The Signaling Cascades of Ginkgolide B-Induced Apoptosis in MCF-7 Breast Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
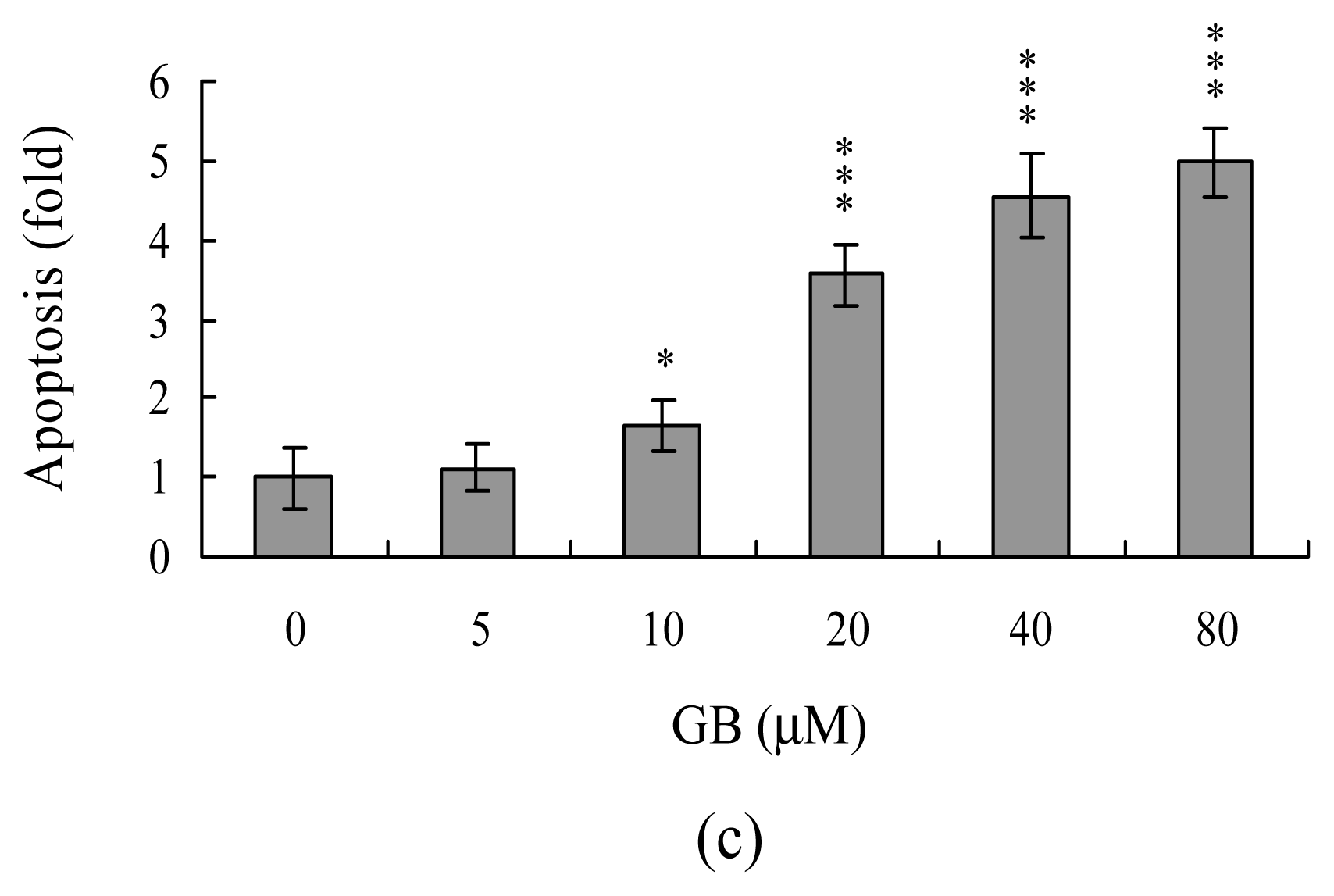
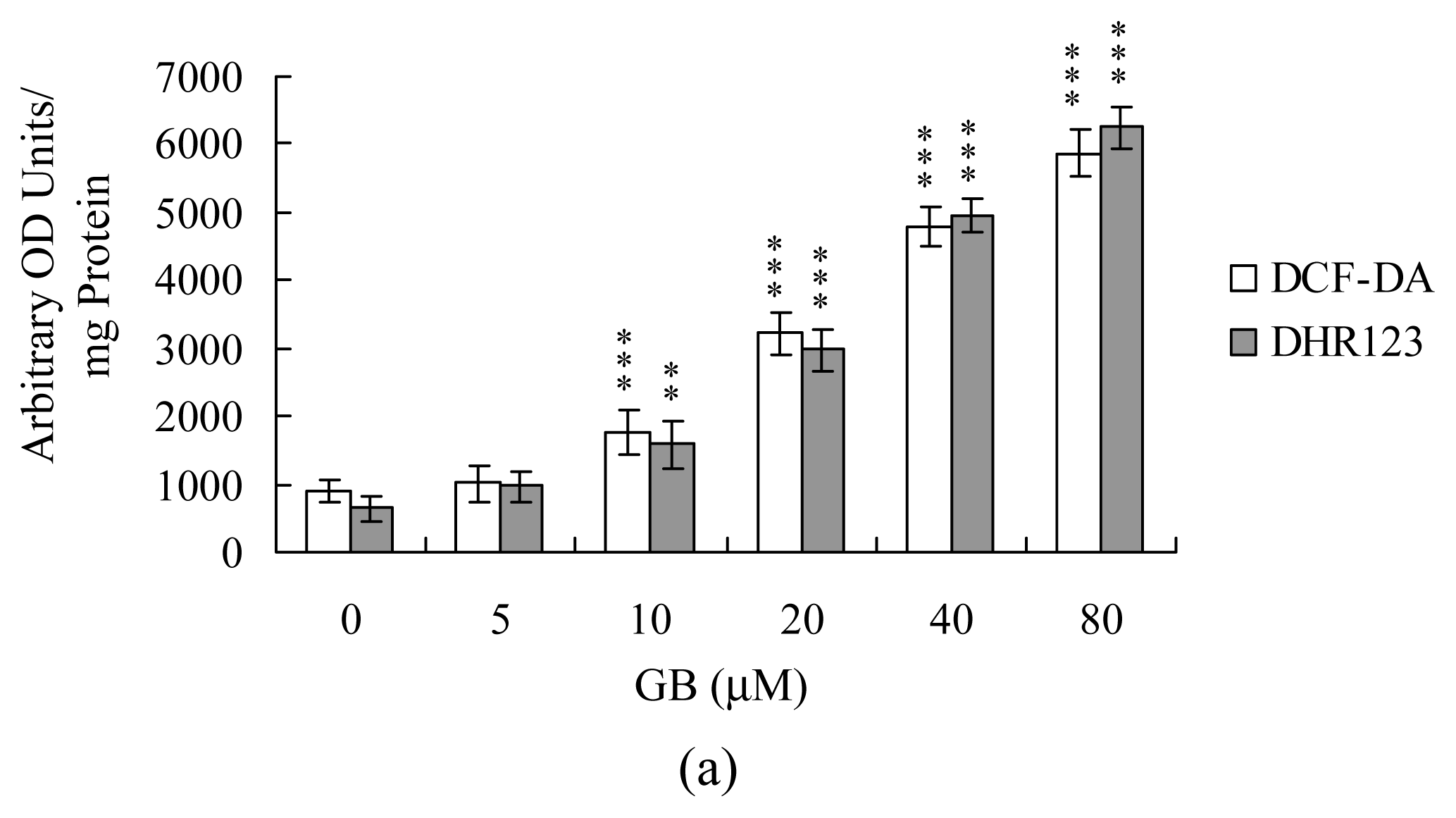
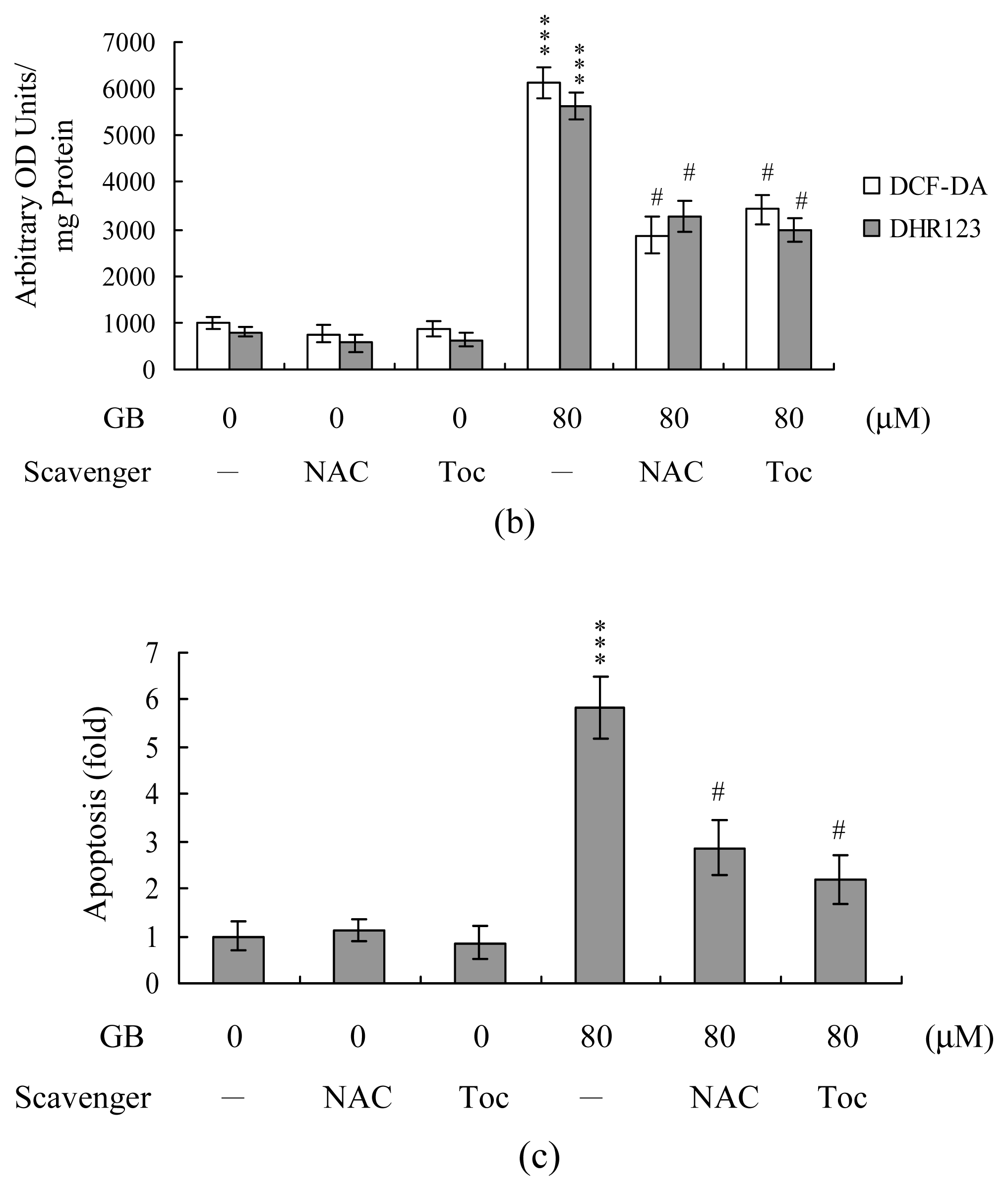
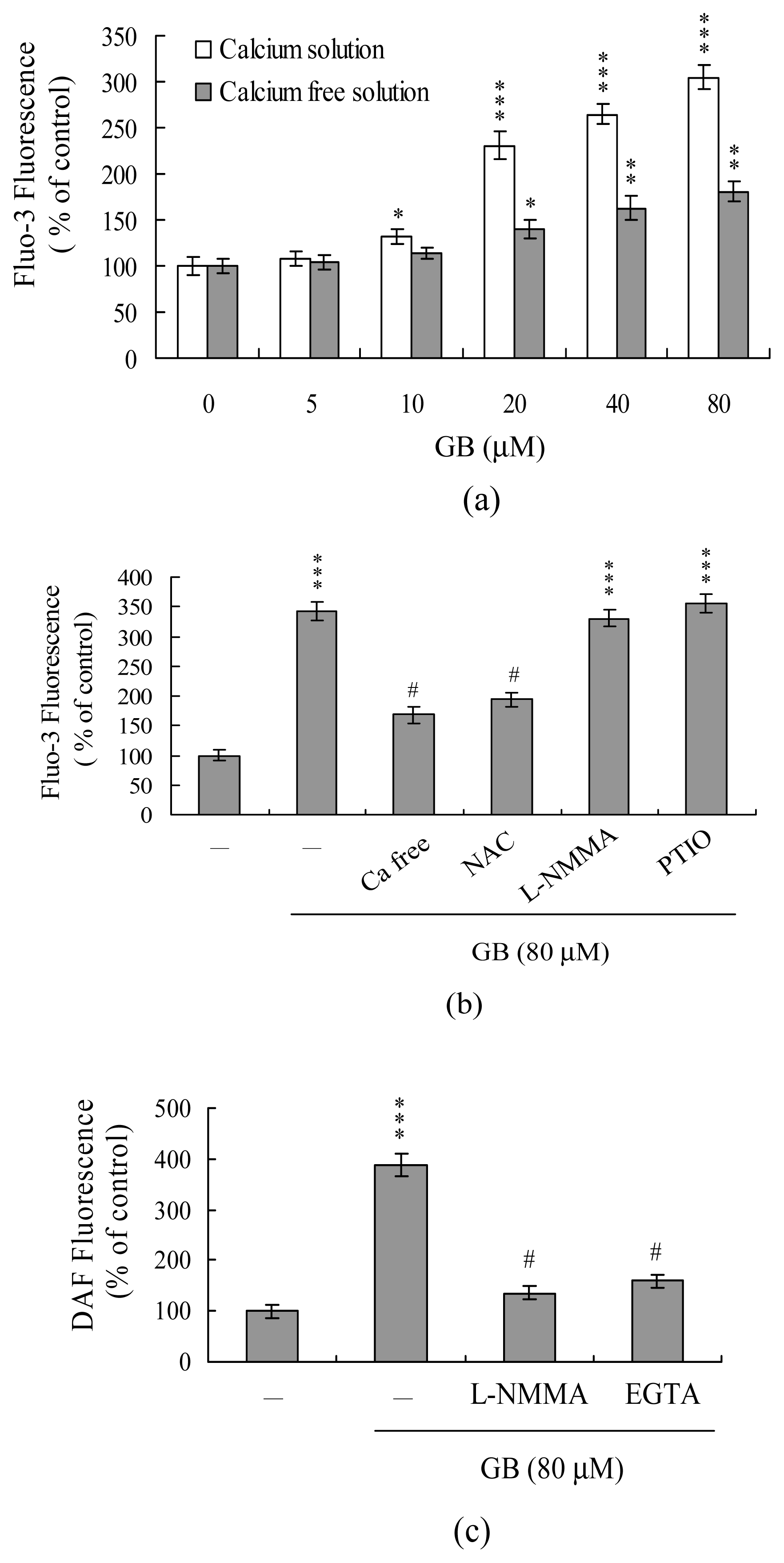
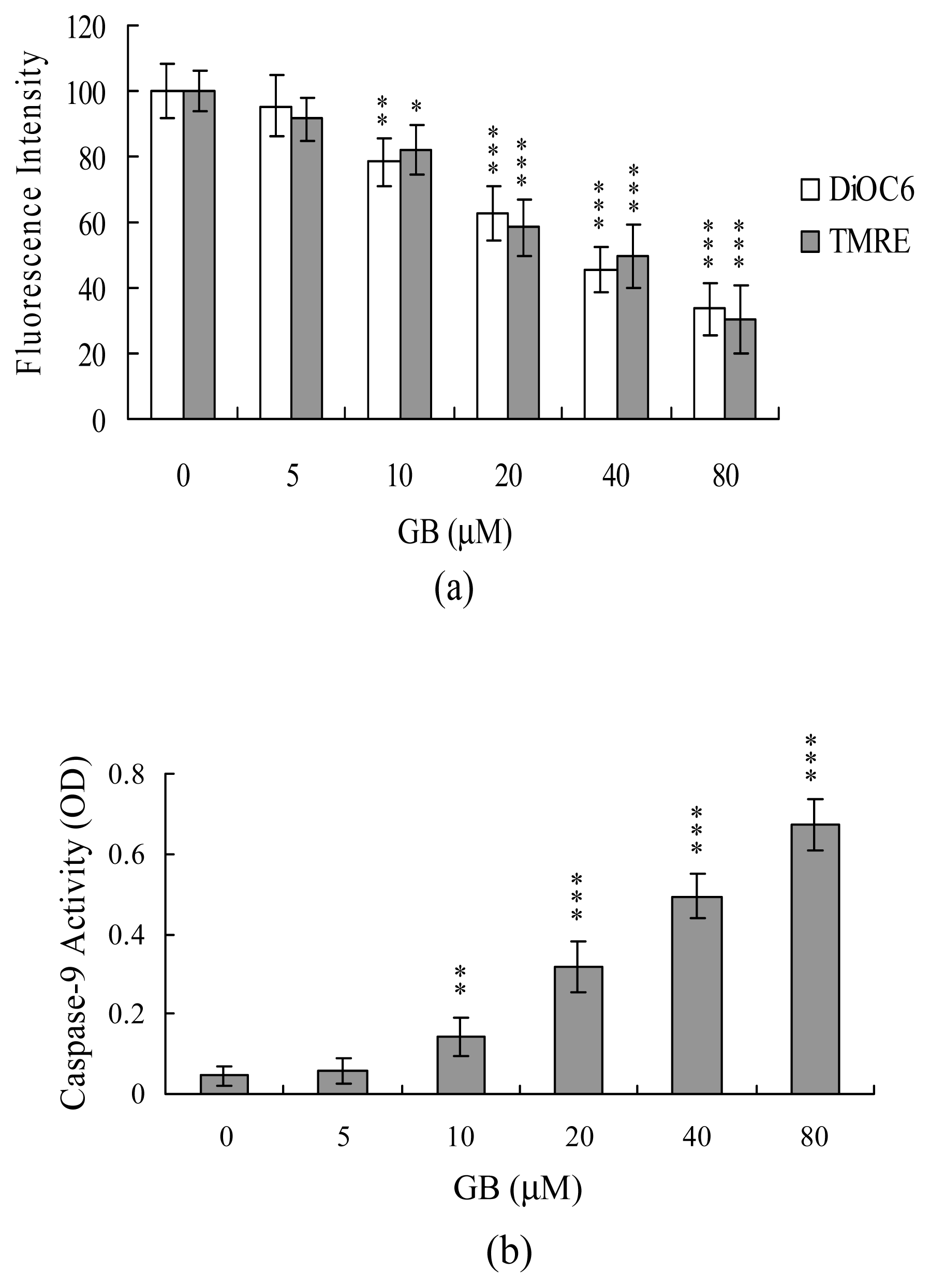
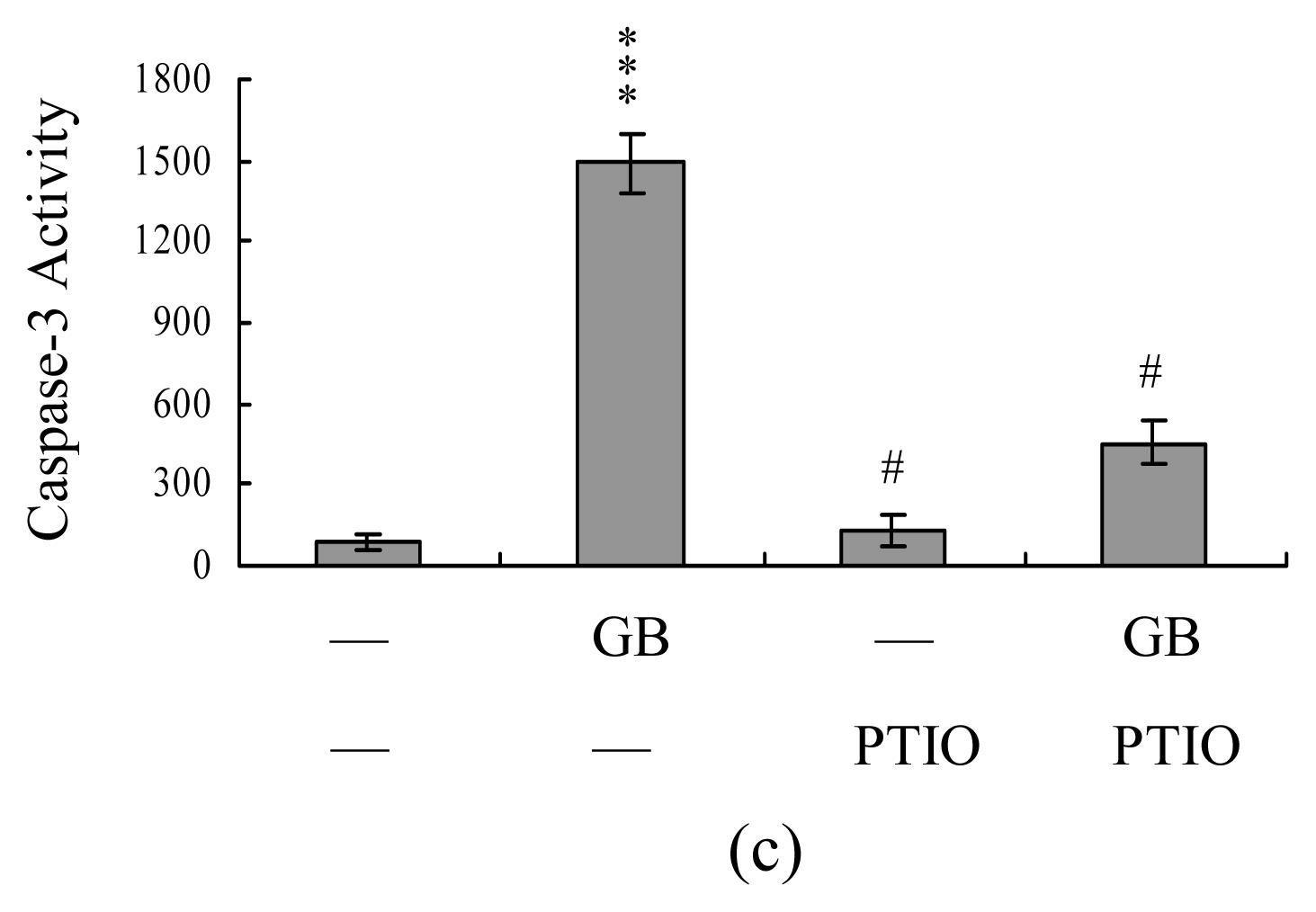
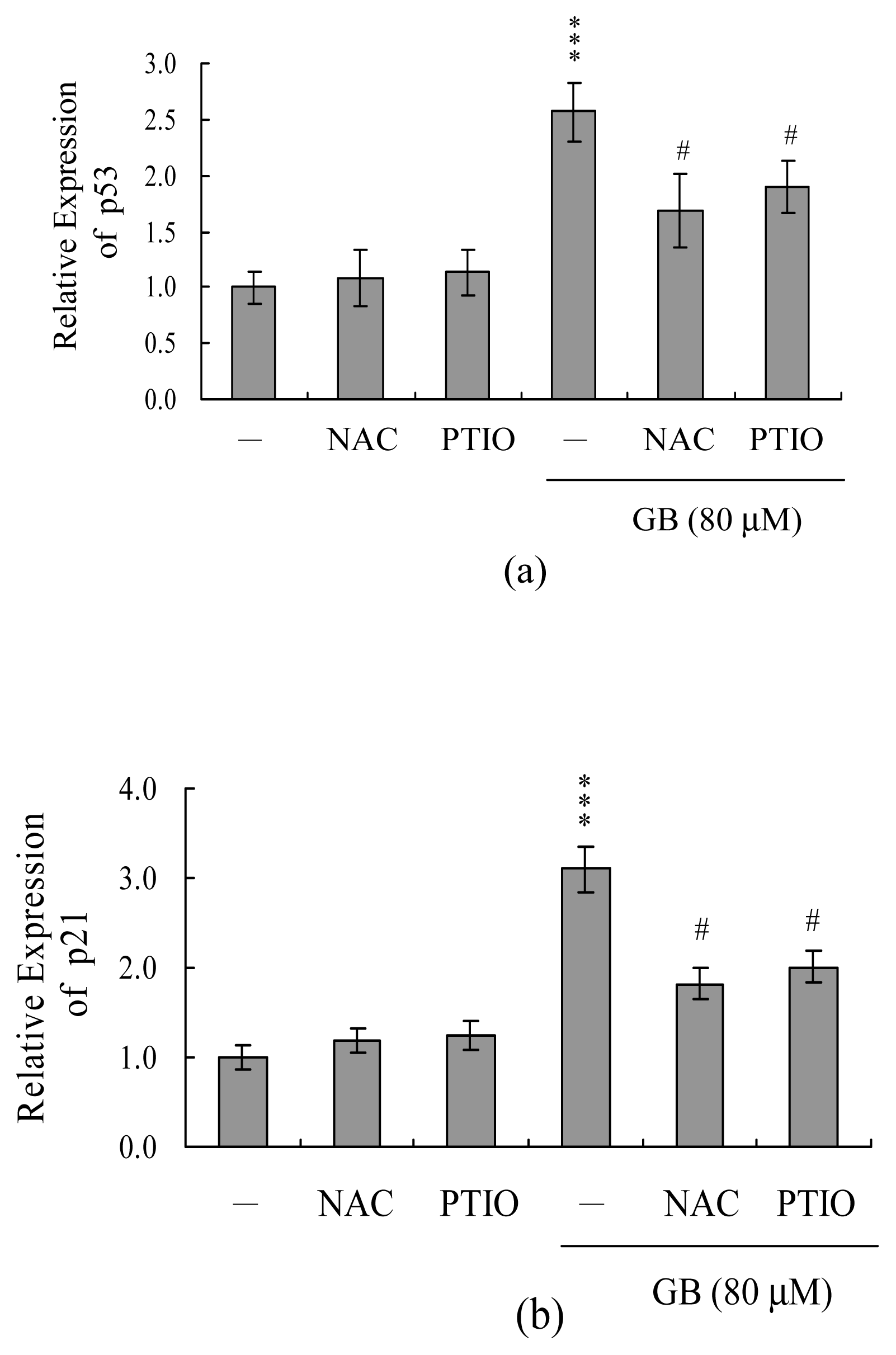
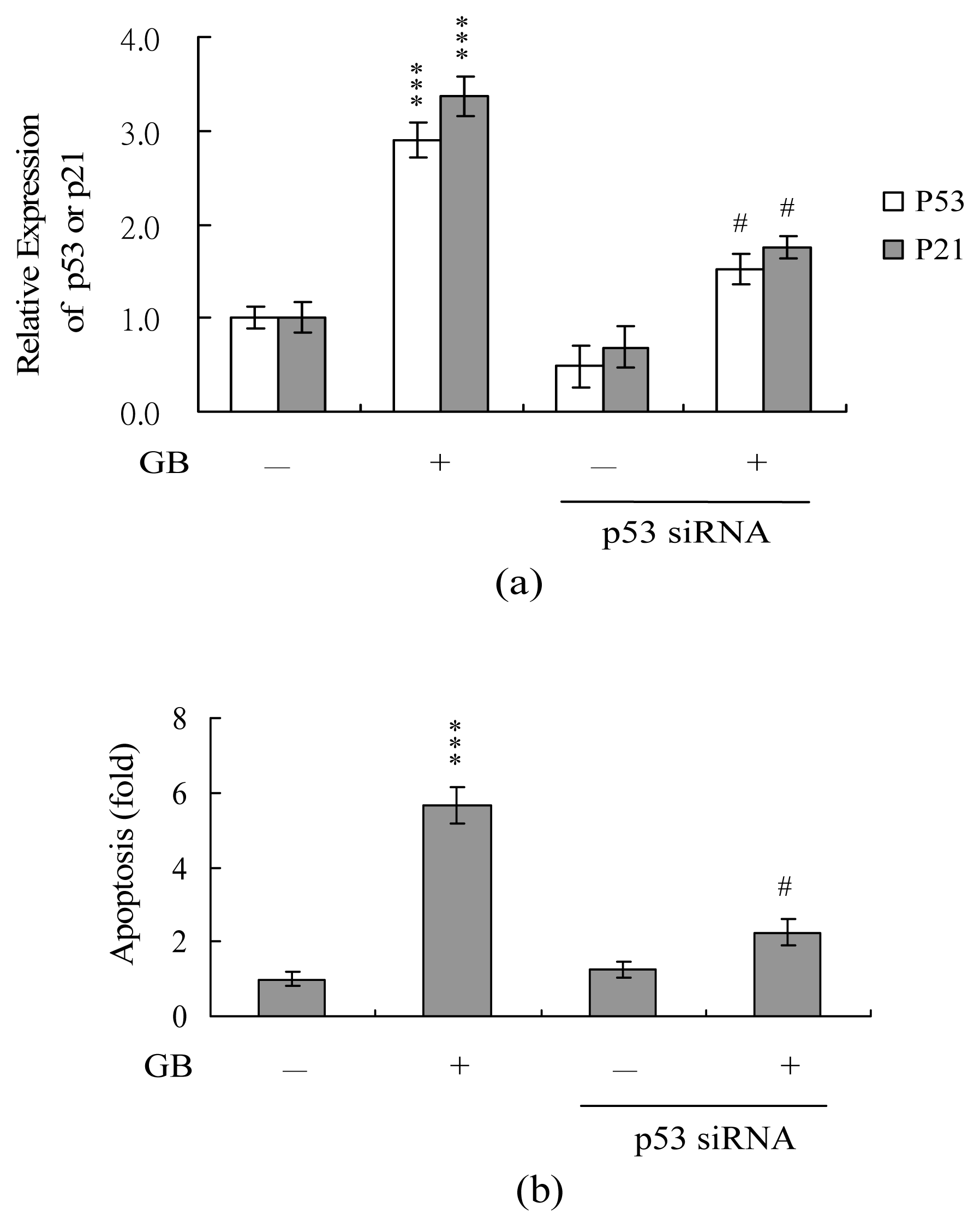
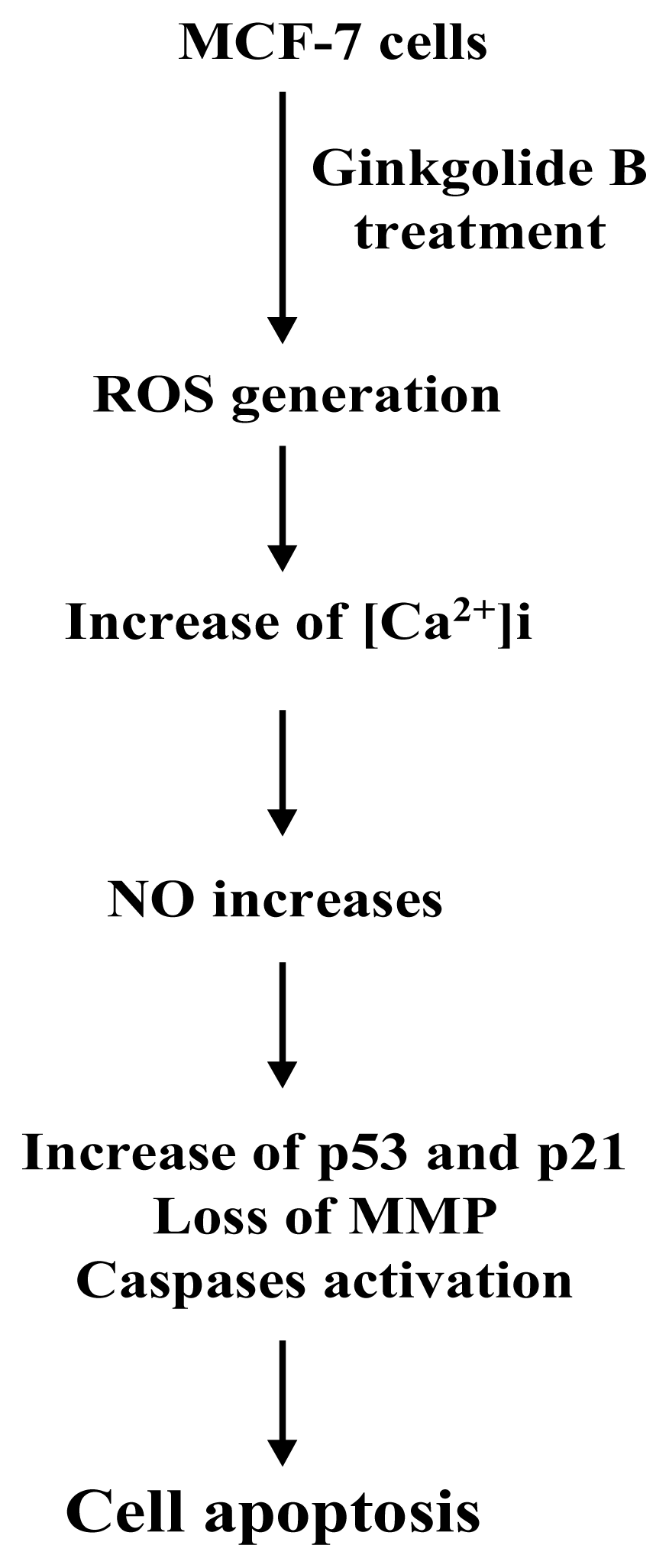
2. Results and Discussion
3. Conclusions
4. Experimental Section
4.1. Chemicals
4.2. Cell culture and ginkgolide B treatment
4.3. MTT assay
4.4. Assessment of necrosis and apoptosis
4.5. ROS assay
4.6. Detection of mitochondrial membrane potential (MMP)
4.7. Caspase activity assays
4.8. Detection of intracellular calcium concentration ([Ca2+]i )
4.9. Detection of intracellular NO content
4.10. Real-time RT-PCR assay
4.11. siRNA knockdown
4.12. Statistics












Acknowledgements
References and Notes
- Ahlemeyer, B.; Krieglstein, J. Pharmacological studies supporting the therapeutic use of Ginkgo biloba extract for Alzheimer’s disease. Pharmacopsychiatry 2003, 36 Suppl 1, S8–14. [Google Scholar]
- DeFeudis, F. V.; Papadopoulos, V.; Drieu, K. Ginkgo biloba extracts and cancer: a research area in its infancy. Fundam. Clin. Pharmacol 2003, 17, 405–417. [Google Scholar]
- Bate, C.; Salmona, M.; Williams, A. Ginkgolide B inhibits the neurotoxicity of prions or amyloid-beta1–42. J. Neuroinflammation 2004, 1, 4. [Google Scholar]
