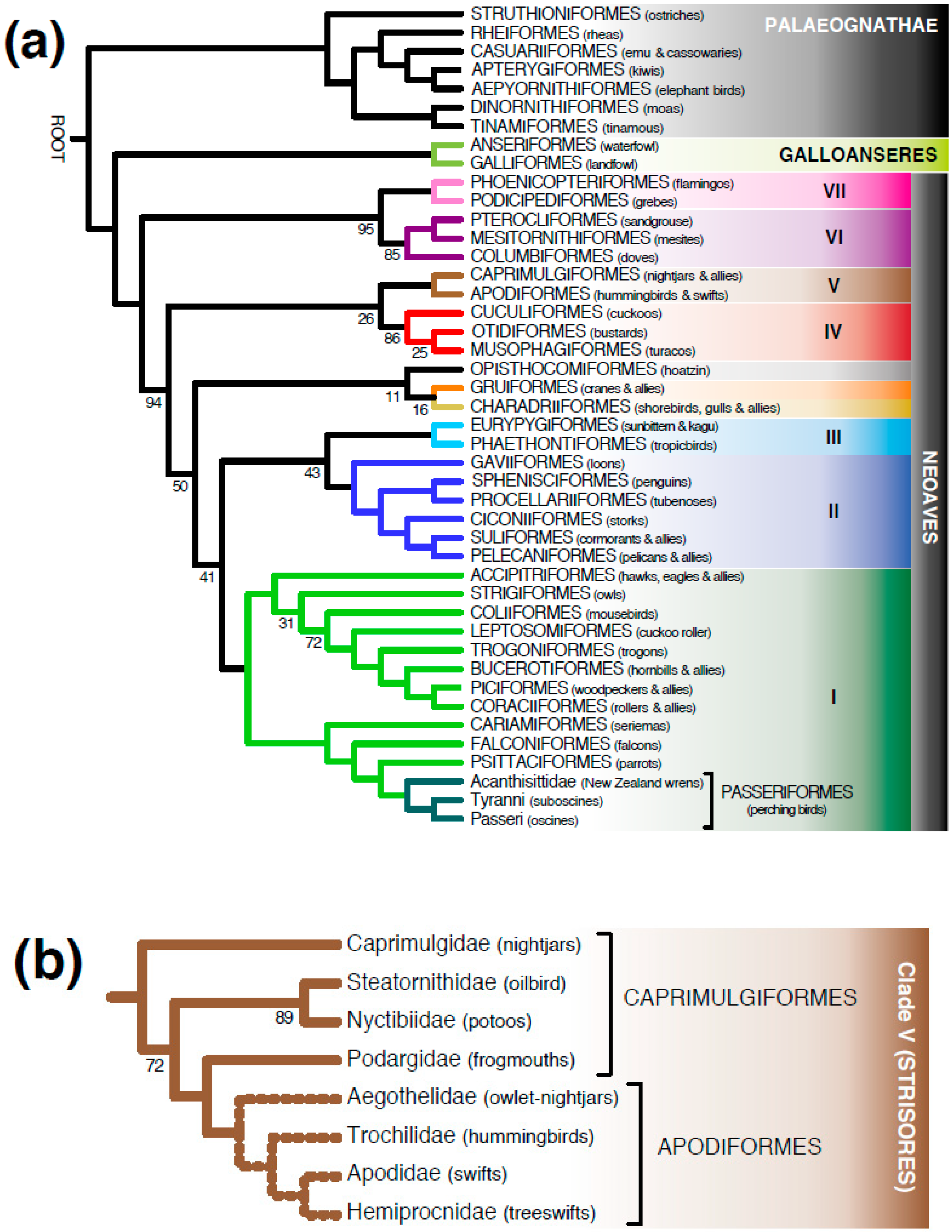
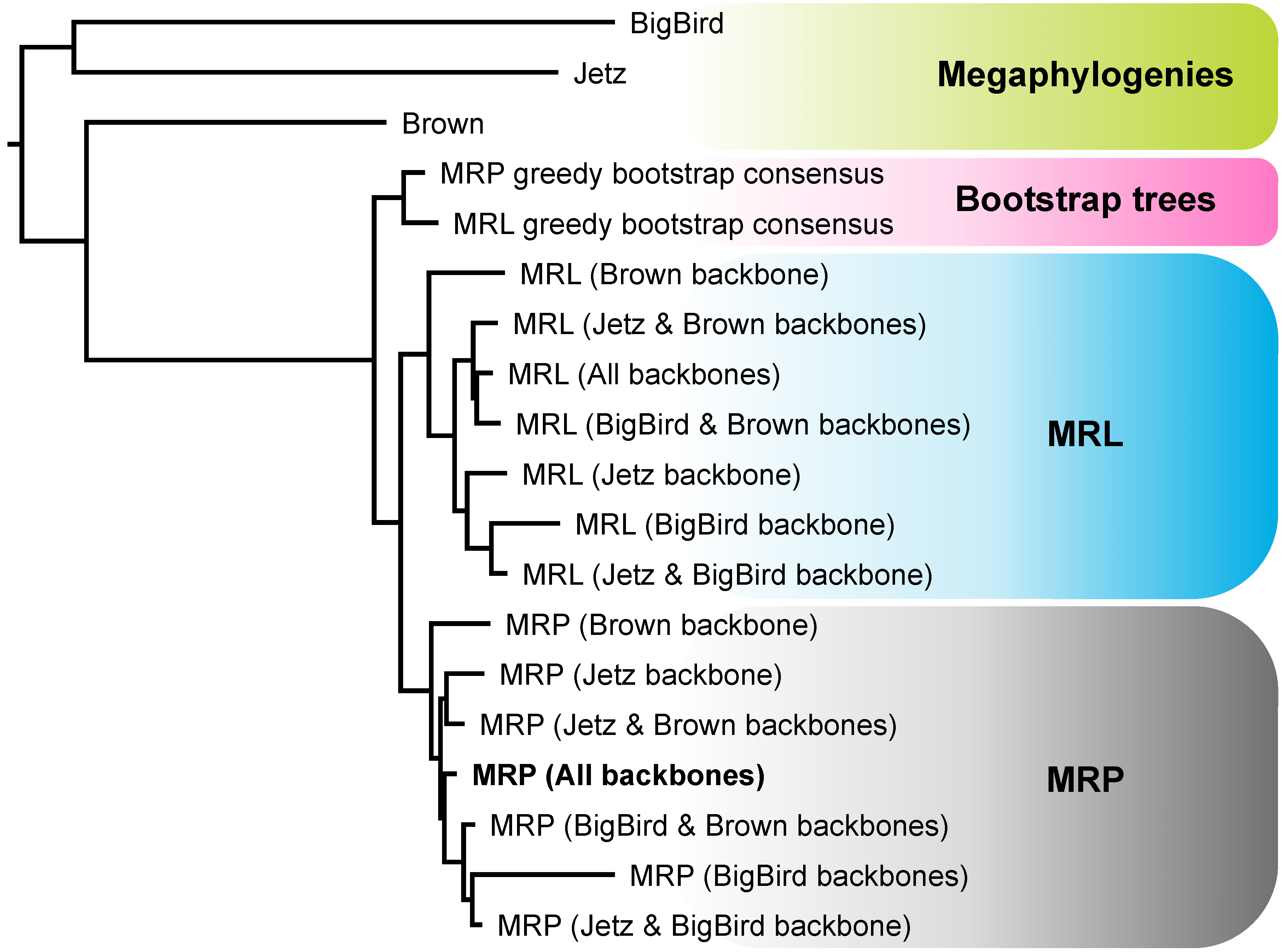
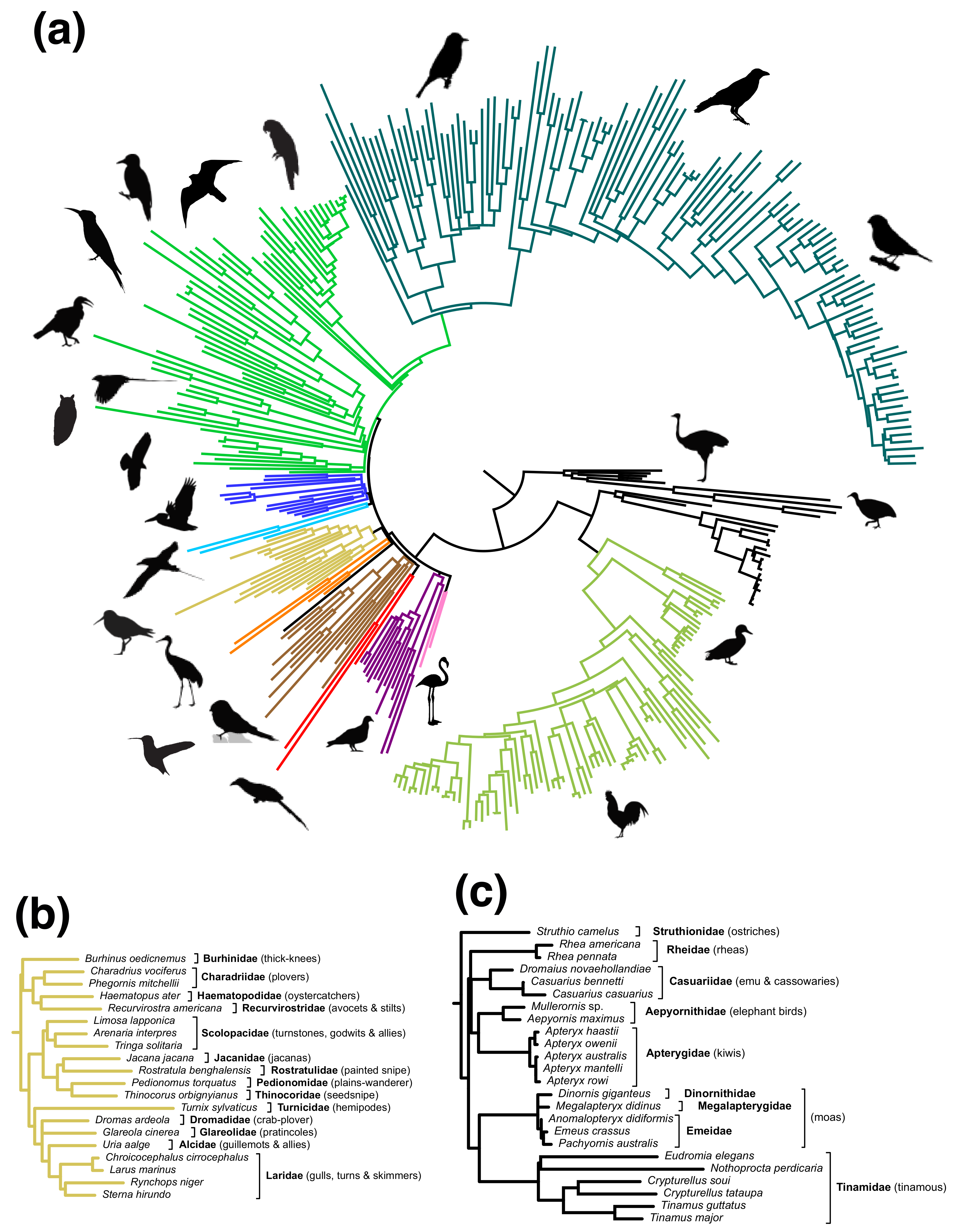
3.1. Meta-Analysis of Phylogenomic Trees Yields A Well-Resolved Supertree
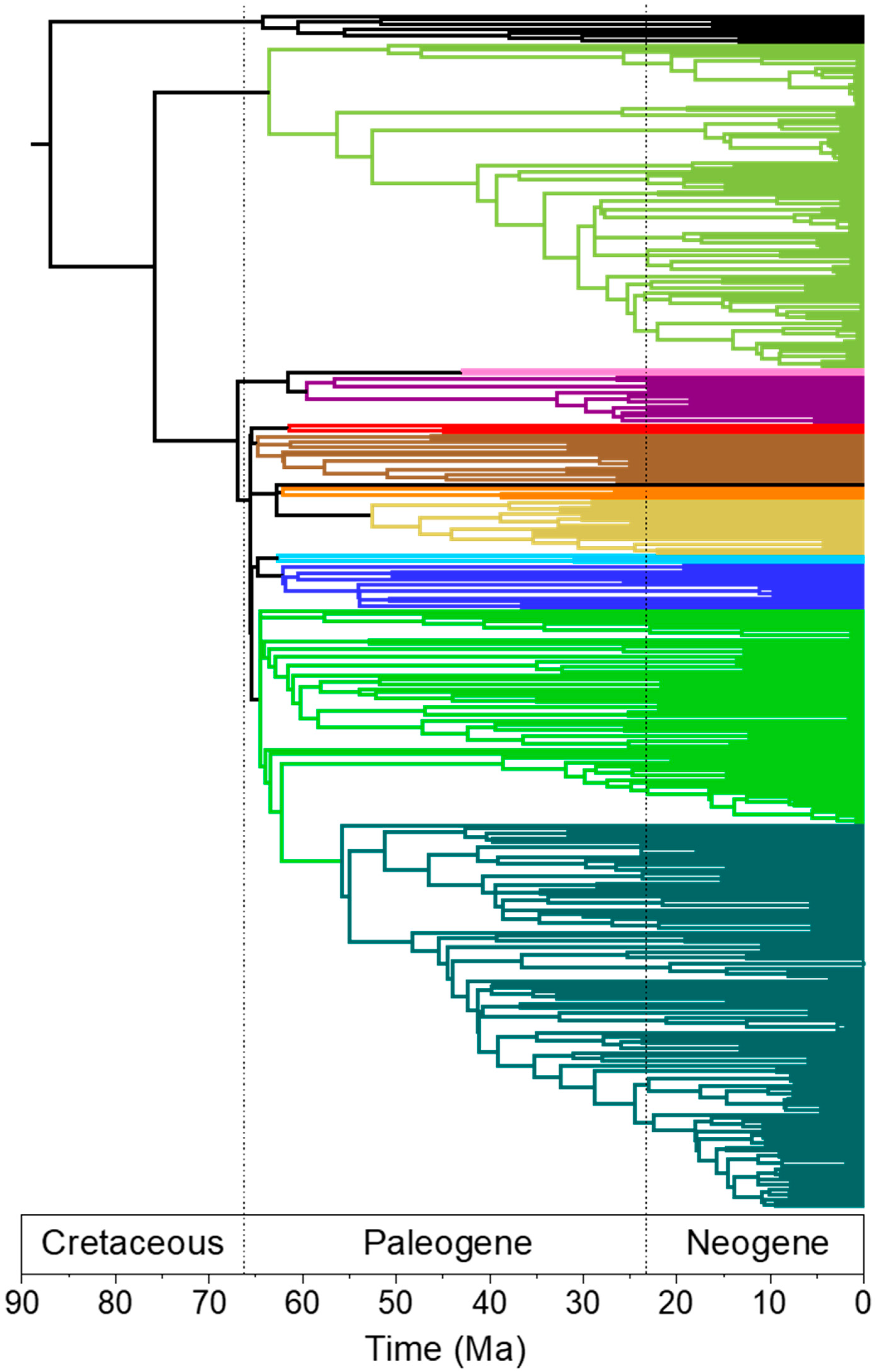
3.2. Rapid Branch Length Estimation and Divergence Time Estimation
4. Discussion
4.1. Strengths and Weaknesses of the Phylogenomic Supertree Approach
4.2. Different Roles for Backbone Trees and Phylogenomic Trees
4.3. MRP and MRL Support Values
4.4. Branch Lengths and Divergence Times
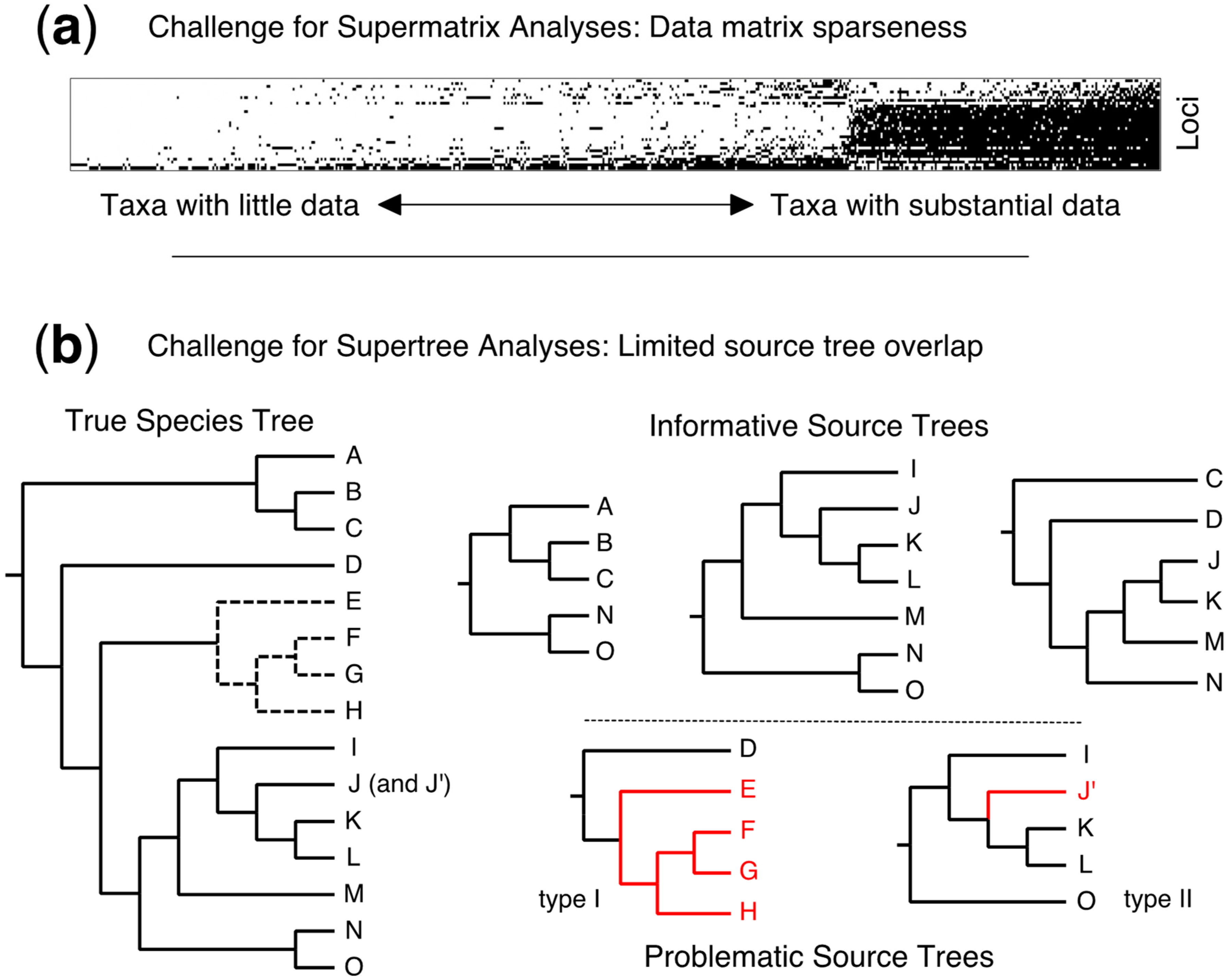
4.5. Taxonomic Flux—A Fundamental Challenge for Supertrees (and Supermatrices)
4.6. Moving Forward: OpenWings, B10K, and Other Phylogenomic Efforts
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- McCormack, J.E.; Hird, S.M.; Zellmer, A.J.; Carstens, B.C.; Brumfield, R.T. Applications of next-generation sequencing to phylogeography and phylogenetics. Mol. Phylogenet. Evol. 2013, 66, 526–538. [Google Scholar] [CrossRef] [PubMed]
- Delsuc, F.; Brinkmann, H.; Philippe, H. Phylogenomics and the reconstruction of the tree of life. Nat. Rev. Genet. 2005, 6, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Philippe, H.; Delsuc, F.; Brinkmann, H.; Lartillot, N. Phylogenomics. Annu. Rev. Ecol. Evol. Syst. 2005, 36, 541–562. [Google Scholar] [CrossRef]
- Eisen, J.A. Phylogenomics: Improving functional predictions for uncharacterized genes by evolutionary analysis. Genome Res. 1998, 8, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Eisen, J.A.; Kaiser, D.; Myers, R.M. Gastrogenomic delights: A movable feast. Nat. Med. 1997, 3, 1076–1078. [Google Scholar] [CrossRef] [PubMed]
- Philippe, H.; Snell, E.A.; Bapteste, E.; Lopez, P.; Holland, P.W.H.; Casane, D. Phylogenomics of eukaryotes: Impact of missing data on large alignments. Mol. Biol. Evol. 2004, 21, 1740–1752. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.W.; Hejnol, A.; Matus, D.Q.; Pang, K.; Browne, W.E.; Smith, S.A.; Seaver, E.; Rouse, G.W.; Obst, M.; Edgecombe, G.D.; et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 2008, 452, 745–749. [Google Scholar] [CrossRef]
- Hackett, S.J.; Kimball, R.T.; Reddy, S.; Bowie, R.C.K.; Braun, E.L.; Braun, M.J.; Chojnowski, J.L.; Cox, W.A.; Han, K.L.; Harshman, J.; et al. A phylogenomic study of birds reveals their evolutionary history. Science 2008, 320, 1763–1768. [Google Scholar] [CrossRef]
- Shen, X.X.; Zhou, X.F.; Kominek, J.; Kurtzman, C.P.; Hittinger, C.T.; Rokas, A. Reconstructing the backbone of the Saccharomycotina yeast phylogeny using genome-scale data. G3 Genes Genomes Genet. 2016, 6, 3927–3939. [Google Scholar] [CrossRef]
- Ascunce, M.S.; Huguet-Tapia, J.C.; Ortiz-Urquiza, A.; Keyhani, N.O.; Braun, E.L.; Goss, E.M. Phylogenomic analysis supports multiple instances of polyphyly in the oomycete peronosporalean lineage. Mol. Phylogenet. Evol. 2017, 114, 199–211. [Google Scholar] [CrossRef]
- Wu, G.A.; Terol, J.; Ibanez, V.; Lopez-Garcia, A.; Perez-Roman, E.; Borreda, C.; Domingo, C.; Tadeo, F.R.; Carbonell-Caballero, J.; Alonso, R.; et al. Genomics of the origin and evolution of citrus. Nature 2018, 554, 311. [Google Scholar] [CrossRef] [PubMed]
- Harvey, M.G.; Smith, B.T.; Glenn, T.C.; Faircloth, B.C.; Brumfield, R.T. Sequence capture versus restriction site associated dna sequencing for shallow systematics. Syst. Biol. 2016, 65, 910–924. [Google Scholar] [CrossRef] [PubMed]
- Misof, B.; Liu, S.L.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G.; et al. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Wickett, N.J.; Mirarab, S.; Nguyen, N.; Warnow, T.; Carpenter, E.; Matasci, N.; Ayyampalayam, S.; Barker, M.S.; Burleigh, J.G.; Gitzendanner, M.A.; et al. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Acad. Sci. USA 2014, 111, E4859–E4868. [Google Scholar] [CrossRef] [PubMed]
- Gayral, P.; Weinert, L.; Chiari, Y.; Tsagkogeorga, G.; Ballenghien, M.; Galtier, N. Next-generation sequencing of transcriptomes: A guide to RNA isolation in nonmodel animals. Mol. Ecol. Resour. 2011, 11, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 2016, 17, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.E.R.; Ree, R.H.; Moreau, C.S. Inferring phylogenies from RADsequence data. PLoS ONE 2012, 7, e33394. [Google Scholar] [CrossRef]
- Eaton, D.A.R.; Spriggs, E.L.; Park, B.; Donoghue, M.J. Misconceptions on missing data in RAD-seq phylogenetics with a deep-scale example from flowering plants. Syst. Biol. 2017, 66, 399–412. [Google Scholar] [CrossRef]
- Tin, M.M.Y.; Rheindt, F.E.; Cros, E.; Mikheyev, A.S. Degenerate adaptor sequences for detecting PCR duplicates in reduced representation sequencing data improve genotype calling accuracy. Mol. Ecol. Resour. 2015, 15, 329–336. [Google Scholar] [CrossRef]
