
Crosstalk Between Sickle Cell Disease and Ferroptosis

 ,
,  ,
,  and
and {kind=link}
{kind=link}
Abstract
1. Introduction
2. Pathophysiology of Sickle Cell Disease
2.1. Polymerization of HbS and Deformation of sRBCs
2.2. Occlusion of Vessels and Endothelial Dysfunction
2.3. Oxidative Stress
2.3.1. Oxidase Activity
2.3.2. Autoxidation of HbS and Decrease in NO
2.4. Iron
3. Ferroptosis
3.1. Iron Metabolism and Mitochondrial Dysfunction
3.2. Biochemical Pathway Involved in Ferroptosis and Antioxidants System
4. Correlation Between Ferroptosis and Sickle Cell Disease
4.1. Ferroptosis and HbS Auto-Oxidation in SCD
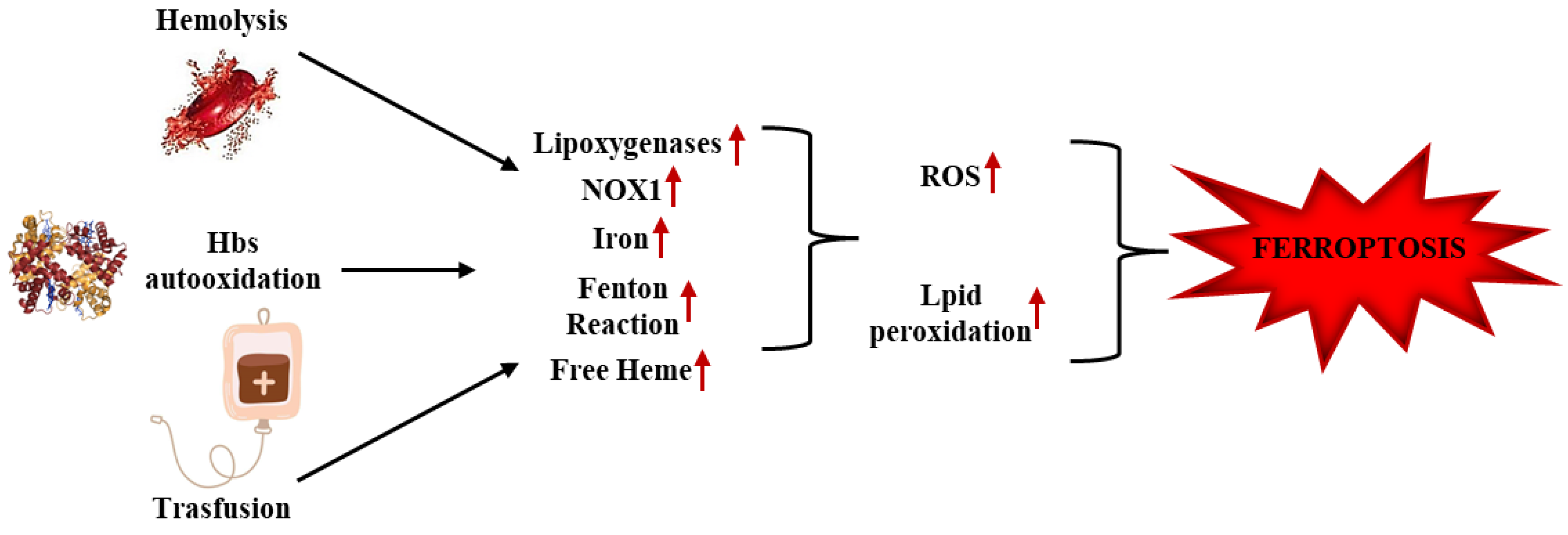
4.2. Ferroptosis, Vaso-Occlusion, and Hemolysis in SCD
4.3. Ferroptosis and IRI in SCD
4.4. Ferroptosis and Inflammation in SCD
4.5. Ferroptosis and Transfusion in SCD
5. Potential Therapeutic Strategies for Ferroptosis Involved in SCD
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SCD | Sickle cell disease |
| HbS | Sickle cell hemoglobin |
| sRBCs | Sickle cell red blood cells |
| Hb | Hemoglobin |
| RBCs | Red blood cells |
| HBB | β-globin |
| glutamic acid | Glu |
| valine | Val |
| adult hemoglobin | HbA |
| ROS | Free oxygen radicals |
| HbF | Fetal hemoglobin |
| B-CAM-1/Lu | Basal cell adhesion molecule-1/Lutheran |
| IAP | Integrin-associated protein |
| ICAM-4 | Intercellular adhesion molecule-4 |
| VOC | Vaso-occlusive crisis |
| VCAM-1 | Vascular cell adhesion molecules-1 |
| RNS | Reactive nitrogen species |
| NADPH | Nicotinamide-adenine dinucleotide phosphate |
| XO | Xanthine oxidase |
| ADMA | Asymmetric dimethylarginine |
| NOS | Nitric oxide synthase |
| MetHb | Methemoglobin |
| BH4 | Tetrahydrobiopterin |
| Hp | Haptoglobin |
| TF | Transferrin |
| TFR1 | Transferrin receptor 1 |
| SLC11A2 | Member 2 of the solute transporter family 11 |
| LIPs | Labile iron complexes |
| FTH1 | Ferritin heavy chain 1 |
| FTL | Ferritin light chain |
| SLC40A1 | Member 1 of the solute transporter family 40 |
| NCOA4 | Nuclear receptor 4 |
| SLC25A37/mitoferrin-1 | Solute transporter family 25 member 37 |
| SLC25A28/mitoferrin-2 | Solute transporter family 25 member 28 |
| VDAC | Voltage-dependent anion channels |
| NTBI | Non-transferrin-bound iron |
| PUFAs | Polyunsaturated fatty acids |
| PUFAs-OOH | Lipid peroxides |
| HNE | 4-hydroxynonenal |
| MDA | Malondialdehyde |
| LPCAT3 | Lysophosphatidylcholine acyltransferase 3 |
| LOX | Lipoxygenases |
| LOOH | Lipid peroxides |
| SLC7A11 | Solute Carrier Family 7 Member 11 |
| SLC3A2 | Solute Carrier Family 3 Member 2 |
| MBOAT1 | PL lysophospholipid acyltransferase 1 |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| FTH1 | Ferritin heavy chain 1 |
| NQO1 | Quinone oxidoreductase 1 |
| HO-1 | Heme oxygenase-1 |
| TSS | Transulfuration pathway |
| DMF | Dimethyl fumarate |
| CBS | De novo cysteine synthesis |
References
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat. Rev. Dis. Prim. 2018, 15, 18010. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef] [PubMed]
- Ballas, S.K. Sickle cell anaemia: Progress in pathogenesis and treatment. Drugs 2002, 62, 1143–1172. [Google Scholar] [CrossRef]