- Maitra, I.; Marcocci, L.; Droy-Lefaix, M. T.; Packer, L. Peroxyl radical scavenging activity of Ginkgo biloba extract EGb 761. Biochem. Pharmacol 1995, 49, 1649–1655. [Google Scholar]
- Pincemail, J.; Thirion, A.; Dupuis, M.; Braquet, P.; Drieu, K.; Deby, C. Ginkgo biloba extract inhibits oxygen species production generated by phorbol myristate acetate stimulated human leukocytes. Experientia 1987, 43, 181–184. [Google Scholar]
- Shen, J. G.; Zhou, D. Y. Efficiency of Ginkgo biloba extract (EGb 761) in antioxidant protection against myocardial ischemia and reperfusion injury. Biochem Mol. Biol. Int 1995, 35, 125–134. [Google Scholar]
- Pietri, S.; Maurelli, E.; Drieu, K.; Culcasi, M. Cardioprotective and anti-oxidant effects of the terpenoid constituents of Ginkgo biloba extract (EGb 761). J. Mol. Cell Cardiol 1997, 29, 733–742. [Google Scholar]
- Luo, Y.; Smith, J. V.; Paramasivam, V.; Burdick, A.; Curry, K. J.; Buford, J. P.; Khan, I.; Netzer, W. J.; Xu, H.; Butko, P. Inhibition of amyloid-beta aggregation and caspase-3 activation by the Ginkgo biloba extract EGb761. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 12197–12202. [Google Scholar]
- Kim, K. S.; Rhee, K. H.; Yoon, J. H.; Lee, J. G.; Lee, J. H.; Yoo, J. B. Ginkgo biloba extract (EGb 761) induces apoptosis by the activation of caspase-3 in oral cavity cancer cells. Oral. Oncol 2005, 41, 383–389. [Google Scholar]
- Thompson, C. B. Apoptosis in the pathogenesis and treatment of disease. Science 1995, 267, 1456–1462. [Google Scholar]
- Halliwell, B.; Gutteridge, J. M. Role of free radicals and catalytic metal ions in human disease: an overview. Methods Enzymol 1990, 186, 1–85. [Google Scholar]