- Jones, M.R.; Good, J.M. Targeted capture in evolutionary and ecological genomics. Mol. Ecol. 2016, 25, 185–202. [Google Scholar] [CrossRef]
- Hosner, P.A.; Faircloth, B.C.; Glenn, T.C.; Braun, E.L.; Kimball, R.T. Avoiding missing data biases in phylogenomic inference: An empirical study in the landfowl. Mol. Biol. Evol. 2016, 33, 1110–1125. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.E.; Tsai, W.L.E.; Faircloth, B.C. Sequence capture of ultraconserved elements from bird museum specimens. Mol. Ecol. Resour. 2016, 16, 1189–1203. [Google Scholar] [CrossRef] [PubMed]
- Ruane, S.; Austin, C.C. Phylogenomics using formalin-fixed and 100+ year-old intractable natural history specimens. Mol. Ecol. Resour. 2017, 17, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Bi, K.; Vanderpool, D.; Singhal, S.; Linderoth, T.; Moritz, C.; Good, J.M. Transcriptome-based exon capture enables highly cost-effective comparative genomic data collection at moderate evolutionary scales. BMC Genom. 2012, 13, 403. [Google Scholar] [CrossRef] [PubMed]
- Ali, O.A.; O’Rourke, S.M.; Amish, S.J.; Meek, M.H.; Luikart, G.; Jeffres, C.; Miller, M.R. RAD capture (Rapture): Flexible and efficient sequence-based genotyping. Genetics 2016, 202, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Hoffberg, S.L.; Kieran, T.J.; Catchen, J.M.; Devault, A.; Faircloth, B.C.; Mauricio, R.; Glenn, T.C. RADcap: Sequence capture of dual-digest RADseq libraries with identifiable duplicates and reduced missing data. Mol. Ecol. Resour. 2016, 16, 1264–1278. [Google Scholar] [CrossRef] [PubMed]
- Glenn, T.C.; Faircloth, B.C. Capturing Darwin’s dream. Mol. Ecol. Resour. 2016, 16, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.J. Bird sequencing project takes off. Nature 2015, 522, 34. [Google Scholar] [CrossRef]
- Lamichhaney, S.; Berglund, J.; Almen, M.S.; Maqbool, K.; Grabherr, M.; Martinez-Barrio, A.; Promerova, M.; Rubin, C.J.; Wang, C.; Zamani, N.; et al. Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature 2015, 518, 371–375. [Google Scholar] [CrossRef]
- Seki, R.; Li, C.; Fang, Q.; Hayashi, S.; Egawa, S.; Hu, J.; Xu, L.H.; Pan, H.L.; Kondo, M.; Sato, T.; et al. Functional roles of Aves class-specific cis-regulatory elements on macroevolution of bird-specific features. Nat. Commun. 2017, 8, 14229. [Google Scholar] [CrossRef]
- Thomas, G.H. Evolution: An avian explosion. Nature 2015, 526, 516–517. [Google Scholar] [CrossRef] [PubMed]
- Moyle, R.G.; Filardi, C.E.; Smith, C.E.; Diamond, J. Explosive Pleistocene diversification and hemispheric expansion of a “Great speciator”. Proc. Natl. Acad. Sci. USA 2009, 106, 1863–1868. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, K.A.; Fabre, P.H.; Ricklefs, R.E.; Fjeldsa, J. Major global radiation of corvoid birds originated in the proto-Papuan archipelago. Proc. Natl. Acad. Sci. USA 2011, 108, 2328–2333. [Google Scholar] [CrossRef] [PubMed]
- Kimball, R.T.; Braun, E.L. Does more sequence data improve estimates of galliform phylogeny? Analyses of a rapid radiation using a complete data matrix. PeerJ 2014, 2, e361. [Google Scholar] [CrossRef] [PubMed]
- Provost, K.L.; Joseph, L.; Smith, B.T. Resolving a phylogenetic hypothesis for parrots: Implications from systematics to conservation. Emu 2018, 118, 7–21. [Google Scholar] [CrossRef]
- McCormack, J.E.; Harvey, M.G.; Faircloth, B.C.; Crawford, N.G.; Glenn, T.C.; Brumfield, R.T. A phylogeny of birds based on over 1,500 loci collected by target enrichment and high-throughput sequencing. PLoS ONE 2013, 8, e54848. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.W.; Faircloth, B.C.; Nabholz, B.; Howard, J.T.; et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef]
- Prum, R.O.; Berv, J.S.; Dornburg, A.; Field, D.J.; Townsend, J.P.; Lemmon, E.M.; Lemmon, A.R. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature 2015, 526, 569–573. [Google Scholar] [CrossRef]
- Reddy, S.; Kimball, R.T.; Pandey, A.; Hosner, P.A.; Braun, M.J.; Hackett, S.J.; Han, K.L.; Harshman, J.; Huddleston, C.J.; Kingston, S.; et al. Why do phylogenomic data sets yield conflicting trees? Data type influences the avian tree of life more than taxon sampling. Syst. Biol. 2017, 66, 857–879. [Google Scholar] [CrossRef]
- Sun, K.P.; Meiklejohn, K.A.; Faircloth, B.C.; Glenn, T.C.; Braun, E.L.; Kimball, R.T. The evolution of peafowl and other taxa with ocelli (eyespots): A phylogenomic approach. Proc. R. Soc. B Biol. Sci. 2014, 281, 20140823. [Google Scholar] [CrossRef]
