
Impact of Ruxolitinib Interactions on JAK2 JH1 Domain Dynamics
 , and
, and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
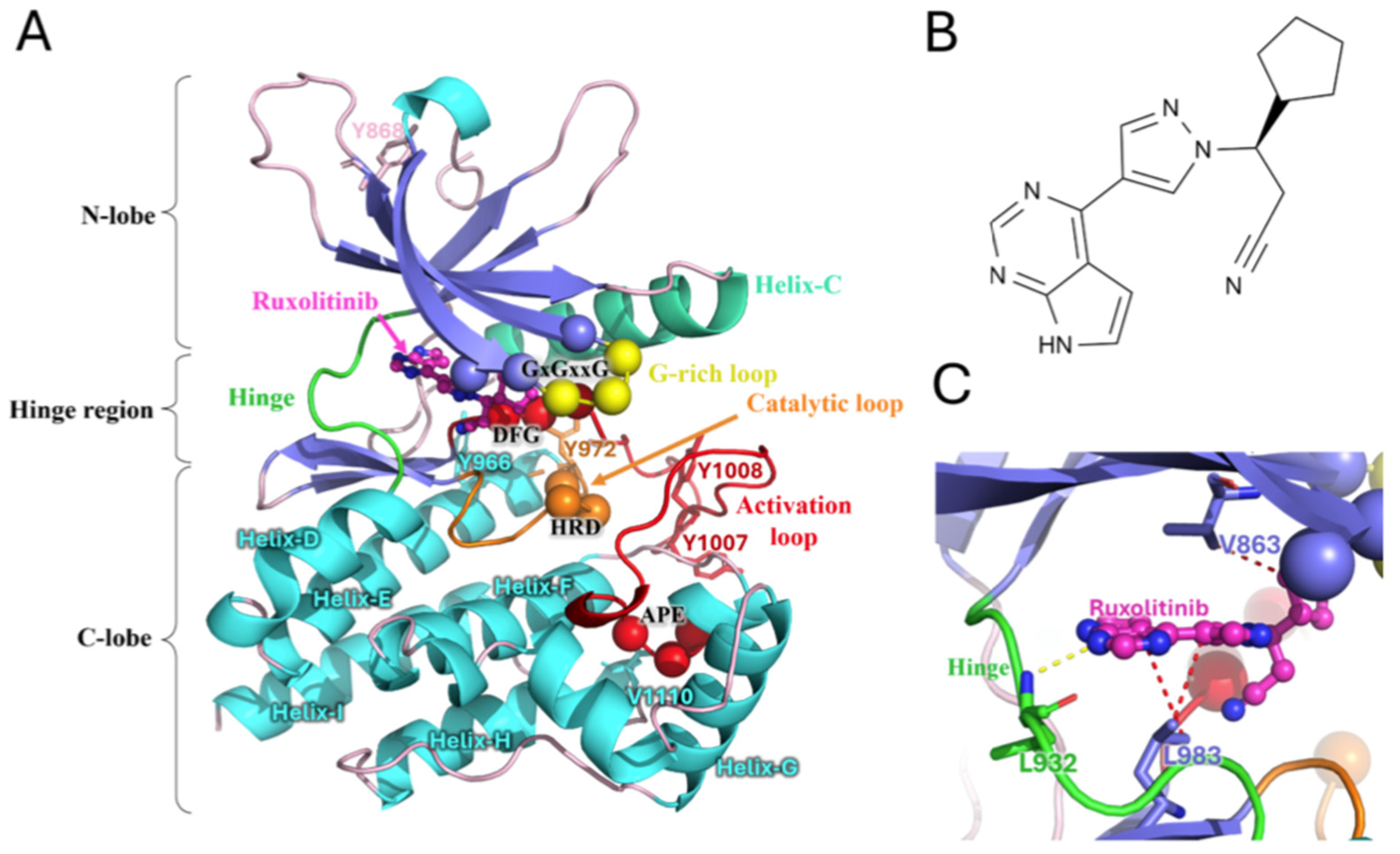
2.1. Overview of Systems
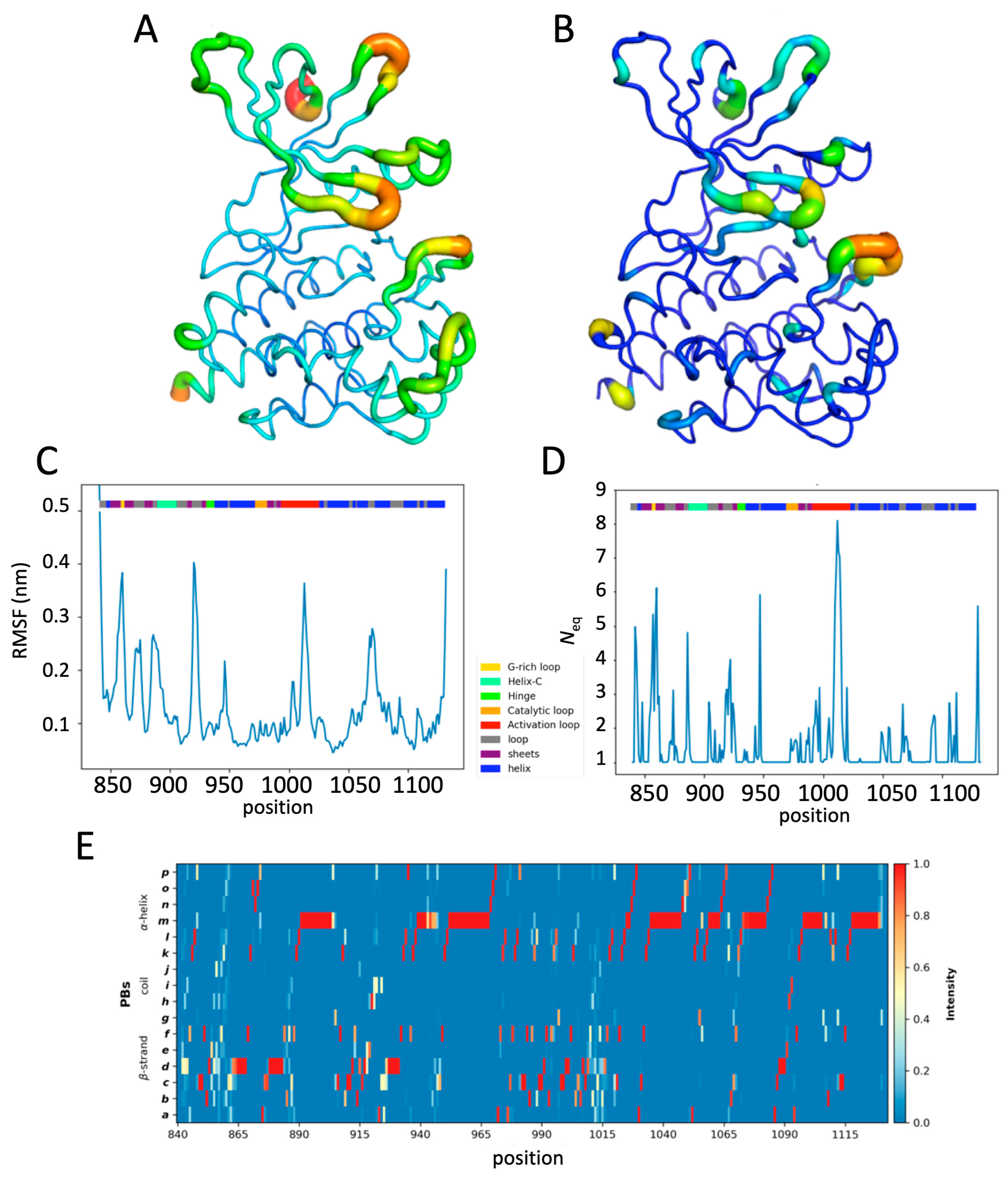
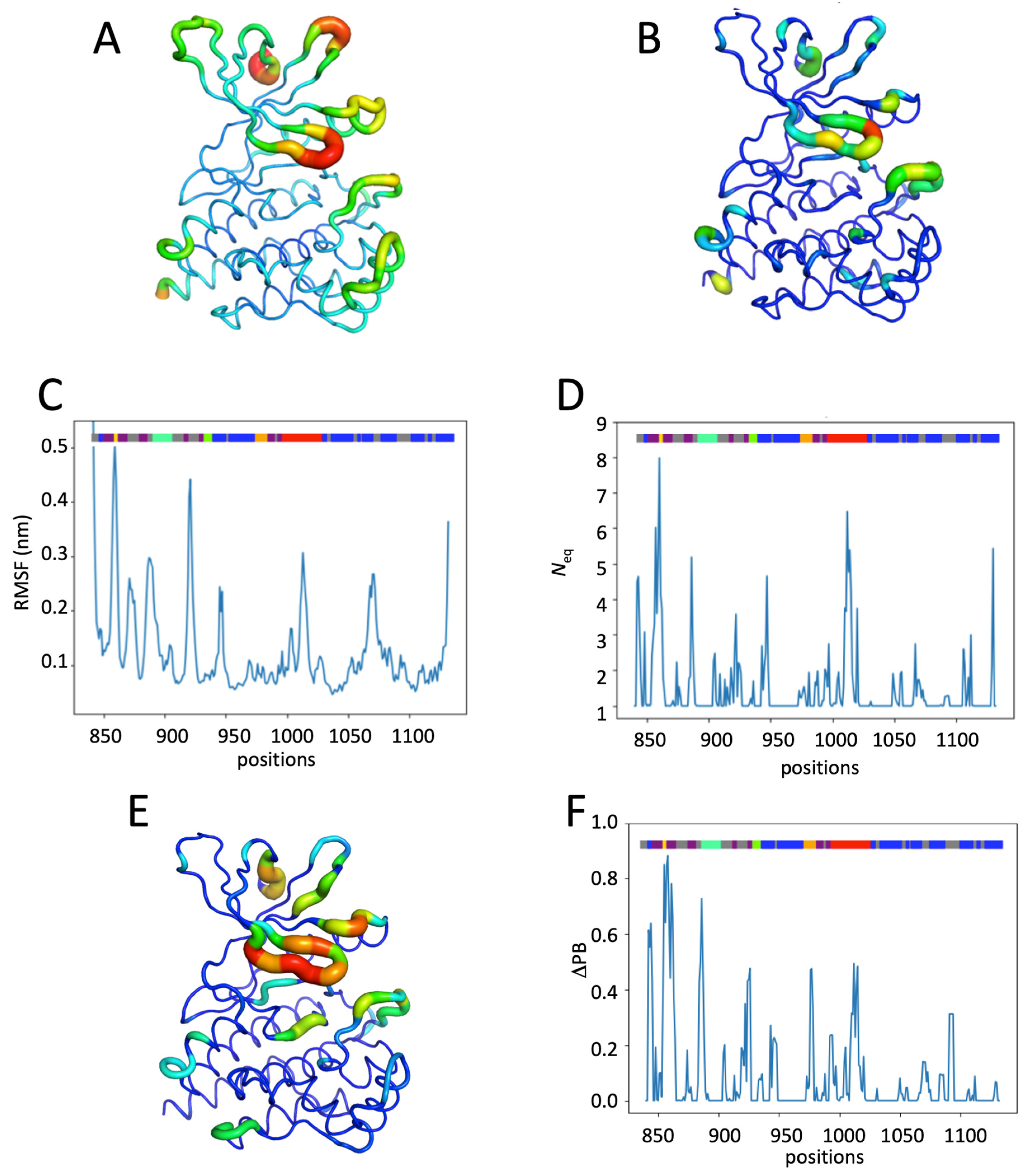
2.2. JAK2 JH1 Apo
2.3. Impact of the Phosphorylation of pTyr1007 on JH1 Dynamics
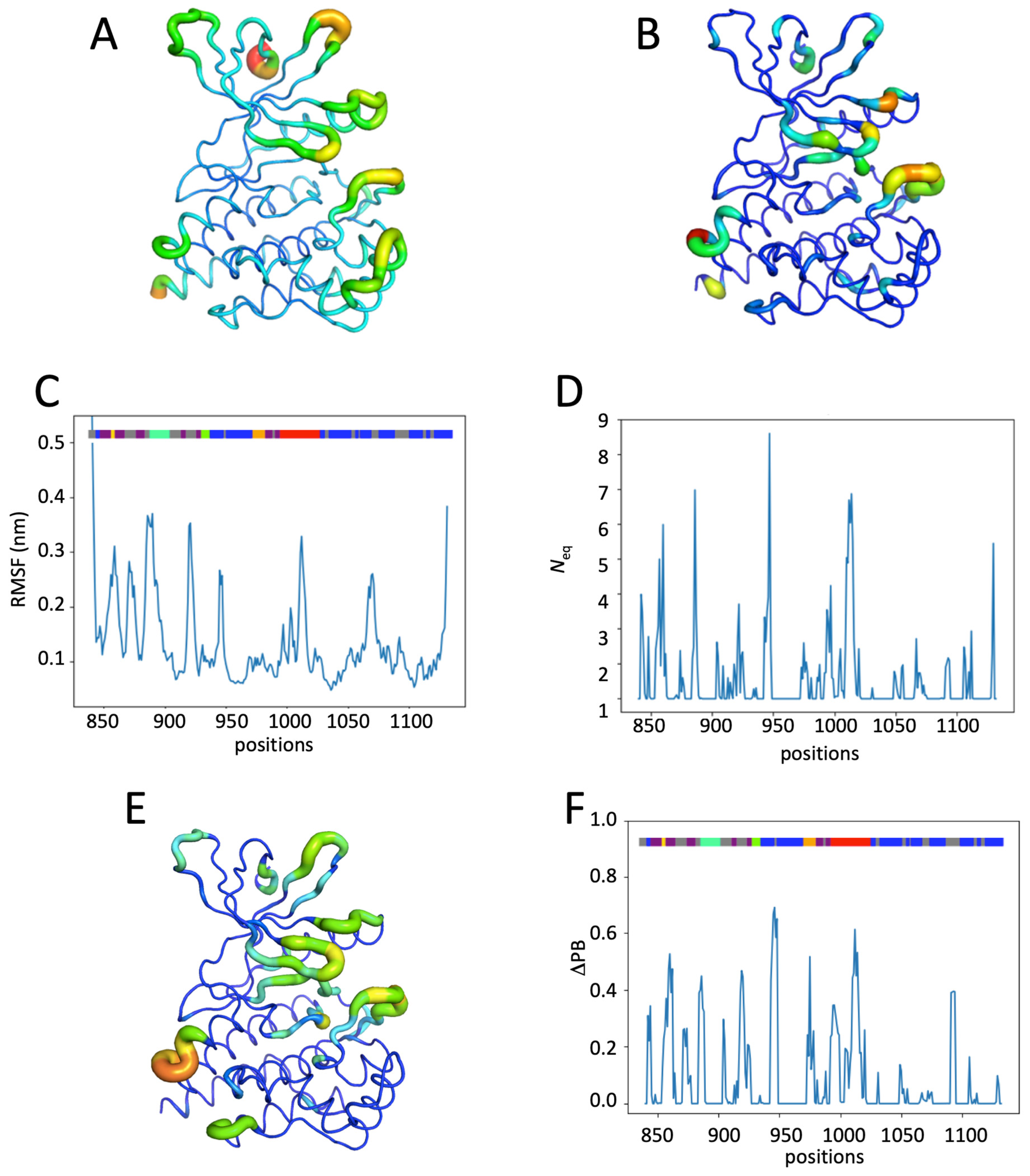
2.4. Impact of the Addition of Ruxolitinib on JH1 Apo-pTyr1007 Dynamics
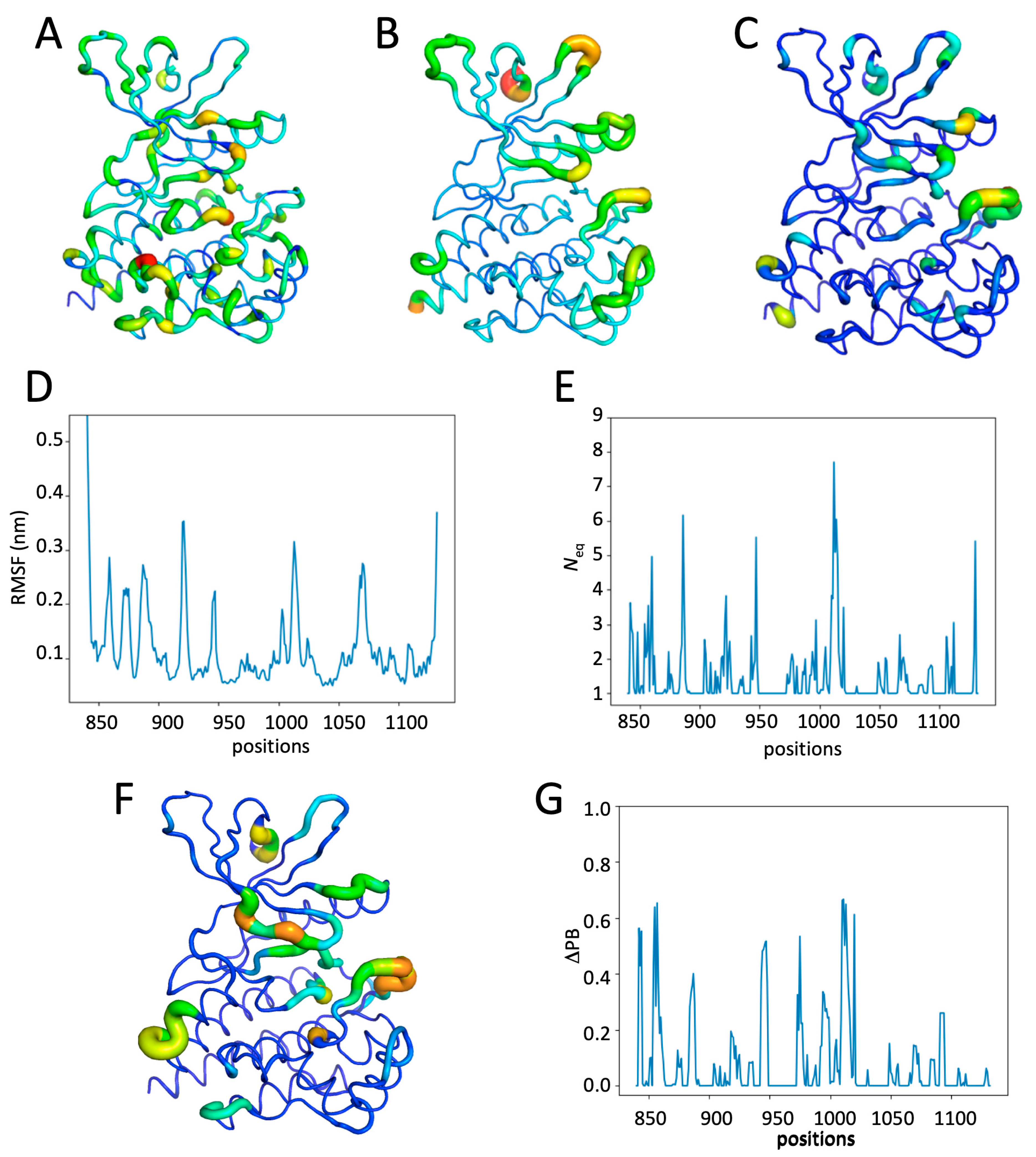
2.5. Impact of Multiple Phosphorylations and Ruxolitinib on JH1 Dynamics
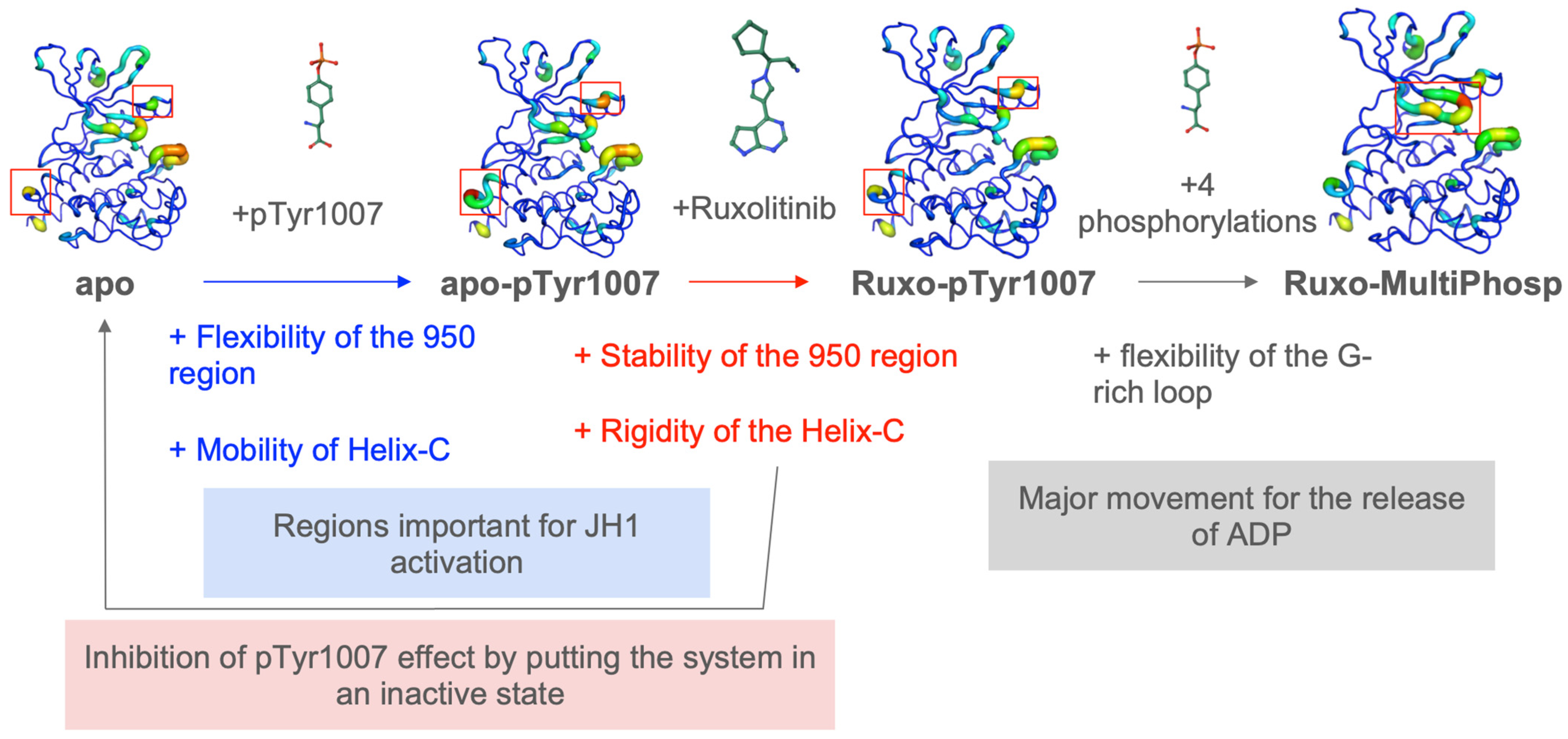
3. Discussion
4. Materials and Methods
4.1. Selection of the JH1 Domain and the Different Systems
4.2. Molecular Dynamics
4.3. Molecular Dynamics Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deshmukh, K.; Anamika, K.; Srinivasan, N. Evolution of domain combinations in protein kinases and its implications for functional diversity. Prog. Biophys. Mol. Biol. 2010, 102, 1–15. [Google Scholar] [CrossRef]
- Janaki, C.; Srinivasan, N.; Manoharan, M. Classification of protein kinases influenced by conservation of substrate binding residues. Methods Mol. Biol. 2016, 1415, 301–313. [Google Scholar]
- Kalaivani, R.; Narwani, T.J.; de Brevern, A.G.; Srinivasan, N. Long-range molecular dynamics show that inactive forms of protein kinase a are more dynamic than active forms. Protein Sci. A Publ. Protein Soc. 2019, 28, 543–560. [Google Scholar] [CrossRef]