- Chan, W. H. Ginkgolide B induces apoptosis and developmental injury in mouse embryonic stem cells and blastocysts. Hum. Reprod 2006, 21, 2985–2995. [Google Scholar]
- Chan, W. H.; Shiao, N. H.; Lu, P. Z. CdSe quantum dots induce apoptosis in human neuroblastoma cells via mitochondrial-dependent pathways and inhibition of survival signals. Toxicol. Lett 2006, 167, 191–200. [Google Scholar]
- Martins, L. M.; Kottke, T.; Mesner, P. W.; Basi, G. S.; Sinha, S.; Frigon, N., Jr; Tatar, E.; Tung, J. S.; Bryant, K.; Takahashi, A.; Svingen, P. A.; Madden, B. J.; McCormick, D. J.; Earnshaw, W. C.; Kaufmann, S. H. Activation of multiple interleukin-1beta converting enzyme homologues in cytosol and nuclei of HL-60 cells during etoposide-induced apoptosis. J. Biol. Chem 1997, 272, 7421–7430. [Google Scholar]
- Nicholson, D. W.; Thornberry, N. A. Caspases: killer proteases. Trends Biochem. Sci 1997, 22, 299–306. [Google Scholar]
- Tsujimoto, Y.; Shimizu, S. Bcl-2 family: life-or-death switch. FEBS Lett 2000, 466, 6–10. [Google Scholar]
- Ekmekcioglu, S.; Tang, C. H.; Grimm, E. A. NO news is not necessarily good news in cancer. Curr. Cancer Drug Targets 2005, 5, 103–115. [Google Scholar]
- Zhou, J.; Brune, B. NO and transcriptional regulation: from signaling to death. Toxicology 2005, 208, 223–233. [Google Scholar]
- Rao, C. V. Nitric oxide signaling in colon cancer chemoprevention. Mutat. Res 2004, 555, 107–119. [Google Scholar]