- Moyle, R.G.; Oliveros, C.H.; Andersen, M.J.; Hosner, P.A.; Benz, B.W.; Manthey, J.D.; Travers, S.L.; Brown, R.M.; Faircloth, B.C. Tectonic collision and uplift of Wallacea triggered the global songbird radiation. Nat. Commun. 2016, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hosner, P.A.; Tobias, J.A.; Braun, E.L.; Kimball, R.T. How do seemingly non-vagile clades accomplish trans-marine dispersal? Trait and dispersal evolution in the landfowl. Proc. R. Soc. B Biol. Sci. 2017, 284, 20170210. [Google Scholar] [CrossRef] [PubMed]
- Burleigh, J.G.; Kimball, R.T.; Braun, E.L. Building the avian tree of life using a large-scale sparse supermatrix. Mol. Phylogenet. Evol. 2015, 84, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.W.; Wang, N.; Smith, S.A. The development of scientific consensus: Analyzing conflict and concordance among avian phylogenies. Mol. Phylogenet. Evol. 2017, 116, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Driskell, A.C.; Ane, C.; Burleigh, J.G.; McMahon, M.M.; O’Meara, B.C.; Sanderson, M.J. Prospects for building the tree of life from large sequence databases. Science 2004, 306, 1172–1174. [Google Scholar] [CrossRef] [PubMed]
- de Queiroz, A.; Gatesy, J. The supermatrix approach to systematics. Trends Ecol. Evol. 2007, 22, 34–41. [Google Scholar] [CrossRef]
- Goloboff, P.A.; Catalano, S.A.; Miranda, J.M.; Szumik, C.A.; Arias, J.S.; Kallersjo, M.; Farris, J.S. Phylogenetic analysis of 73 060 taxa corroborates major eukaryotic groups. Cladistics 2009, 25, 211–230. [Google Scholar] [CrossRef]
- Sanderson, M.J.; Purvis, A.; Henze, C. Phylogenetic supertrees: Assembling the trees of life. Trends Ecol. Evol. 1998, 13, 105–109. [Google Scholar] [CrossRef]
- Bininda-Emonds, O.R.P.; Cardillo, M.; Jones, K.E.; MacPhee, R.D.E.; Beck, R.M.D.; Grenyer, R.; Price, S.A.; Vos, R.A.; Gittleman, J.L.; Purvis, A. The delayed rise of present-day mammals. Nature 2007, 446, 507–512. [Google Scholar] [CrossRef]
- Cotton, J.A.; Wilkinson, M. Supertrees join the mainstream of phylogenetics. Trends Ecol. Evol. 2009, 24, 1–3. [Google Scholar] [CrossRef]
- Warnow, T. Supertree construction: Opportunities and challenges. arXiv 2018, arXiv:1805.03530. [Google Scholar]
- Smith, S.A.; Beaulieu, J.M.; Donoghue, M.J. Mega-phylogeny approach for comparative biology: An alternative to supertree and supermatrix approaches. BMC Evol. Biol. 2009, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.H.; Hartmann, K.; Jetz, W.; Joy, J.B.; Mimoto, A.; Mooers, A.O. Pastis: An R package to facilitate phylogenetic assembly with soft taxonomic inferences. Methods Ecol. Evol. 2013, 4, 1011–1017. [Google Scholar] [CrossRef]
- Jetz, W.; Thomas, G.H.; Joy, J.B.; Hartmann, K.; Mooers, A.O. The global diversity of birds in space and time. Nature 2012, 491, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Faircloth, B.C.; McCormack, J.E.; Crawford, N.G.; Harvey, M.G.; Brumfield, R.T.; Glenn, T.C. Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Syst. Biol. 2012, 61, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.J.; Haddrath, O.; McPherson, J.D.; Cloutier, A. Genomic support for a moa-tinamou clade and adaptive morphological convergence in flightless ratites. Mol. Biol. Evol. 2014, 31, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- Bryson, R.W.; Faircloth, B.C.; Tsai, W.L.E.; McCormack, J.E.; Klicka, J. Target enrichment of thousands of ultraconserved elements sheds new light on early relationships within new world sparrows (Aves: Passerellidae). Auk 2016, 133, 451–458. [Google Scholar] [CrossRef]
- Hosner, P.A.; Braun, E.L.; Kimball, R.T. Rapid and recent diversification of curassows, guans, and chachalacas (Galliformes: Cracidae) out of Mesoamerica: Phylogeny inferred from mitochondrial, intron, and ultraconserved element sequences. Mol. Phylogenet. Evol. 2016, 102, 320–330. [Google Scholar] [CrossRef]
- Manthey, J.D.; Campillo, L.C.; Burns, K.J.; Moyle, R.G. Comparison of target-capture and restriction-site associated DNA sequencing for phylogenomics: A test in cardinalid tanagers (Aves, genus: Piranga). Syst. Biol. 2016, 65, 640–650. [Google Scholar] [CrossRef]
- Meiklejohn, K.A.; Faircloth, B.C.; Glenn, T.C.; Kimball, R.T.; Braun, E.L. Analysis of a rapid evolutionary radiation using ultraconserved elements: Evidence for a bias in some multispecies coalescent methods. Syst. Biol. 2016, 65, 612–627. [Google Scholar] [CrossRef]