- Agashe, R.P.; Lippman, S.M.; Kurzrock, R. Jak: Not just another kinase. Mol. Cancer Ther. 2022, 21, 1757–1764. [Google Scholar] [CrossRef]
- Abraham, B.G.; Haikarainen, T.; Vuorio, J.; Girych, M.; Virtanen, A.T.; Kurttila, A.; Karathanasis, C.; Heilemann, M.; Sharma, V.; Vattulainen, I.; et al. Molecular basis of jak2 activation in erythropoietin receptor and pathogenic jak2 signaling. Sci. Adv. 2024, 10, eadl2097. [Google Scholar] [CrossRef]
- Groner, B.; von Manstein, V. Jak stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition. Mol. Cell. Endocrinol. 2017, 451, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Yao, Q.; Gu, X.; Shi, Q.; Yuan, X.; Chu, Q.; Bao, Z.; Lu, J.; Li, L. Evolving cognition of the jak-stat signaling pathway: Autoimmune disorders and cancer. Signal Transduct. Target. Ther. 2023, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Witthuhn, B.A.; Matsuda, T.; Kohlhuber, F.; Kerr, I.M.; Ihle, J.N. Activation of jak2 catalytic activity requires phosphorylation of y1007 in the kinase activation loop. Mol. Cell. Biol. 1997, 17, 2497–2501. [Google Scholar] [CrossRef]
- Caveney, N.A.; Saxton, R.A.; Waghray, D.; Glassman, C.R.; Tsutsumi, N.; Hubbard, S.R.; Garcia, K.C. Structural basis of janus kinase trans-activation. Cell Rep. 2023, 42, 112201. [Google Scholar] [CrossRef]