- Thein, S.L. The molecular basis of β-thalassemia. Cold Spring Harb Perspect. Med. 2013, 3, a011700. [Google Scholar] [CrossRef]
- Tarasev, M.; Muchnik, M.; Light, L.; Alfano, K.; Chakraborty, S. Individual variability in response to a single sickling event for normal, sickle cell, and sickle trait erythrocytes. Transl. Res. 2017, 181, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Migotsky, M.; Beestrum, M.; Badawy, S.M. Recent Advances in Sickle-Cell Disease Therapies: A Review of Voxelotor, Crizanlizumab, and L-glutamine. Pharmacy 2022, 10, 123. [Google Scholar] [CrossRef]
- Zhang, J.; Abiraman, K.; Jones, S.M.; Lykotrafitis, G.; Andemariam, B. Regulation of Active ICAM-4 on Normal and Sickle Cell Disease RBCs via AKAPs Is Revealed by AFM. Biophys. J. 2017, 112, 143–152. [Google Scholar] [CrossRef] [PubMed]
- An, R.; Man, Y.; Cheng, K.; Zhang, T.; Chen, C.; Wang, F.; Abdulla, F.; Kucukal, E.; Wulftange, W.J.; Goreke, U.; et al. Sickle red blood cell-derived extracellular vesicles activate endothelial cells and enhance sickle red cell adhesion mediated by von Willebrand factor. Br. J Haematol. 2023, 201, 552–563. [Google Scholar] [CrossRef]
- Pavitra, E.; Acharya, R.K.; Gupta, V.K.; Verma, H.K.; Kang, H.; Lee, J.H.; Sahu, T.; Bhaskar, L.; Raju, G.S.R.; Huh, Y.S. Impacts of oxidative stress and anti-oxidants on the development, pathogenesis, and therapy of sickle cell disease: A comprehensive review. Biomed. Pharmacother. 2024, 176, 116849. [Google Scholar] [CrossRef]
- Elendu, C.; Amaechi, D.C.; Alakwe-Ojimba, C.E.; Elendu, T.C.; Elendu, R.C.; Ayabazu, C.P.; Aina, T.O.; Aborisade, O.; Adenikinju, J.S. Understanding Sickle cell disease: Causes, symptoms, and treatment options. Medicine 2023, 102, e35237. [Google Scholar] [CrossRef]
- Patanè, G.T.; Putaggio, S.; Tellone, E.; Barreca, D.; Ficarra, S.; Maffei, C.; Calderaro, A.; Laganà, G. Ferroptosis: Emerging Role in Diseases and Potential Implication of Bioactive Compounds. Int. J. Mol. Sci. 2023, 24, 17279. [Google Scholar] [CrossRef] [PubMed]
- Manwani, D.; Frenette, P.S. Vaso-occlusion in sickle cell disease: Pathophysiology and novel targeted therapies. Blood 2013, 122, 3892–3898. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.V. Sickle Cell Disease: Advances in Treatment. Ochsner J. 2018, 18, 377–389. [Google Scholar] [CrossRef]
- Frenette, P.S.; Atweh, G.F. Sickle cell disease: Old discoveries, new concepts, and future promise. J. Clin. Investig. 2007, 117, 850–858. [Google Scholar] [CrossRef]
- Bunn, H.F. Pathogenesis and treatment of sickle cell disease. N. Engl. J. Med. 1997, 337, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Barabino, G.A.; Platt, M.O.; Kaul, D.K. Sickle cell biomechanics. Annu. Rev. Biomed. Eng. 2010, 12, 345–367. [Google Scholar] [CrossRef]
- Brittenham, G.M.; Schechter, A.N.; Noguchi, C.T. Hemoglobin S polymerization: Primary determinant of the hemolytic and clinical severity of the sickling syndromes. Blood 1985, 65, 183–189. [Google Scholar] [CrossRef]
- Akinsheye, I.; Alsultan, A.; Solovieff, N.; Ngo, D.; Baldwin, C.T.; Sebastiani, P.; Chui, D.H.; Steinberg, M.H. Fetal hemoglobin in sickle cell anemia. Blood 2011, 118, 19–27. [Google Scholar] [CrossRef]