- Lu, Z.; Tao, Y.; Zhou, Z.; Zhang, J.; Li, C.; Ou, L.; Zhao, B. Mitochondrial reactive oxygen species and nitric oxide-mediated cancer cell apoptosis in 2-butylamino-2-demethoxyhypocrellin B photodynamic treatment. Free Radic. Biol. Med 2006, 41, 1590–1605. [Google Scholar]
- Nazarewicz, R. R.; Zenebe, W. J.; Parihar, A.; Larson, S. K.; Alidema, E.; Choi, J.; Ghafourifar, P. Tamoxifen induces oxidative stress and mitochondrial apoptosis via stimulating mitochondrial nitric oxide synthase. Cancer Res 2007, 67, 1282–1290. [Google Scholar]
- Ghafourifar, P.; Cadenas, E. Mitochondrial nitric oxide synthase. Trends Pharmacol. Sci 2005, 26, 190–195. [Google Scholar]
- Brookes, P. S. Mitochondrial nitric oxide synthase. Mitochondrion 2004, 3, 187–204. [Google Scholar]
- Dennis, J.; Bennett, J. P., Jr. Interactions among nitric oxide and Bcl-family proteins after MPP+ exposure of SH-SY5Y neural cells I: MPP+ increases mitochondrial NO and Bax protein. J. Neurosci. Res 2003, 72, 76–88. [Google Scholar]
- Elfering, S. L.; Sarkela, T. M.; Giulivi, C. Biochemistry of mitochondrial nitric-oxide synthase. J. Biol. Chem 2002, 277, 38079–38086. [Google Scholar]
- Dedkova, E. N.; Ji, X.; Lipsius, S. L.; Blatter, L. A. Mitochondrial calcium uptake stimulates nitric oxide production in mitochondria of bovine vascular endothelial cells. Am. J. Physiol. Cell Physiol 2004, 286, C406–415. [Google Scholar]
- Chan, W. H. Effect of resveratrol on high glucose-induced stress in human leukemia K562 cells. J. Cell. Biochem 2005, 94, 1267–1279. [Google Scholar]