- Sayed, W.S.; Al-Bayati, A.; Elzubair, L.G.; Mohamed, S.; Alharthi, M. The association between janus kinase 2 and factor v leiden mutations and thrombotic complications in patients with myeloproliferative disorders: A study from saudi arabia. Cureus 2024, 16, e74401. [Google Scholar] [CrossRef]
- Tevet, M.; Ionescu, R.; Dragan, C.; Lupu, A.R. Influence of the jak2 v617f mutation and inherited thrombophilia on the thrombotic risk among patients with myeloproliferative disorders. Maedica 2015, 10, 27–32. [Google Scholar]
- Langabeer, S.E. The jak2 v617f mutation in lung cancer: Caveat emptor. Exp. Oncol. 2018, 40, 343–344. [Google Scholar] [CrossRef]
- Naeem, M.A. The jak2 v617f mutation in breast cancer? Breast J. 2020, 26, 593. [Google Scholar] [CrossRef]
- Perner, F.; Perner, C.; Ernst, T.; Heidel, F.H. Roles of jak2 in aging, inflammation, hematopoiesis and malignant transformation. Cells 2019, 8, 854. [Google Scholar] [CrossRef] [PubMed]
- James, C.; Ugo, V.; Le Couédic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal jak2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of jak2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef]
- Wolanskyj, A.P.; Lasho, T.L.; Schwager, S.M.; McClure, R.F.; Wadleigh, M.; Lee, S.J.; Gilliland, D.G.; Tefferi, A. Jak2 mutation in essential thrombocythaemia: Clinical associations and long-term prognostic relevance. Br. J. Haematol. 2005, 131, 208–213. [Google Scholar] [CrossRef]