- Gutierrez, M.; Shamoun, M.; Seu, K.G.; Tanski, T.; Kalfa, T.A.; Eniola-Adefeso, O. Characterizing bulk rigidity of rigid red blood cell populations in sickle-cell disease patients. Sci. Rep. 2021, 11, 7909. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, V.; Manwani, D.; Frenette, P.S. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood 2016, 127, 801–809. [Google Scholar] [CrossRef]
- Sundd, P.; Gladwin, M.T.; Novelli, E.M. Pathophysiology of Sickle Cell Disease. Annu. Rev. Pathol. 2019, 14, 263–292. [Google Scholar] [CrossRef] [PubMed]
- Ballas, S.K.; Kuypers, F.A.; Gordeuk, V.R.; Hankins, J.S.; Thompson, A.A.; Vichinsky, E. Time to rethink haemoglobin threshold guidelines in sickle cell disease. Br. J. Haematol. 2021, 195, 518–522. [Google Scholar] [CrossRef]
- Jimenez, M.A.; Kato, G.J.; Sundd, P. Neutrophil-Platelet Aggregation Enables Vaso-Occlusion in Sickle Cell Disease. Blood 2016, 128, 1295. [Google Scholar] [CrossRef]
- Veluswamy, S.; Shah, P.; Denton, C.C.; Chalacheva, P.; Khoo, M.C.K.; Coates, T.D. Vaso-Occlusion in Sickle Cell Disease: Is Autonomic Dysregulation of the Microvasculature the Trigger? J. Clin. Med. 2019, 8, 1690. [Google Scholar] [CrossRef] [PubMed]
- Wongtong, N.; Jones, S.; Deng, Y.; Cai, J.; Ataga, K.I. Monocytosis is associated with hemolysis in sickle cell disease. Hematology 2015, 20, 593–597. [Google Scholar] [CrossRef]
- Hidalgo, A.; Chang, J.; Jang, J.E.; Peired, A.J.; Chiang, E.Y.; Frenette, P.S. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat. Med. 2009, 15, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef]
- Wallace, K.L.; Linden, J. Adenosine A2A receptors induced on iNKT and NK cells reduce pulmonary inflammation and injury in mice with sickle cell disease. Blood 2010, 116, 5010–5020. [Google Scholar] [CrossRef]
- Bennewitz, M.F.; Jimenez, M.A.; Vats, R.; Tutuncuoglu, E.; Jonassaint, J.; Kato, G.J.; Gladwin, M.T.; Sundd, P. Lung vaso-occlusion in sickle cell disease mediated by arteriolar neutrophil-platelet microemboli. JCI Insight 2017, 12, e89761. [Google Scholar] [CrossRef]
- Vona, R.; Sposi, N.M.; Mattia, L.; Gambardella, L.; Straface, E.; Pietraforte, D. Sickle Cell Disease: Role of Oxidative Stress and Antioxidant Therapy. Antioxidants 2021, 10, 296. [Google Scholar] [CrossRef]
- Aslan, M.; Ryan, T.M.; Adler, B.; Townes, T.M.; Parks, D.A.; Thompson, J.A.; Tousson, A.; Gladwin, M.T.; Patel, R.P.; Tarpey, M.M.; et al. Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proc. Natl. Acad. Sci. USA 2001, 18, 15215–15220. [Google Scholar] [CrossRef]
- Stuart, M.J.; Nagel, R.L. Sickle-cell disease. Lancet 2004, 364, 1343–1360. [Google Scholar] [CrossRef] [PubMed]
- Chirico, E.N.; Pialoux, V. Role of oxidative stress in the pathogenesis of sickle cell disease. IUBMB Life 2012, 64, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Dosunmu-Ogunbi, A.M.; Wood, K.C.; Novelli, E.M.; Straub, A.C. Decoding the role of SOD2 in sickle cell disease. Blood Adv. 2019, 3, 2679–2687. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, M.H.; Sebastiani, P. Genetic modifiers of sickle cell disease. Am. J. Hematol. 2012, 87, 795–803. [Google Scholar] [CrossRef]