- Hsuuw, Y. D.; Chang, C. K.; Chan, W. H.; Yu, J. S. Curcumin prevents methylglyoxal-induced oxidative stress and apoptosis in mouse embryonic stem cells and blastocysts. J. Cell. Physiol 2005, 205, 379–386. [Google Scholar]
- Shimizu, S.; Konishi, A.; Kodama, T.; Tsujimoto, Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc. Natl. Acad. Sci. U. S. A 2000, 97, 3100–3105. [Google Scholar]
- Webster, R. P.; Gawde, M. D.; Bhattacharya, R. K. Protective effect of rutin, a flavonol glycoside, on the carcinogen-induced DNA damage and repair enzymes in rats. Cancer Lett 1996, 109, 185–191. [Google Scholar]
- Mutoh, M.; Takahashi, M.; Fukuda, K.; Matsushima-Hibiya, Y.; Mutoh, H.; Sugimura, T.; Wakabayashi, K. Suppression of cyclooxygenase-2 promoter-dependent transcriptional activity in colon cancer cells by chemopreventive agents with a resorcin-type structure. Carcinogenesis 2000, 21, 959–963. [Google Scholar]
- Chan, W. H. Ginkgolides induce apoptosis and decrease cell numbers in mouse blastocysts. Biochem. Biophys. Res. Commun 2005, 338, 1263–1267. [Google Scholar]
- Buttke, T. M.; Sandstrom, P. A. Oxidative stress as a mediator of apoptosis. Immunol. Today 1994, 15, 7–10. [Google Scholar]
- Slater, A. F.; Nobel, C. S.; Maellaro, E.; Bustamante, J.; Kimland, M.; Orrenius, S. Nitrone spin traps and a nitroxide antioxidant inhibit a common pathway of thymocyte apoptosis. Biochem. J 1995, 306 Pt 3, 771–778. [Google Scholar]
- Chan, W. H.; Wu, C. C.; Yu, J. S. Curcumin inhibits UV irradiation-induced oxidative stress and apoptotic biochemical changes in human epidermoid carcinoma A431 cells. J. Cell. Biochem 2003, 90, 327–338. [Google Scholar]