- Luque Paz, D.; Kralovics, R.; Skoda, R.C. Genetic basis and molecular profiling in myeloproliferative neoplasms. Blood 2023, 141, 1909–1921. [Google Scholar] [CrossRef]
- Regimbeau, M.; Mary, R.; Hermetet, F.; Girodon, F. Genetic background of polycythemia vera. Genes 2022, 13, 637. [Google Scholar] [CrossRef]
- Spivak, J.L. Polycythemia vera. Curr. Treat. Options Oncol. 2018, 19, 12. [Google Scholar] [CrossRef]
- Tefferi, A.; Gangat, N.; Loscocco, G.G.; Guglielmelli, P.; Szuber, N.; Pardanani, A.; Orazi, A.; Barbui, T.; Vannucchi, A.M. Essential thrombocythemia: A review. JAMA 2025, 333, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Barbui, T. Polycythemia vera: 2024 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2023, 98, 1465–1487. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, D. Cytoreduction for et and pv: Who, what, when, and how? Hematol. Am. Soc. Hematol. Educ. Program 2023, 2023, 660–666. [Google Scholar] [CrossRef]
- Harrison, C.N.; Vannucchi, A.M.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Knoops, L.; Cervantes, F.; Jones, M.M.; Sun, K.; McQuitty, M.; et al. Long-term findings from comfort-ii, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 2016, 30, 1701–1707. [Google Scholar] [CrossRef]
- Masarova, L.; Mascarenhas, J.; Rampal, R.; Hu, W.; Livingston, R.A.; Pemmaraju, N. Ten years of experience with ruxolitinib since approval for polycythemia vera: A review of clinical efficacy and safety. Cancer 2025, 131, e35661. [Google Scholar] [CrossRef] [PubMed]