- Ware, R.E.; de Montalembert, M.; Tshilolo, L.; Abboud, M.R. Sickle cell disease. Lancet 2017, 390, 311–323. [Google Scholar] [CrossRef]
- Cardenes, N.; Corey, C.; Geary, L.; Jain, S.; Zharikov, S.; Barge, S.; Novelli, E.M.; Shiva, S. Platelet bioenergetic screen in sickle cell patients reveals mitochondrial complex V inhibition, which contributes to platelet activation. Blood 2014, 123, 2864–2872. [Google Scholar] [CrossRef]
- Wood, K.C.; Granger, D.N. Sickle cell disease: Role of reactive oxygen and nitrogen metabolites. Clin. Exp. Pharmacol. Physiol. 2007, 34, 926–932. [Google Scholar] [CrossRef]
- George, A.; Pushkaran, S.; Konstantinidis, D.G.; Koochaki, S.; Malik, P.; Mohandas, N.; Zheng, Y.; Joiner, C.H.; Kalfa, T.A. Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood 2013, 121, 2099–2107. [Google Scholar] [CrossRef]
- Möller, M.N.; Orrico, F.; Villar, S.F.; López, A.C.; Silva, N.; Donzé, M.; Thomson, L.; Denicola, A. Oxidants and Antioxidants in the Redox Biochemistry of Human Red Blood Cells. ACS Omega 2022, 8, 147–168. [Google Scholar] [CrossRef]
- Schaer, D.J.; Buehler, P.W.; Alayash, A.I.; Belcher, J.D.; Vercellotti, G.M. Hemolysis and free hemoglobin revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 2013, 121, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.; Faustino, P. From Stress to Sick(le) and Back Again-Oxidative/Antioxidant Mechanisms, Genetic Modulation, and Cerebrovascular Disease in Children with Sickle Cell Anemia. Antioxidants 2023, 12, 1977. [Google Scholar] [CrossRef] [PubMed]
- Kassa, T.; Jana, S.; Strader, M.B.; Meng, F.; Jia, Y.; Wilson, M.T.; Alayash, A.I. Sickle Cell Hemoglobin in the Ferryl State Promotes βCys-93 Oxidation and Mitochondrial Dysfunction in Epithelial Lung Cells (E10). J. Biol. Chem. 2015, 290, 27939–27958. [Google Scholar] [CrossRef] [PubMed]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O.; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef]
- Aslan, M.; Freeman, B.A. Oxidant-mediated impairment of nitric oxide signaling in sickle cell disease—Mechanisms and consequences. Cell. Mol. Biol. 2004, 50, 95–105. [Google Scholar]
- Jeffers, A.; Gladwin, M.T.; Kim-Shapiro, D.B. Computation of plasma hemoglobin nitric oxide scavenging in hemolytic anemias. Free Radic. Biol. Med. 2006, 41, 1557–1565. [Google Scholar] [CrossRef]
- Meyerstein, D. What Are the Oxidizing Intermediates in the Fenton and Fenton-like Reactions? A Perspective. Antioxidants 2022, 11, 1368. [Google Scholar] [CrossRef]
- Wu, G.; Meininger, C.J.; McNeal, C.J.; Bazer, F.W.; Rhoads, J.M. Role of L-Arginine in Nitric Oxide Synthesis and Health in Humans. Adv. Exp. Med. Biol. 2021, 1332, 167–187. [Google Scholar]
- Morris, C.R.; Brown, L.A.S.; Reynolds, M.; Dampier, C.D.; Lane, P.A.; Watt, A.; Kumari, P.; Harris, F.; Manoranjithan, S.; Mendis, R.D.; et al. Impact of arginine therapy on mitochondrial function in children with sickle cell disease during vaso-occlusive pain. Blood 2020, 136, 1402–1406. [Google Scholar] [CrossRef]