- Ottenburghs, J.; Megens, H.J.; Kraus, R.H.S.; Madsen, O.; van Hooft, P.; van Wieren, S.E.; Crooijmans, R.P.M.A.; Ydenberg, R.C.; Groenen, M.A.M.; Prins, H.H.T. A tree of geese: A phylogenomic perspective on the evolutionary history of true geese. Mol. Phylogenet. Evol. 2016, 101, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Persons, N.W.; Hosner, P.A.; Meiklejohn, K.A.; Braun, E.L.; Kimball, R.T. Sorting out relationships among the grouse and ptarmigan using intron, mitochondrial, and ultra-conserved element sequences. Mol. Phylogenet. Evol. 2016, 98, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Zarza, E.; Faircloth, B.C.; Tsai, W.L.E.; Bryson, R.W.; Klicka, J.; Mccormack, J.E. Hidden histories of gene flow in highland birds revealed with genomic markers. Mol. Ecol. 2016, 25, 5144–5157. [Google Scholar] [CrossRef] [PubMed]
- Burga, A.; Wang, W.G.; Ben-David, E.; Wolf, P.C.; Ramey, A.M.; Verdugo, C.; Lyons, K.; Parker, P.G.; Kruglyak, L. A genetic signature of the evolution of loss of flight in the Galapagos cormorant. Science 2017, 356, eaal3345. [Google Scholar] [CrossRef]
- Wang, N.; Hosner, P.A.; Liang, B.; Braun, E.L.; Kimball, R.T. Historical relationships of three enigmatic phasianid genera (Aves: Galliformes) inferred using phylogenomic and mitogenomic data. Mol. Phylogenet. Evol. 2017, 109, 217–225. [Google Scholar] [CrossRef]
- White, N.D.; Mitter, C.; Braun, M.J. Ultraconserved elements resolve the phylogeny of potoos (Aves: Nyctibiidae). J. Avian Biol. 2017, 48, 872–880. [Google Scholar] [CrossRef]
- Yonezawa, T.; Segawa, T.; Mori, H.; Campos, P.F.; Hongoh, Y.; Endo, H.; Akiyoshi, A.; Kohno, N.; Nishida, S.; Wu, J.Q.; et al. Phylogenomics and morphology of extinct paleognaths reveal the origin and evolution of the ratites. Curr. Biol. 2017, 27, 68–77. [Google Scholar] [CrossRef]
- Andersen, M.J.; McCullough, J.M.; Mauck, W.M.; Smith, B.T.; Moyle, R.G. A phylogeny of kingfishers reveals an Indomalayan origin and elevated rates of diversification on oceanic islands. J. Biogeogr. 2018, 45, 269–281. [Google Scholar] [CrossRef]
- Bruxaux, J.; Gabrielli, M.; Ashari, H.; Prys-Jones, R.; Joseph, L.; Mila, B.; Besnard, G.; Thebaud, C. Recovering the evolutionary history of crowned pigeons (Columbidae: Goura): Implications for the biogeography and conservation of New Guinean lowland birds. Mol. Phylogenet. Evol. 2018, 120, 248–258. [Google Scholar] [CrossRef]
- Campillo, L.C.; Oliveros, C.H.; Sheldon, F.H.; Moyle, R.G. Genomic data resolve gene tree discordance in spiderhunters (Nectariniidae, Arachnothera). Mol. Phylogenet. Evol. 2018, 120, 151–157. [Google Scholar] [CrossRef]
- Chen, D.; Braun, E.L.; Forthman, M.; Kimball, R.T.; Zhang, Z.W. A simple strategy for recovering ultraconserved elements, exons, and introns from low coverage shotgun sequencing of museum specimens: Placement of the partridge genus Tropicoperdix within the Galliformes. Mol. Phylogenet. Evol. 2018, 129, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Musher, L.J.; Cracraft, J. Phylogenomics and species delimitation of a complex radiation of Neotropical suboscine birds (Pachyramphus). Mol. Phylogenet. Evol. 2018, 118, 204–221. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.T.; Mauck III, W.M.; Benz, B.; Andersen, M.J. Uneven missing data skews phylogenomic relationships within the lories and lorikeets. Biorxiv 2018. [Google Scholar] [CrossRef]
- Younger, J.L.; Strozier, L.; Maddox, J.D.; Nyari, A.S.; Bonfitto, M.T.; Raherilalao, M.J.; Goodman, S.M.; Reddy, S. Hidden diversity of forest birds in Madagascar revealed using integrative taxonomy. Mol. Phylogenet. Evol. 2018, 124, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Sackton, T.; Grayson, P.; Cloutier, A.; Hu, Z.; Liu, J.; Wheeler, N.; Gardner, P.; Clarke, J.; Baker, A.; Clamp, M.; et al. Convergent regulatory evolution and loss of flight in paleognathous birds. Science 2019, 364, 74. [Google Scholar] [CrossRef] [PubMed]
- Dobrin, B.H.; Zwickl, D.J.; Sanderson, M.J. The prevalence of terraced treescapes in analyses of phylogenetic data sets. BMC Evol. Biol. 2018, 18, 46. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, M.J.; McMahon, M.M.; Stamatakis, A.; Zwickl, D.J.; Steel, M. Impacts of terraces on phylogenetic inference. Syst. Biol. 2015, 64, 709–726. [Google Scholar] [CrossRef]
- Sanderson, M.J.; McMahon, M.M.; Steel, M. Terraces in phylogenetic tree space. Science 2011, 333, 448–450. [Google Scholar] [CrossRef]
- Kimball, R.T.; Braun, E.L.; Barker, F.K.; Bowie, R.C.K.; Braun, M.J.; Chojnowski, J.L.; Hackett, S.J.; Han, K.L.; Harshman, J.; Heimer-Torres, V.; et al. A well-tested set of primers to amplify regions spread across the avian genome. Mol. Phylogenet. Evol. 2009, 50, 654–660. [Google Scholar] [CrossRef]
- Gill, F.; Donsker, D. IOC World Bird List, 7.3. Available online: https://www.worldbirdnames.org/ (accessed on 5 August 2017).