- Kondratyev, M.; Rudnev, V.R.; Nikolsky, K.S.; Stepanov, A.A.; Petrovsky, D.V.; Kulikova, L.I.; Kopylov, A.T.; Malsagova, K.A.; Kaysheva, A.L. Atomic simulation of the binding of jak1 and jak2 with the selective inhibitor ruxolitinib. Int. J. Mol. Sci. 2022, 23, 10466. [Google Scholar] [CrossRef]
- King, B.; Ohyama, M.; Kwon, O.; Zlotogorski, A.; Ko, J.; Mesinkovska, N.A.; Hordinsky, M.; Dutronc, Y.; Wu, W.S.; McCollam, J.; et al. Two phase 3 trials of baricitinib for alopecia areata. N. Engl. J. Med. 2022, 386, 1687–1699. [Google Scholar] [CrossRef]
- Taylor, P.C.; Keystone, E.C.; van der Heijde, D.; Weinblatt, M.E.; Del Carmen Morales, L.; Reyes Gonzaga, J.; Yakushin, S.; Ishii, T.; Emoto, K.; Beattie, S.; et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N. Engl. J. Med. 2017, 376, 652–662. [Google Scholar] [CrossRef]
- Davis, R.R.; Li, B.; Yun, S.Y.; Chan, A.; Nareddy, P.; Gunawan, S.; Ayaz, M.; Lawrence, H.R.; Reuther, G.W.; Lawrence, N.J.; et al. Structural insights into jak2 inhibition by ruxolitinib, fedratinib, and derivatives thereof. J. Med. Chem. 2021, 64, 2228–2241. [Google Scholar] [CrossRef]
- Talpaz, M.; Kiladjian, J.J. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm-associated myelofibrosis. Leukemia 2021, 35, 1–17. [Google Scholar] [CrossRef]