- Lewis, C.V.; Sellak, H.; Hansen, L.; Joseph, G.; Hurtado, J.; Archer, D.R.; Jun, H.W.; Brown, L.A.; Taylor, W.R. Increasing nitric oxide bioavailability fails to improve collateral vessel formation in humanized sickle cell mice. Lab. Investig. 2022, 102, 805–813. [Google Scholar] [CrossRef]
- di Masi, A.; De Simone, G.; Ciaccio, C.; D’Orso, S.; Coletta, M.; Ascenzi, P. Haptoglobin: From hemoglobin scavenging to human health. Mol. Asp. Med. 2020, 73, 100851. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, J.H.; Etzerodt, A.; Svendsen, P.; Moestrup, S.K. The haptoglobin-CD163-heme oxygenase-1 pathway for hemoglobin scavenging. Oxid. Med. Cell. Longev. 2013, 2013, 523652. [Google Scholar] [CrossRef] [PubMed]
- Sesti-Costa, R.; Costa, F.F.; Conran, N. Role of macrophages in sickle cell disease erythrophagocytosis and erythropoiesis. Int. J. Mol. Sci. 2023, 24, 6333. [Google Scholar] [CrossRef] [PubMed]
- Koskenkorva-Frank, T.S.; Weiss, G.; Koppenol, W.H.; Burckhardt, S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: Insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic. Biol. Med. 2013, 65, 1174–1194. [Google Scholar] [CrossRef]
- Rifkind, J.M.; Mohanty, J.G.; Nagababu, E. The pathophysiology of extracellular hemoglobin associated with enhanced oxidative reactions. Front. Physiol. 2015, 5, 500. [Google Scholar] [CrossRef]
- Shah, F.T.; Porter, J.B.; Sadasivam, N.; Kaya, B.; Moon, J.C.; Velangi, M.; Ako, E.; Pancham, S. Guidelines for the monitoring and management of iron overload in patients with haemoglobinopathies and rare anaemias. Br. J. Haematol. 2022, 196, 336–350. [Google Scholar] [CrossRef]
- Ballas, S.K. Iron overload is a determinant of morbidity and mortality in adult patients with sickle cell disease. Semin. Hematol. 2001, 38, 30–36. [Google Scholar] [CrossRef]
- van Beers, E.J.; Yang, Y.; Raghavachari, N.; Tian, X.; Allen, D.T.; Nichols, J.S.; Mendelsohn, L.; Nekhai, S.; Gordeuk, V.R.; Taylor, J.G.; et al. Iron, inflammation, and early death in adults with sickle cell disease. Circ. Res. 2015, 116, 298–306. [Google Scholar] [CrossRef]
- Coates, T.D.; Wood, J.C. How we manage iron overload in sickle cell patients. Br. J. Haematol. 2017, 177, 703–716. [Google Scholar] [CrossRef]
- Canli, Ö.; Alankuş, Y.B.; Grootjans, S.; Vegi, N.; Hültner, L.; Hoppe, P.S.; Schroeder, T.; Vandenabeele, P.; Bornkamm, G.W.; Greten, F.R. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood 2016, 127, 139–148. [Google Scholar] [CrossRef]
- Andrews, N.C.; Schmidt, P.J. Iron homeostasis. Annu. Rev. Physiol. 2007, 69, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Ponka, P. The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim. Biophys. Acta 1997, 1331, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Kuang, F.; Liu, J.; Tang, D.; Kang, R. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 586578. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020, 30, 3411–3423.e7. [Google Scholar] [CrossRef]
- El Hout, M.; Dos Santos, L.; Hamaï, A.; Mehrpour, M. A promising new approach to cancer therapy: Targeting iron metabolism in cancer stem cells. Semin. Cancer Biol. 2018, 53, 125–138. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron metabolism in ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef]