- Baum, B.R. Combining trees as a way of combining data sets for phylogenetic inference, and the desirability of combining gene trees. Taxon 1992, 41, 3–10. [Google Scholar] [CrossRef]
- Ragan, M.A. Matrix representation in reconstructing phylogenetic-relationships among the eukaryotes. Biosystems 1992, 28, 47–55. [Google Scholar] [CrossRef]
- Creevey, C.J.; McInerney, J.O. Clann: Investigating phylogenetic information through supertree analyses. Bioinformatics 2005, 21, 390–392. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. PAUP*. Available online: http://paup.phylosolutions.com/ (accessed on 2 August 2018).
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Nixon, K.C. The parsimony ratchet, a new method for rapid parsimony analysis. Cladistics 1999, 15, 407–414. [Google Scholar] [CrossRef]
- Yuri, T.; Kimball, R.T.; Harshman, J.; Bowie, R.C.; Braun, M.J.; Chojnowski, J.L.; Han, K.L.; Hackett, S.J.; Huddleston, C.J.; Moore, W.S.; et al. Parsimony and model-based analyses of indels in avian nuclear genes reveal congruent and incongruent phylogenetic signals. Biology 2013, 2, 419–444. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.; Mirarab, S.; Warnow, T. MRL and superfine plus MRL: New supertree methods. Algorithms Mol. Biol. 2012, 7, 3. [Google Scholar]
- Cavender, J.A. Taxonomy with confidence. Math. Biosci. 1978, 40, 271–280. [Google Scholar] [CrossRef]
- Farris, J.S. Probability model for inferring evolutionary trees. Syst. Zool. 1973, 22, 250–256. [Google Scholar] [CrossRef]
- Neyman, J. A source of novel statistical problems. In Molecular Studies of Evolution: A Source of Novel Statistical Problems; Gupta, S.S., Yackel, J., Eds.; Academic Press: New York, NY, USA, 1971; pp. 1–27. [Google Scholar]
- Bininda-Emonds, O.R.P. Novel versus unsupported clades: Assessing the qualitative support for clades in MRP supertrees. Syst. Biol. 2003, 52, 839–848. [Google Scholar] [PubMed]
- Wilkinson, M.; Pisani, D.; Cotton, J.A.; Corfe, I. Measuring support and finding unsupported relationships in supertrees. Syst. Biol. 2005, 54, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Burleigh, J.G.; Driskell, A.C.; Sanderson, M.J. Supertree bootstrapping methods for assessing phylogenetic variation among genes in genome-scale data sets. Syst. Biol. 2006, 55, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.R.; Smith, S.A.; Donoghue, M.J. Increasing data transparency and estimating phylogenetic uncertainty in supertrees: Approaches using nonparametric bootstrapping. Syst. Biol. 2006, 55, 662–676. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Gil, M.; Dufayard, J.F.; Dessimoz, C.; Gascuel, O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst. Biol. 2011, 60, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.F.; Foulds, L.R. Comparison of phylogenetic trees. Math. Biosci. 1981, 53, 131–147. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Ostell, J.; Pruitt, K.D.; Sayers, E.W. Genbank. Nucleic Acids Res. 2018, 46, D41–D47. [Google Scholar] [CrossRef] [PubMed]
- Meiklejohn, K.A.; Danielson, M.J.; Faircloth, B.C.; Glenn, T.C.; Braun, E.L.; Kimball, R.T. Incongruence among different mitochondrial regions: A case study using complete mitogenomes. Mol. Phylogenet. Evol. 2014, 78, 314–323. [Google Scholar] [CrossRef]
- Tamashiro, R.A.; White, N.D.; Braun, M.J.; Faircloth, B.C.; Braun, E.L.; Kimball, R.T. What are the roles of taxon sampling and model fit in tests of cyto-nuclear discordance using avian mitogenomic data? Mol. Phylogenet. Evol. 2019, 130, 132–142. [Google Scholar] [CrossRef]
- Ho, S.Y.W.; Duchene, S. Molecular-clock methods for estimating evolutionary rates and timescales. Mol. Ecol. 2014, 23, 5947–5965. [Google Scholar] [CrossRef]
- Smith, S.A.; O’Meara, B.C. Treepl: Divergence time estimation using penalized likelihood for large phylogenies. Bioinformatics 2012, 28, 2689–2690. [Google Scholar] [CrossRef]
- Sanderson, M.J. Estimating absolute rates of molecular evolution and divergence times: A penalized likelihood approach. Mol. Biol. Evol. 2002, 19, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Parham, J.F.; Donoghue, P.C.J.; Bell, C.J.; Calway, T.D.; Head, J.J.; Holroyd, P.A.; Inoue, J.G.; Irmis, R.B.; Joyce, W.G.; Ksepka, D.T.; et al. Best practices for justifying fossil calibrations. Syst. Biol. 2012, 61, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Claramunt, S.; Cracraft, J. A new time tree reveals earth history’s imprint on the evolution of modern birds. Sci. Adv. 2015, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Cracraft, J.; Houde, P.; Ho, S.Y.W.; Mindell, D.P.; Fjeldsa, J.; Lindow, B.; Edwards, S.V.; Rahbek, C.; Mirarab, S.; Warnow, T.; et al. Response to comment on “Whole-genome analyses resolve early branches in the tree of life of modern birds”. Science 2015, 349, 3. [Google Scholar] [CrossRef] [PubMed]
- Bininda-Emonds, O.R.P. Phylogenetic Supertrees: Combining Information to Reveal the Tree of Life; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2004; Volume 4. [Google Scholar]
- Hinchliff, C.E.; Smith, S.A. Some limitations of public sequence data for phylogenetic inference (in plants). PLoS ONE 2014, 9, e98986. [Google Scholar] [CrossRef]
- Philippe, H.; de Vienne, D.M.; Ranwez, V.; Roure, B.; Baurain, D.; Delsuc, F. Pitfalls in supermatrix phylogenomics. Eur. J. Taxon. 2017, 283, 1–25. [Google Scholar] [CrossRef]
- Goloboff, P.A. Parsimony, likelihood, and simplicity. Cladistics Int. J. Willi Hennig Soc. 2003, 19, 91–103. [Google Scholar] [CrossRef]
- Sanderson, M.J.; Kim, J. Parametric phylogenetics? Syst. Biol. 2000, 49, 817–829. [Google Scholar] [CrossRef]
- Redelings, B.D.; Holder, M.T. A supertree pipeline for summarizing phylogenetic and taxonomic information for millions of species. PeerJ 2017, 5, e3058. [Google Scholar] [CrossRef]
- Swenson, M.S.; Suri, R.; Linder, C.R.; Warnow, T. Superfine: Fast and accurate supertree estimation. Syst. Biol. 2012, 61, 214–227. [Google Scholar] [CrossRef]
- Gatesy, J.; Matthee, C.; DeSalle, R.; Hayashi, C. Resolution of a supertree/supermatrix paradox. Syst. Biol. 2002, 51, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Gatesy, J.; Baker, R.H. Hidden likelihood support in genomic data: Can forty-five wrongs make a right? Syst. Biol. 2005, 54, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Gatesy, J.; O’Grady, P.; Baker, R.H. Corroboration among data sets in simultaneous analysis: Hidden support for phylogenetic relationships among higher level artiodactyl taxa. Cladistics 1999, 15, 271–313. [Google Scholar] [CrossRef]