- Waksal, J.A.; Tremblay, D.; Mascarenhas, J. Clinical utility of fedratinib in myelofibrosis. OncoTargets Ther. 2021, 14, 4509–4521. [Google Scholar] [CrossRef] [PubMed]
- Lancet, J.E. New agents: Great expectations not realized. Best Pract. Res. Clin. Haematol. 2013, 26, 269–274. [Google Scholar] [CrossRef]
- Miao, Y.; Virtanen, A.; Zmajkovic, J.; Hilpert, M.; Skoda, R.C.; Silvennoinen, O.; Haikarainen, T. Functional and structural characterization of clinical-stage janus kinase 2 inhibitors identifies determinants for drug selectivity. J. Med. Chem. 2024, 67, 10012–10024. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Min, J.; Jarusiewicz, J.A.; Actis, M.; Yu-Chen Bradford, S.; Mayasundari, A.; Yang, L.; Chepyala, D.; Alcock, L.J.; Roberts, K.G.; et al. Degradation of janus kinases in crlf2-rearranged acute lymphoblastic leukemia. Blood 2021, 138, 2313–2326. [Google Scholar] [CrossRef] [PubMed]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. Plip: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Hemmer, W.; McGlone, M.; Tsigelny, I.; Taylor, S.S. Role of the glycine triad in the atp-binding site of camp-dependent protein kinase. J. Biol. Chem. 1997, 272, 16946–16954. [Google Scholar] [CrossRef]
- Kanev, G.K.; de Graaf, C.; de Esch, I.J.P.; Leurs, R.; Würdinger, T.; Westerman, B.A.; Kooistra, A.J. The landscape of atypical and eukaryotic protein kinases. Trends Pharmacol. Sci. 2019, 40, 818–832. [Google Scholar] [CrossRef]