- Yambire, K.F.; Rostosky, C.; Watanabe, T.; Pacheu-Grau, D.; Torres-Odio, S.; Sanchez-Guerrero, A.; Senderovich, O.; Meyron-Holtz, E.G.; Milosevic, I.; Frahm, J.; et al. Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo. Elife 2019, 8, e51031. [Google Scholar] [CrossRef]
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells 2020, 9, 1505. [Google Scholar] [CrossRef]
- Galy, B.; Conrad, M.; Muckenthaler, M. Mechanisms controlling cellular and systemic iron homeostasis. Nat. Rev. Mol. Cell Biol. 2024, 25, 133–155. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, S.; Wu, L.L.; Yang, L.; Yang, L.; Wang, J. The diversified role of mitochondria in ferroptosis in cancer. Cell Death Dis. 2023, 14, 519. [Google Scholar] [CrossRef]
- Oh, S.J.; Ikeda, M.; Ide, T.; Hur, K.Y.; Lee, M.S. Correction: Mitochondrial event as an ultimate step in ferroptosis. Cell Death Discov. 2022, 8, 422. [Google Scholar] [CrossRef] [PubMed]
- Le, Y.; Zhang, Z.; Wang, C.; Lu, D. Ferroptotic cell death: New regulatory mechanisms for metabolic diseases. Endocr. Metab. Immune Disord. Drug Targets 2021, 21, 785–800. [Google Scholar]
- Brissot, P.; Ropert, M.; Le Lan, C.; Loréal, O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim. Biophys. Acta 2012, 1820, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.S.; Ruiz, J.; Watts, J.L. Polyunsaturated Fatty Acids Drive Lipid Peroxidation during Ferroptosis. Cells 2023, 12, 804. [Google Scholar] [CrossRef]
- Sun, S.; Shen, J.; Jiang, J.; Wang, F.; Min, J. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal Transduct. Target. Ther. 2023, 8, 372. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Cheu, J.W.; Lee, D.; Li, Q.; Goh, C.C.; Bao, M.H.; Yuen, V.W.; Zhang, M.S.; Yang, C.; Chan, C.Y.; Tse, A.P.; et al. Ferroptosis Suppressor Protein 1 Inhibition Promotes Tumor Ferroptosis and Anti-tumor Immune Responses in Liver Cancer. Cell. Mol. Gastroenterol. Hepatol. 2023, 16, 133–159. [Google Scholar] [CrossRef]
- Dai, Q.; Wei, X.; Zhao, J.; Zhang, D.; Luo, Y.; Yang, Y.; Xiang, Y.; Liu, X. Inhibition of FSP1: A new strategy for the treatment of tumors (Review). Oncol. Rep. 2024, 52, 105. [Google Scholar] [CrossRef]
- Liang, D.; Feng, Y.; Zandkarimi, F.; Wang, H.; Zhang, Z.; Kim, J.; Cai, Y.; Gu, W.; Stockwell, B.R.; Jiang, X. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell 2023, 186, 2748–2764.e22. [Google Scholar] [CrossRef] [PubMed]
- Cronin, S.J.F.; Seehus, C.; Weidinger, A.; Talbot, S.; Reissig, S.; Seifert, M.; Pierson, Y.; McNeill, E.; Longhi, M.S.; Turnes, B.L.; et al. The metabolite BH4 controls T cell proliferation in autoimmunity and cancer. Nature 2018, 563, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Lin, B.; Jin, W.; Tang, L.; Hu, S.; Cai, R. NRF2, a Superstar of Ferroptosis. Antioxidants 2023, 12, 1739. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, V.; Lima, J.; Oliveira, G.F.; Oliveira, Y.S.; Getachew, B.; Nekhai, S.; Aschner, M.; Tizabi, Y. Ferroptosis as an emerging target in sickle cell disease. Curr. Res. Toxicol. 2024, 7, 100181. [Google Scholar] [CrossRef]