- Hinchliff, C.E.; Smith, S.A.; Allman, J.F.; Burleigh, J.G.; Chaudhary, R.; Coghill, L.M.; Crandall, K.A.; Deng, J.; Drew, B.T.; Gazis, R.; et al. Synthesis of phylogeny and taxonomy into a comprehensive tree of life. Proc. Natl. Acad. Sci. USA 2015, 112, 12764–12769. [Google Scholar] [CrossRef] [PubMed]
- Barker, F.K.; Burns, K.J.; Klicka, J.; Lanyon, S.M.; Lovette, I.J. New insights into new world biogeography: An integrated view from the phylogeny of blackbirds, cardinals, sparrows, tanagers, warblers, and allies. Auk 2015, 132, 333–348. [Google Scholar] [CrossRef]
- Hosner, P.A.; Braun, E.L.; Kimball, R.T. Land connectivity changes and global cooling shaped the colonization history and diversification of New World quail (Aves: Galliformes: Odontophoridae). J. Biogeogr. 2015, 42, 1883–1895. [Google Scholar] [CrossRef]
- Wang, N.; Kimball, R.T.; Braun, E.L.; Liang, B.; Zhang, Z.W. Ancestral range reconstruction of Galliformes: The effects of topology and taxon sampling. J. Biogeogr. 2017, 44, 122–135. [Google Scholar] [CrossRef]
- Townsend, J.P. Profiling phylogenetic informativeness. Syst. Biol. 2007, 56, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Duchêne, D.A.; Duchêne, S.; Ho, S.Y. Differences in performance among test statistics for assessing phylogenomic model adequacy. Genome Biol. Evol. 2018, 10, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Sayyari, E.; Mirarab, S. Fast coalescent-based computation of local branch support from quartet frequencies. Mol. Biol. Evol. 2016, 33, 1654–1668. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.V.; Xi, Z.X.; Janke, A.; Faircloth, B.C.; McCormack, J.E.; Glenn, T.C.; Zhong, B.J.; Wu, S.Y.; Lemmon, E.M.; Lemmon, A.R.; et al. Implementing and testing the multispecies coalescent model: A valuable paradigm for phylogenomics. Mol. Phylogenet. Evol. 2016, 94, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Mirarab, S.; Bayzid, M.S.; Warnow, T. Evaluating summary methods for multilocus species tree estimation in the presence of incomplete lineage sorting. Syst. Biol. 2016, 65, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Bininda-Emonds, O.R.P.; Gittleman, J.L.; Purvis, A. Building large trees by combining phylogenetic information: A complete phylogeny of the extant Carnivora (Mammalia). Biol. Rev. 1999, 74, 143–175. [Google Scholar] [CrossRef] [PubMed]
- Purvis, A. A composite estimate of primate phylogeny. Philos. Trans. R. Soc. Lond. 1995, 348, 405–421. [Google Scholar]
- Moles, A.T.; Ackerly, D.D.; Webb, C.O.; Tweddle, J.C.; Dickie, J.B.; Westoby, M. A brief history of seed size. Science 2005, 307, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Torices, R. Adding time-calibrated branch lengths to the Asteraceae supertree. J. Syst. Evol. 2010, 48, 271–278. [Google Scholar] [CrossRef]
- Webb, C.O.; Ackerly, D.D.; Kembel, S.W. Phylocom: Software for the analysis of phylogenetic community structure and trait evolution. Bioinformatics 2008, 24, 2098–2100. [Google Scholar] [CrossRef]
- do Amaral, F.R.; Neves, L.G.; Resende, M.F.R.; Mobili, F.; Miyaki, C.Y.; Pellegrino, K.C.M.; Biondo, C. Ultraconserved elements sequencing as a low-cost source of complete mitochondrial genomes and microsatellite markers in non-model amniotes. PLoS ONE 2015, 10, e0138446. [Google Scholar] [CrossRef]
- Barker, F.K.; Oyler-McCance, S.; Tomback, D.F. Blood from a turnip: Tissue origin of low-coverage shotgun sequencing libraries affects recovery of mitogenome sequences. Mitochondrial DNA 2015, 26, 384–388. [Google Scholar] [CrossRef]
- Reddy, S. What’s missing from avian global diversification analyses? Mol. Phylogenet. Evol. 2014, 77, 159–165. [Google Scholar] [CrossRef]
- Berv, J.S.; Field, D.J. Genomic signature of an avian Lilliput effect across the K-Pg extinction. Syst. Biol. 2018, 67, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ksepka, D.T.; Phillips, M.J. Avian diversification patterns across the K-Pg boundary: Influence of calibrations, datasets, and model misspecification. Ann. Mo. Bot. Gard. 2015, 100, 300–328. [Google Scholar] [CrossRef]
- Cooper, A.; Penny, D. Mass survival of birds across the Cretaceous-Tertiary boundary: Molecular evidence. Science 1997, 275, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.J.; Cooper, A.; Phillips, M.J. Comment on “Whole-genome analyses resolve early branches in the tree of life of modern birds”. Science 2015, 349, 1460. [Google Scholar] [CrossRef] [PubMed]
- Field, D.J.; Bercovici, A.; Berv, J.S.; Dunn, R.; Fastovsky, D.E.; Lyson, T.R.; Vajda, V.; Gauthier, J.A. Early evolution of modern birds structured by global forest collapse at the end-Cretaceous mass extinction. Curr. Biol. 2018, 28, 1825–1831. [Google Scholar] [CrossRef]
- Longrich, N.R.; Tokaryk, T.; Field, D.J. Mass extinction of birds at the Cretaceous-Paleogene (K-Pg) boundary. Proc. Natl. Acad. Sci. USA 2011, 108, 15253–15257. [Google Scholar] [CrossRef]
- Barrowclough, G.F.; Cracraft, J.; Klicka, J.; Zink, R.M. How many kinds of birds are there and why does it matter? PLoS ONE 2016, 11, e0166307. [Google Scholar] [CrossRef]
- Gill, F.B. Species taxonomy of birds: Which null hypothesis? Auk 2014, 131, 150–161. [Google Scholar] [CrossRef]
- Matasci, N.; Hung, L.H.; Yan, Z.X.; Carpenter, E.J.; Wickett, N.J.; Mirarab, S.; Nguyen, N.; Warnow, T.; Ayyampalayam, S.; Barker, M.; et al. Data access for the 1,000 plants (1KP) project. Gigascience 2014, 3, 17. [Google Scholar] [CrossRef]
- Robinson, G.E.; Hackett, K.J.; Purcell-Miramontes, M.; Brown, S.J.; Evans, J.D.; Goldsmith, M.R.; Lawson, D.; Okamuro, J.; Robertson, H.M.; Schneider, D.J. Creating a buzz about insect genomes. Science 2011, 331, 1386. [Google Scholar] [CrossRef]
- Sun, Y.; Huang, Y.; Li, X.F.; Baldwin, C.C.; Zhou, Z.C.; Yan, Z.X.; Crandall, K.A.; Zhang, Y.; Zhao, X.M.; Wang, M.; et al. Fish-T1K (transcriptomes of 1,000 fishes) project: Large-scale transcriptome data for fish evolution studies. Gigascience 2016, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, E. Bigger, better bird tree of life will soon fly into view. Available online: https://www.sciencemag.org/news/2018/04/bigger-better-bird-tree-life-will-soon-fly-view/ (accessed on 16 April 2018).