- Lucet, I.S.; Fantino, E.; Styles, M.; Bamert, R.; Patel, O.; Broughton, S.E.; Walter, M.; Burns, C.J.; Treutlein, H.; Wilks, A.F.; et al. The structural basis of janus kinase 2 inhibition by a potent and specific pan-janus kinase inhibitor. Blood 2006, 107, 176–183. [Google Scholar] [CrossRef]
- de Brevern, A.G.; Etchebest, C.; Hazout, S. Bayesian probabilistic approach for predicting backbone structures in terms of protein blocks. Proteins 2000, 41, 271–287. [Google Scholar] [CrossRef]
- Akhila, M.V.; Narwani, T.J.; Floch, A.; Maljković, M.; Bisoo, S.; Shinada, N.K.; Kranjc, A.; Gelly, J.-C.; Srinivasan, N.; Mitić, N.; et al. A structural entropy index to analyse local conformations in intrinsically disordered proteins. J. Struct. Biol. 2020, 210, 107464. [Google Scholar] [CrossRef]
- Goguet, M.; Narwani, T.J.; Petermann, R.; Jallu, V.; de Brevern, A.G. In silico analysis of glanzmann variants of calf-1 domain of α(iib)β(3) integrin revealed dynamic allosteric effect. Sci. Rep. 2017, 7, 8001. [Google Scholar] [CrossRef] [PubMed]
- Bornot, A.; Etchebest, C.; de Brevern, A.G. Predicting protein flexibility through the prediction of local structures. Proteins 2011, 79, 839–852. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. Molprobity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Delano, W.L. The Pymol Molecular Graphics System on World Wide Web. 2013. Available online: http://www.pymol.org (accessed on 15 June 2024).