- Pirenne, F.; Floch, A.; Habibi, A. How to avoid the problem of erythrocyte alloimmunization in sickle cell disease. Hematol. Am. Soc. Hematol. Educ. Program 2021, 2021, 689–695. [Google Scholar] [CrossRef]
- Arthur, C.M.; Stowell, S.R. The Development and Consequences of Red Blood Cell Alloimmunization. Annu. Rev. Pathol. 2023, 18, 537–564. [Google Scholar] [CrossRef]
- Ashouri, R.; Fangman, M.; Burris, A.; Ezenwa, M.O.; Wilkie, D.J.; Doré, S. Critical role of hemopexin mediated cytoprotection in the pathophysiology of sickle cell disease. Int. J. Mol. Sci. 2021, 22, 6408. [Google Scholar] [CrossRef]
- Gbotosho, O.T.; Kapetanaki, M.G.; Kato, G.J. The worst things in life are free: The role of free heme in sickle cell disease. Front. Immunol. 2021, 11, 561917. [Google Scholar] [CrossRef]
- Menon, A.V.; Liu, J.; Tsai, H.P.; Zeng, L.; Yang, S.; Asnani, A.; Kim, J. Excess heme upregulates heme oxygenase 1 and promotes cardiac ferroptosis in mice with sickle cell disease. Blood 2022, 139, 936–941. [Google Scholar] [CrossRef]
- Yu, Y.; Jiang, L.; Wang, H.; Shen, Z.; Cheng, Q.; Zhang, P.; Wang, J.; Wu, Q.; Fang, X.; Duan, L.; et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood 2020, 136, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; An, P.; Xie, E.; Wu, Q.; Fang, X.; Gao, H.; Zhang, Z.; Li, Y.; Wang, X.; Zhang, J.; et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017, 66, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Xi, C.; Pang, J.; Zhi, W.; Chang, C.S.; Siddaramappa, U.; Shi, H.; Horuzsko, A.; Pace, B.S.; Zhu, X. Nrf2 sensitizes ferroptosis through l-2-hydroxyglutarate-mediated chromatin modifications in sickle cell disease. Blood 2023, 142, 382–396. [Google Scholar] [CrossRef]
- Reid, M.; Badaloo, A.; Forrester, T.; Jahoor, F. In vivo rates of erythrocyte glutathione synthesis in adults with sickle cell disease. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E73–E79. [Google Scholar] [CrossRef] [PubMed]
- Xi, C.; Pang, J.; Xue, W.; Cui, Y.; Jiang, N.; Zhi, W.; Shi, H.; Horuzsko, A.; Pace, B.S.; Zhu, X. Transsulfuration pathway activation attenuates oxidative stress and ferroptosis in sickle primary erythroblasts and transgenic mice. Commun. Biol. 2025, 8, 15. [Google Scholar] [CrossRef]
- Geneen, L.J.; Dorée, C.; Estcourt, L.J. Interventions for improving adherence to iron chelation therapy in people with sickle cell disease or thalassaemia. Cochrane Database Syst. Rev. 2023, 3, CD012349. [Google Scholar]
- Nur, E.; Biemond, B.J.; Otten, H.M.; Brandjes, D.P.; Schnog, J.J.; CURAMA Study Group. Oxidative stress in sickle cell disease; pathophysiology and potential implications for disease management. Am. J. Hematol. 2011, 86, 484–489. [Google Scholar] [CrossRef]
- Nader, E.; Romana, M.; Connes, P. The Red Blood Cell-Inflammation Vicious Circle in Sickle Cell Disease. Front. Immunol. 2020, 11, 454. [Google Scholar] [CrossRef]
- Biswal, S.; Rizwan, H.; Pal, S.; Sabnam, S.; Parida, P.; Pal, A. Oxidative stress, antioxidant capacity, biomolecule damage, and inflammation symptoms of sickle cell disease in children. Hematology 2019, 24, 1–9. [Google Scholar] [CrossRef]