- Long, L. H.; Clement, M. V.; Halliwell, B. Artifacts in cell culture: rapid generation of hydrogen peroxide on addition of (−)-epigallocatechin, (−)-epigallocatechin gallate, (+)-catechin, and quercetin to commonly used cell culture media. Biochem. Biophys. Res. Commun 2000, 273, 50–53. [Google Scholar]
- Halliwell, B. Oxidative stress in cell culture: an under-appreciated problem? FEBS Lett 2003, 540, 3–6. [Google Scholar]
- Kroemer, G.; Zamzami, N.; Susin, S. A. Mitochondrial control of apoptosis. Immunol. Today 1997, 18, 44–51. [Google Scholar]
- Green, D. R.; Reed, J. C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar]
- Liu, G.; Hinch, B.; Davatol-Hag, H.; Lu, Y.; Powers, M.; Beavis, A. D. Temperature dependence of the mitochondrial inner membrane anion channel. The relationship between temperature and inhibition by protons. J. Biol. Chem 1996, 271, 19717–19723. [Google Scholar]
- Almeida, R. D.; Manadas, B. J.; Carvalho, A. P.; Duarte, C. B. Intracellular signaling mechanisms in photodynamic therapy. Biochim. Biophys. Acta 2004, 1704, 59–86. [Google Scholar]
- Inanami, O.; Yoshito, A.; Takahashi, K.; Hiraoka, W.; Kuwabara, M. Effects of BAPTA-AM and forskolin on apoptosis and cytochrome c release in photosensitized Chinese hamster V79 cells. Photochem. Photobiol 1999, 70, 650–655. [Google Scholar]
- Monteiro, H. P.; Silva, E. F.; Stern, A. Nitric oxide: a potential inducer of adhesion-related apoptosis--anoikis. Nitric Oxide 2004, 10, 1–10. [Google Scholar]
- Li, C. Q.; Wogan, G. N. Nitric oxide as a modulator of apoptosis. Cancer Lett 2005, 226, 1–15. [Google Scholar]