- Schrodinger, LLC. The Pymol Molecular Graphics System, version 1.7.2.2.; Schrodinger, LLC: San Diego, NY, USA, 2015. [Google Scholar]
- Schrödinger, LLC. Pymol; 2.4.0; Schrodinger, LLC: San Diego, NY, USA, 2020. [Google Scholar]
- Jo, S.; Cheng, X.; Lee, J.; Kim, S.; Park, S.J.; Patel, D.S.; Beaven, A.H.; Lee, K.I.; Rui, H.; Park, S.; et al. Charmm-gui 10 years for biomolecular modeling and simulation. J. Comput. Chem. 2017, 38, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. Gromacs: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. Charmm36 all-atom additive protein force field: Validation based on comparison to nmr data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. Charmm general force field: A force field for drug-like molecules compatible with the charmm all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Narwani, T.J.; Craveur, P.; Shinada, N.K.; Floch, A.; Santuz, H.; Vattekatte, A.M.; Srinivasan, N.; Rebehmed, J.; Gelly, J.C.; Etchebest, C.; et al. Discrete analyses of protein dynamics. J. Biomol. Struct. Dyn. 2020, 38, 2988–3002. [Google Scholar] [CrossRef]
- Barnoud, J.; Santuz, H.; Craveur, P.; Joseph, A.P.; Jallu, V.; de Brevern, A.G.; Poulain, P. Pbxplore: A tool to analyze local protein structure and deformability with protein blocks. PeerJ 2017, 5, e4013. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Touw, W.G.; Baakman, C.; Black, J.; te Beek, T.A.; Krieger, E.; Joosten, R.P.; Vriend, G. A series of pdb-related databanks for everyday needs. Nucleic Acids Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef]
- de Brevern, A.G. Analysis of protein disorder predictions in the light of a protein structural alphabet. Biomolecules 2020, 10, 1080. [Google Scholar] [CrossRef]
- Offmann, B.; Tyagi, M.; de Brevern, A.G. Local protein structures. Curr. Bioinform. 2007, 3, 165–202. [Google Scholar] [CrossRef]
- Joseph, A.P.; Agarwal, G.; Mahajan, S.; Gelly, J.C.; Swapna, L.S.; Offmann, B.; Cadet, F.; Bornot, A.; Tyagi, M.; Valadié, H. A short survey on protein blocks. Biophys. Rev. 2010, 2, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.P.; Srinivasan, N.; de Brevern, A.G. Improvement of protein structure comparison using a structural alphabet. Biochimie 2011, 93, 1434–1445. [Google Scholar] [CrossRef]
- Léonard, S.; Joseph, A.P.; Srinivasan, N.; Gelly, J.C.; de Brevern, A.G. Mulpba: An efficient multiple protein structure alignment method based on a structural alphabet. J. Biomol. Struct. Dyn. 2014, 32, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Craveur, P.; Joseph, A.P.; Esque, J.; Narwani, T.J.; Noël, F.; Shinada, N.; Goguet, M.; Leonard, S.; Poulain, P.; Bertrand, O.; et al. Protein flexibility in the light of structural alphabets. Front. Mol. Biosci. 2015, 2, 20. [Google Scholar] [CrossRef]
- Radjasandirane, R.; de Brevern, A.G. Structural and dynamic differences between calreticulin mutants associated with essential thrombocythemia. Biomolecules 2023, 13, 509. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. Weblogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vu, H.N.; Radjasandirane, R.; Diharce, J.; de Brevern, A.G. Impact of Ruxolitinib Interactions on JAK2 JH1 Domain Dynamics. Int. J. Mol. Sci. 2025, 26, 3727. https://doi.org/10.3390/ijms26083727
Vu HN, Radjasandirane R, Diharce J, de Brevern AG. Impact of Ruxolitinib Interactions on JAK2 JH1 Domain Dynamics. International Journal of Molecular Sciences. 2025; 26(8):3727. https://doi.org/10.3390/ijms26083727
Chicago/Turabian StyleVu, Hong Nhung, Ragousandirane Radjasandirane, Julien Diharce, and Alexandre G. de Brevern. 2025. "Impact of Ruxolitinib Interactions on JAK2 JH1 Domain Dynamics" International Journal of Molecular Sciences 26, no. 8: 3727. https://doi.org/10.3390/ijms26083727
APA StyleVu, H. N., Radjasandirane, R., Diharce, J., & de Brevern, A. G. (2025). Impact of Ruxolitinib Interactions on JAK2 JH1 Domain Dynamics. International Journal of Molecular Sciences, 26(8), 3727. https://doi.org/10.3390/ijms26083727