- Worthy, T.H.; Hand, S.J.; Archer, M. Phylogenetic relationships of the Australian Oligo-Miocene ratite Emuarius gidju Casuariidae. Integr. Zool. 2014, 9, 148–166. [Google Scholar] [CrossRef] [PubMed]
- Woodburne, M.O.; Macfadden, B.J.; Case, J.A.; Springer, M.S.; Pledge, N.S.; Power, J.D.; Woodburne, J.M.; Springer, K.B. Land mammal biostratigraphy and magnetostratigraphy of the Etadunna Formation (late Oligocene) of south Australia. J. Vertebr. Paleontol. 1994, 13, 483–515. [Google Scholar] [CrossRef]
- Gradstein, F.; Ogg, J.; Smith, A. A Geologic Time Scale 2004; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Houde, P.W. Paleognathous Birds from the Early Tertiary of the Northern Hemisphere; Nuttall Ornithologcal Club: Cambridge, UK, 1988; Volume 22. [Google Scholar]
- Alvarenga, H.M.F. Uma ave ratite do Paleoceno brasileiro: Bacia calcária de Itaboraí, estado do Rio de Janeiro, Brasil. Bol. Mus. Nac. Rio J. 1983, 41, 1–47. [Google Scholar]
- Mayr, G.; Poschmann, M.; Wuttke, M. A nearly complete skeleton of the fossil galliform bird Palaeortyx from the late Oligocene of Germany. Acta Ornithol. 2006, 41, 129–135. [Google Scholar]
- Storch, G.; Engesser, B.; Wuttke, M. Oldest fossil record of gliding in rodents. Nature 1996, 379, 439–441. [Google Scholar] [CrossRef]
- Mayr, G. The Eocene Juncitarsus—Its phylogenetic position and significance for the evolution and higher-level affinities of flamingos and grebes. C. R. Palevol 2014, 13, 9–18. [Google Scholar] [CrossRef]
- Mertz, D.F.; Harms, F.-J.; Gabriel, G.; Felder, M. Arbeitstreffen in der Forschungsstation Grube Messel mit neven Egebrissen aus der Messel-Forschung. Nat. Und Mus. 2004, 134, 289–290. [Google Scholar]
- Franzen, J.F. The implications of the numerical dating of the Messel fossil deposit (Eocene, Germany) for mammalian biochronology. Ann. Paléontol. 2005, 91, 329–335. [Google Scholar] [CrossRef]
- Olson, S.L. A lower Eocene frigatebird from the Green River formation of Wyoming (Pelecaniformes, Fregatidae). Smithson. Contrib. Paleontol. 1977, 35, 1–33. [Google Scholar] [CrossRef]
- Mayr, G. The Palaeogene Old World potoo Paraprefica Mayr, 1999 (Aves, Nyctibiidae): Its osteology and affinities to the New World Preficinae Olson, 1987. J. Syst. Palaeontol. 2005, 3, 359–370. [Google Scholar] [CrossRef]
- Smith, M.E.; Chamberlain, K.R.; Singer, B.S.; Carroll, A.R. Eocene clocks agree: Coeval Ar-40/Ar-39, U-Pb, and astronomical ages from the Green River formation. Geology 2010, 38, 527–530. [Google Scholar] [CrossRef]
- Ksepka, D.T.; Clarke, J.A.; Nesbitt, S.J.; Kulp, F.B.; Grande, L. Fossil evidence of wing shape in a stem relative of swifts and hummingbirds (Aves, Pan-Apodiformes). Proc. R. Soc. B Biol. Sci. 2013, 280, 20130580. [Google Scholar] [CrossRef] [PubMed]
- Mayr, G. A new Eocene swift-like bird with a peculiar feathering. Ibis 2003, 145, 382–391. [Google Scholar] [CrossRef]
- Mayr, G. A new cypselomorph bird from the middle Eocene of Germany and the early diversification of avian aerial insectivores. Condor 2005, 107, 342–352. [Google Scholar] [CrossRef]
- Thiede, J.; Nielsen, O.B.; Perch-Nielsen, K. Lithofacies, mineralogy and biostratigraphy of Eocene sediments in northern Denmark (Deep test Viborg 1). Neues Jahrb. Geol. Paläontol. Abh. 1980, 160, 149–172. [Google Scholar]
- Milkovsky, J. Tertiary avian localities of Denmark. Acta Univ. Carol. Geol. 1996, 39, 559–562. [Google Scholar]
- Gradstein, F.M.; Ogg, J.G.; Hilgen, F.J. On the geologic time scale. Newsl. Stratigr. 2012, 45, 171–188. [Google Scholar] [CrossRef]
- Mayr, G. Phylogenetic relationships of the early tertiary Messel rails (Aves, Messelornithidae). Senckenberg. Lethaea 2004, 84, 317–322. [Google Scholar] [CrossRef]
- Bertelli, S.; Chiappe, L.M.; Mayr, G. A new Messel rail from the early Eocene Fur Formation of Denmark (Aves, Messelornithidae). J. Syst. Palaeontol. 2011, 9, 551–562. [Google Scholar] [CrossRef]
- Musser, G.; Ksepka, D.T.; Field, D.J. New material of Palaeocene-Eocene Pellornis (Aves: Gruiformes) clarifies pattern and timing of the extant gruiform radiation. Diversity 2019, 11, 102. [Google Scholar] [CrossRef]
- Chambers, L.; Pringle, M.; Fitton, G.; Larsen, L.M.; Pedersen, A.K.; Parrish, R. Recalibration of the Palaeocene-Eocene boundary (P-E) using high precision U-Pb and Ar-Ar isotopic datingIn Proceedings of the EGS-AGU-EUG Joint Assembly, Abstracts from the meeting, Nice, France, 6–11 April 2003.
- Feduccia, A.; Voorhies, M.R. Crowned cranes (Gruidae: Balearica) in the Miocene of Nebraska. Nat. Hist. Mus. Los Angel. Cty. Sci. Ser. 1992, 36, 239–248. [Google Scholar]
- Mayr, G. A survey of casques, frontal humps, and other extravagant bony cranial protuberances in birds. Zoomorphology 2018, 137, 457–472. [Google Scholar] [CrossRef]
- Boellstorf, J. Chronology of some late Cenozoic deposits from the central United States and the ice ages. Trans. Neb. Acad. Sci. 1978, 6, 35–49. [Google Scholar]
- De Pietri, V.L.; Costeur, L.; Guntert, M.; Mayr, G. A revision of the Lari (Aves: Charadriiformes) from the early Miocene of Saint-Gérand-le-Puy (Allier, France). J. Vertebr. Paleontol. 2011, 31, 812–828. [Google Scholar] [CrossRef]
- Smith, N.A. Sixteen vetted fossil calibrations for divergence dating of Charadriiformes (Aves, Neognathae). Palaeontol. Electron. 2015, 18, 1–18. [Google Scholar] [CrossRef]
- Mayr, G. Charadriiform birds from the early Oligocene of Cereste (France) and the middle Eocene of Messel (Hessen, Germany). Geobios 2000, 33, 625–636. [Google Scholar] [CrossRef]
- Bourdon, E.; Mourer-Chauvire, C.; Amaghzaz, M.; Bouya, B. New specimens of Lithoptila abdounensis (Aves, Prophaethontidae) from the lower Paleogene of Morocco. J. Vertebr. Paleontol. 2008, 28, 751–761. [Google Scholar] [CrossRef]
- Smith, N.D. Phylogenetic analysis of Pelecaniformes (Aves) based on osteological data: Implications for waterbird phylogeny and fossil calibration studies. PLoS ONE 2010, 5, e13354. [Google Scholar] [CrossRef]
- Mayr, G.; Scofield, R.P. New avian remains from the Paleocene of New Zealand: The first early Cenozoic Phaethontiformes (tropicbirds) from the Southern Hemisphere. J. Vertebr. Paleontol. 2016, 36, e1031343. [Google Scholar] [CrossRef]
- Mayr, G. A new skeleton of the late Oligocene “Enspel cormorant” from Oligocorax to Borvocarbo, and back again. Palaeobiodivers. Palaeoenviron. 2015, 95, 87–101. [Google Scholar] [CrossRef]
- Slack, K.E.; Jones, C.M.; Ando, T.; Harrison, G.L.; Fordyce, R.E.; Arnason, U.; Penny, D. Early penguin fossils, plus mitochondrial genomes, calibrate avian evolution. Mol. Biol. Evol. 2006, 23, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Ksepka, D.; Bertelli, S.; Norberto, G. The phylogeny of living and fossil Sphenisciformes (penguins). Cladistics 2006, 22, 412–441. [Google Scholar] [CrossRef]
- Cooper, R.A. The New Zealand Geological Timescale; Institute of Geological and Nuclear Sciences: Lower Hutt, New Zealand, 2004. [Google Scholar]
- Ogg, J.G.; Ogg, G.; Gradstein, F.M. The Concise Geological Time Scale; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]
- Smith, M.E.; Singer, B.S.; Carroll, A.R.; Fournelle, J.H. Precise dating of biotite in distal volcanic ash: Isolating subtle alteration using 40Ar/39Ar laser incremental heating and electron microprobe techniques. Am. Mineral. 2008, 93, 784–795. [Google Scholar] [CrossRef]
- Stidham, T.A. A new species of Limnofregata (Pelecaniformes: Fregatidae) from the early Eocene Wasatch Formation of Wyoming: Implications for palaeoecology and palaeobiology. Palaeontology 2015, 58, 239–249. [Google Scholar] [CrossRef]
- Mayr, G. The world’s smallest owl, the earliest unambiguous charadriiform bird, and other avian remains from the early Eocene Nanjemoy Formation of Virginia (USA). Palaeontol. Z. 2016, 90, 747–763. [Google Scholar] [CrossRef]
- Acosta Hospitaleche, C.; Tambussi, C.; Donato, M.; Cozzuol, M. A new Miocene penguin from Patagonia and its phylogenetic relationships. Acta Palaeontol. Pol. 2007, 52, 299–314. [Google Scholar]
- Ksepka, D.T.; Clarke, J.A. The basal penguin (Aves: Sphenisciformes) Perudyptes devriesi and a phylogenetic evaluation of the penguin fossil record. Bull. Am. Mus. Nat. Hist. 2010, 337, 1–77. [Google Scholar] [CrossRef]
- Chavez Hoffmeister, M. Phylogenetic characters in the humerus and tarsometatarsus of penguins. Pol. Polar Res. 2014, 35, 469–496. [Google Scholar] [CrossRef]
- Chavez Hoffmeister, M.; Briceno, J.D.C.; Nielsen, S.N. The evolution of seabirds in the Humboldt Current: New clues from the Pliocene of central Chile. PLoS ONE 2014, 9, e90043. [Google Scholar] [CrossRef]