- Gomes, E. R.; Almeida, R. D.; Carvalho, A. P.; Duarte, C. B. Nitric oxide modulates tumor cell death induced by photodynamic therapy through a cGMP-dependent mechanism. Photochem. Photobiol 2002, 76, 423–430. [Google Scholar]
- Li, C. Q.; Robles, A. I.; Hanigan, C. L.; Hofseth, L. J.; Trudel, L. J.; Harris, C. C.; Wogan, G. N. Apoptotic signaling pathways induced by nitric oxide in human lymphoblastoid cells expressing wild-type or mutant p53. Cancer Res 2004, 64, 3022–3029. [Google Scholar]
- Okada, H.; Mak, T. W. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat. Rev. Cancer 2004, 4, 592–603. [Google Scholar]
- Chan, W. H.; Wu, H. J. Methylglyoxal and high glucose co-treatment induces apoptosis or necrosis in human umbilical vein endothelial cells. J. Cell. Biochem 2007. In Press. [Google Scholar]
- Chan, W. H.; Hsuuw, Y. D. Dosage effects of ginkgolide B on ethanol-induced cell death in human hepatoma G2 cells. Ann. N. Y. Acad. Sci 2007, 1095, 388–398. [Google Scholar]
- Chan, W. H. Citrinin induces apoptosis via a mitochondria-dependent pathway and inhibition of survival signals in embryonic stem cells, and causes developmental injury in blastocysts. Biochem. J 2007, 404, 317–326. [Google Scholar]
- Chan, W. H.; Chang, Y. J. Dosage effects of resveratrol on ethanol-induced cell death in the human K562 cell line. Toxicol. Lett 2006, 161, 1–9. [Google Scholar]