- Degrange, F.J.; Ksepka, D.T.; Tambussi, C.P. Redescription of the oldest crown clade penguin: Cranial osteology, jaw myology, neuroanatomy, and phylogenetic affinities of Madrynornis mirandus. J. Vertebr. Paleontol. 2018, 38, e1445636. [Google Scholar] [CrossRef]
- Scasso, R.A.; McArthur, J.M.; del Rio, C.J.; Martinez, S.; Thirlwall, M.F. 87Sr/86Sr late Miocene age of fossil molluscs in the ‘Entrerriense’ of the Valdés Peninsula (Chubut, Argentina). J. S. Am. Earth Sci. 2001, 14, 319–329. [Google Scholar] [CrossRef]
- Mayr, G. Phylogenetic relationships of the paraphyletic ‘caprimulgiform’ birds (nightjars and allies). J. Zool. Syst. Evol. Res. 2010, 48, 126–137. [Google Scholar] [CrossRef]
- Mayr, G.; Bertelli, S. A record of Rhynchaeites (Aves, Threskiornithidae) from the early Eocene Fur Formation of Denmark, and the affinities of the alleged parrot Mopsitta. Palaeobiodivers. Palaeoenviron. 2011, 91, 229–236. [Google Scholar] [CrossRef]
- Field, D.J.; Hsiang, A.Y. A North American stem turaco, and the complex biogeographic history of modern birds. BMC Evol. Biol. 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Ksepka, D.T.; Stidham, T.A.; Williamson, T.E. Early Paleocene landbird supports rapid phylogenetic and morphological diversification of crown birds after the K-Pg mass extinction. Proc. Natl. Acad. Sci. USA 2017, 114, 8047–8052. [Google Scholar] [CrossRef] [PubMed]
- Mayr, G. A tiny barbet-like bird from the lower Oligocene of Germany: The smallest species and earliest substantial fossil record of the Pici (woodpeckers and allies). Auk 2005, 122, 1055–1063. [Google Scholar] [CrossRef]
- Mayr, G. First fossil skull of a Palaeogene representative of the Pici (woodpeckers and allies) and its evolutionary implications. Ibis 2006, 148, 824–827. [Google Scholar] [CrossRef]
- Micklich, N.; Hildebrandt, L. The Frauenweiler clay pit (“Grube Unterfeld”). Kaupia Darmstädter Beiträge Nat. 2005, 14, 113–118. [Google Scholar]
- Clarke, J.A.; Ksepka, D.T.; Smith, N.A.; Norell, M.A. Combined phylogenetic analysis of a new North American fossil species confirms widespread Eocene distribution for stem rollers (Aves, Coracii). Zool. J. Linn. Soc. 2009, 157, 586–611. [Google Scholar] [CrossRef]
- Mayr, G.; Mourer-Chauvire, C.; Weidig, I. Osteology and systematic position of the Eocene Primobucconidae (Aves, Coraciiformes sensu stricto), with first records from Europe. J. Syst. Palaeontol. 2004, 2, 1–12. [Google Scholar] [CrossRef]
- Houde, P.; Olson, S.L. Small arboreal nonpasserine birds from the early Tertiary of western North America. In Acta XIX Congressus Internationalis Ornithologici; Ouellet, H., Ed.; University of Ottawa Press: Ottawa, ON, Canada, 1989; pp. 2030–2036. [Google Scholar]
- Mayr, G.; Knopf, C.W. A tody (Alcediniformes: Todidae) from the early Oligocene of Germany. Auk 2007, 124, 1294–1304. [Google Scholar] [CrossRef]
- Mayr, G. A reassessment of Eocene parrotlike fossils indicates a previously undetected radiation of zygodactyl stem group representatives of passerines (Passeriformes). Zool. Scr. 2015, 44, 587–602. [Google Scholar] [CrossRef]
- Mayr, G. Phylogenetic affinities of the enigmatic avian taxon Zygodactylus based on new material from the early oligocene of France. J. Syst. Palaeontol. 2008, 6, 333–344. [Google Scholar] [CrossRef]
- Harrison, C.J.O.; Walker, C.A. Birds of the British Lower Eocene; Tertiary Research Special Paper; BRILL: Leiden, The Netherlands, 1977; pp. 1–52. [Google Scholar]
- Kristoffersen, A.V. The Avian Diversity in the Latest Paleocene Earliest Eocene Fur Formation, Denmark: A Synopsis; University of Copenhagen: Copenhagen, Denmark, 2002. [Google Scholar]
- Mayr, G. Paleogene Fossil Birds; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Mayr, G.; Manegold, A. A small suboscine-like passeriform bird from the early Oligocene of France. Condor 2006, 108, 717–720. [Google Scholar] [CrossRef]
- Manegold, A. Passerine diversity in the late Oligocene of Germany: Earliest evidence for the sympatric coexistence of suboscines and oscines. Ibis 2008, 150, 377–387. [Google Scholar] [CrossRef]
- Mayr, G.; Manegold, A. The oldest European fossil songbird from the early Oligocene of Germany. Naturwissenschaften 2004, 91, 173–177. [Google Scholar] [CrossRef]
- Ballmann, P. Die Vögel aus der altburdigalen Spaltenfüllung von Wintershof (West) bei Eichstätt in Bayern. Zitteliana 1969, 1, 5–60. [Google Scholar]
- Manegold, A.; Mayr, G.; Mourer-Chauvire, C. Miocene songbirds and the composition of the European passeriform avifauna. Auk 2004, 121, 1155–1160. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Focal Group | # of Species | Used as Source Tree? | Loci Targeted 2 |
|---|---|---|---|---|
| Faircloth et al. [55] | NEORNITHES | 9 | 2.5K UCE probe set | |
| McCormack et al. [36] | NEOAVES | 33 | YES | 2.5K UCE probe set |
| Baker et al. [56] | PALAEOGNATHAE | 7 | Subset of Faircloth et al. [55] loci | |
| Jarvis et al. [37] | NEORNITHES | 48 | YES | Whole genomes |
| Sun et al. [40] | Phasianidae (peafowl) | 15 | 5k UCE probe set | |
| Prum et al. [38] | NEORNITHES | 197 | YES | AHE probe set |
| Bryson et al. [57] | Passerellidae | 30 | YES | 5k UCE probe set |
| Hosner et al. [58] | Cracidae | 23 | 5k UCE probe set | |
| Hosner et al. [21] | Phasianidae | 90 | 5k UCE probe set | |
| Manthey et al. [59] | Piranga | 11 | YES | 5k UCE probe set |
| McCormack et al. [22] | Aphelocoma | 1 (3) | 5k UCE probe set | |
| Meiklejohn et al. [60] | Phasianidae (gallopheasants) | 18 | 5k UCE probe set | |
| Ottenburghs et al. [61] | Anatidae–Anserini | 19 | YES | Whole genomes |
| Persons et al. [62] | Phasianidae (grouse) | 11 | 5k UCE probe set | |
| Zarza et al. [63] | Aphelocoma | 3 | YES | 5k UCE probe set |
| Burga et al. [64] | Phalacrocorax | 7 | YES | Whole genomes |
| Hosner et al. [42] | Phasianidae | 115 | YES | 5k UCE probe set |
| Reddy et al. [39] | NEORNITHES | 235 | YES | legacy with data mining |
| Wang et al. [65] | Phasianidae | 20 | YES | 5k UCE probe set |
| White et al. [66] | Nyctibiidae | 12 | YES | 5k UCE probe set |
| Yonezawa et al. [67] | PALAEOGNATHAE | YES | legacy with data mining | |
| Andersen et al. [68] | Alcedinidae | 21 | YES | 5k UCE probe set |
| Bruxaux et al. [69] | Goura | 6 | YES | Subset of UCE and AHE loci |
| Campillo et al. [70] | Arachnothera | 17 | YES | 5k UCE probe set |
| Chen et al. [71] | Phasianidae | 27 | YES | 5k UCE probe set |
| Musher & Cracraft [72] | Pachyramphus | 18 | YES | 2.5K/5k UCE probe set |
| Smith et al. 2018 [73] | Psittaculidae–Loriini | 54 | YES | 5k UCE probe set |
| Younger et al. [74] | Newtonia | 4 | YES | 5k UCE probe set |
| Sackton et al. [75] | PALAEOGNATHAE | 15 | YES | Whole genomes |
| Backbone: | Resolved | % Branches | |||
|---|---|---|---|---|---|
| Method | BigBird | Brown | Jetz | Branches | Collapsed |
| MRP | + | + | + | 696 | 1.28% |
| MRP | + | 642 | 8.94% | ||
| MRP | + | 687 | 2.55% | ||
| MRP | + | 698 | 0.99% | ||
| MRP | + | + | 689 | 2.27% | |
| MRP | + | + | 691 | 1.99% | |
| MRP | + | + | 694 | 1.56% | |
| MRL | + | + | + | 704 | 0.14% |
| MRL | + | 690 | 2.13% | ||
| MRL | + | 698 | 0.99% | ||
| MRL | + | 704 | 0.14% | ||
| MRL | + | + | 703 | 0.28% | |
| MRL | + | + | 705 | 0.00% | |
| MRL | + | + | 703 | 0.28% | |
| MRP bootstrap | + | + | + | 703 | 0.28% |
| MRL bootstrap | + | + | + | 705 | 0.00% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimball, R.T.; Oliveros, C.H.; Wang, N.; White, N.D.; Barker, F.K.; Field, D.J.; Ksepka, D.T.; Chesser, R.T.; Moyle, R.G.; Braun, M.J.; et al. A Phylogenomic Supertree of Birds. Diversity 2019, 11, 109. https://doi.org/10.3390/d11070109
Kimball RT, Oliveros CH, Wang N, White ND, Barker FK, Field DJ, Ksepka DT, Chesser RT, Moyle RG, Braun MJ, et al. A Phylogenomic Supertree of Birds. Diversity. 2019; 11(7):109. https://doi.org/10.3390/d11070109
Chicago/Turabian StyleKimball, Rebecca T., Carl H. Oliveros, Ning Wang, Noor D. White, F. Keith Barker, Daniel J. Field, Daniel T. Ksepka, R. Terry Chesser, Robert G. Moyle, Michael J. Braun, and et al. 2019. "A Phylogenomic Supertree of Birds" Diversity 11, no. 7: 109. https://doi.org/10.3390/d11070109
APA StyleKimball, R. T., Oliveros, C. H., Wang, N., White, N. D., Barker, F. K., Field, D. J., Ksepka, D. T., Chesser, R. T., Moyle, R. G., Braun, M. J., Brumfield, R. T., Faircloth, B. C., Smith, B. T., & Braun, E. L. (2019). A Phylogenomic Supertree of Birds. Diversity, 11(7), 109. https://doi.org/10.3390/d11070109