- Hsieh, Y. J.; Wu, C. C.; Chang, C. J.; Yu, J. S. Subcellular localization of Photofrin determines the death phenotype of human epidermoid carcinoma A431 cells triggered by photodynamic therapy: when plasma membranes are the main targets. J. Cell. Physiol 2003, 194, 363–375. [Google Scholar]
- Aoshima, H.; Satoh, T.; Sakai, N.; Yamada, M.; Enokido, Y.; Ikeuchi, T.; Hatanaka, H. Generation of free radicals during lipid hydroperoxide-triggered apoptosis in PC12h cells. Biochim. Biophys. Acta 1997, 1345, 35–42. [Google Scholar]
- Nakatsubo, N.; Kojima, H.; Kikuchi, K.; Nagoshi, H.; Hirata, Y.; Maeda, D.; Imai, Y.; Irimura, T.; Nagano, T. Direct evidence of nitric oxide production from bovine aortic endothelial cells using new fluorescence indicators: diaminofluoresceins. FEBS Lett 1998, 427, 263–266. [Google Scholar]
© 2007 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
Chan, W.-H. The Signaling Cascades of Ginkgolide B-Induced Apoptosis in MCF-7 Breast Cancer Cells. Int. J. Mol. Sci. 2007, 8, 1177-1195. https://doi.org/10.3390/i8111177
Chan W-H. The Signaling Cascades of Ginkgolide B-Induced Apoptosis in MCF-7 Breast Cancer Cells. International Journal of Molecular Sciences. 2007; 8(11):1177-1195. https://doi.org/10.3390/i8111177
Chicago/Turabian StyleChan, Wen-Hsiung. 2007. "The Signaling Cascades of Ginkgolide B-Induced Apoptosis in MCF-7 Breast Cancer Cells" International Journal of Molecular Sciences 8, no. 11: 1177-1195. https://doi.org/10.3390/i8111177
APA StyleChan, W.-H. (2007). The Signaling Cascades of Ginkgolide B-Induced Apoptosis in MCF-7 Breast Cancer Cells. International Journal of Molecular Sciences, 8(11), 1177-1195. https://doi.org/10.3390/i8111177