- Jana, S.; Strader, M.B.; Meng, F.; Hicks, W.; Kassa, T.; Tarandovskiy, I.; Paoli, S.D.; Simak, J.; Heaven, M.R.; Belcher, J.D.; et al. Hemoglobin oxidation–dependent reactions promote interactions with band 3 and oxidative changes in sickle cell–derived microparticles. JCI Insight 2018, 3, e120451. [Google Scholar] [CrossRef]
- Orrico, F.; Laurance, S.; Lopez, A.C.; Lefevre, S.D.; Thomson, L.; Möller, M.N.; Ostuni, M.A. Oxidative stress in healthy and pathological red blood cells. Biomolecules 2023, 13, 1262. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Investig. 2017, 127, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Fujii, J.; Homma, T.; Kobayashi, S.; Warang, P.; Madkaikar, M.; Mukherjee, M.B. Erythrocytes as a preferential target of oxidative stress in blood. Free Radic. Res. 2021, 55, 562–580. [Google Scholar] [CrossRef]
- Vinchi, F.; Sparla, R.; Passos, S.T.; Sharma, R.; Vance, S.Z.; Zreid, H.S.; Juaidi, H.; Manwani, D.; Yazdanbakhsh, K.; Nandi, V.; et al. Vasculo-toxic and pro-inflammatory action of unbound haemoglobin, haem and iron in transfusion-dependent patients with haemolytic anaemias. Br. J. Haematol. 2021, 193, 637–658. [Google Scholar] [CrossRef]
- NaveenKumar, S.K.; SharathBabu, B.N.; Hemshekhar, M.; Kemparaju, K.; Girish, K.S.; Mugesh, G. The role of reactive oxygen species and ferroptosis in heme-mediated activation of human platelets. ACS Chem. Biol. 2018, 13, 1996–2002. [Google Scholar] [CrossRef]
- NaveenKumar, S.K.; Hemshekhar, M.; Kemparaju, K.; Girish, K.S. Hemin-induced platelet activation and ferroptosis is mediated through ROS-driven proteasomal activity and inflammasome activation: Protection by melatonin. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2303–2316. [Google Scholar] [CrossRef] [PubMed]
- Conran, N.; De Paula, E.V. Thromboinflammatory mechanisms in sickle cell disease—Challenging the hemostatic balance. Haematologica 2020, 105, 2380–2390. [Google Scholar] [CrossRef]
- Nader, E.; Romana, M.; Guillot, N.; Fort, R.; Stauffer, E.; Lemonne, N.; Garnier, Y.; Skinner, S.C.; Etienne-Julan, M.; Robert, M.; et al. Association between nitric oxide, oxidative stress, eryptosis, red blood cell microparticles, and vascular function in sickle cell anemia. Front. Immunol. 2020, 11, 551441. [Google Scholar] [CrossRef]
- Jutant, E.M.; Voiriot, G.; Labbé, V.; Savale, L.; Mokrani, H.; Van Dreden, P.; Gerotziafas, G.; Fartoukh, M. Endothelial dysfunction and hypercoagulability in severe sickle-cell acute chest syndrome. Eur. Respir. J. Open Res. 2021, 7, 00496–02021. [Google Scholar] [CrossRef]
- Wang, Q.; Zennadi, R. The role of RBC oxidative stress in sickle cell disease: From the molecular basis to pathologic implications. Antioxidants 2021, 10, 1608. [Google Scholar] [CrossRef]
- Aboderin, F.I.; Oduola, T.; Davison, G.M.; Oguntibeju, O.O. A review of the relationship between the immune response, inflammation, oxidative stress, and the pathogenesis of sickle cell anaemia. Biomedicines 2023, 11, 2413. [Google Scholar] [CrossRef] [PubMed]
- Ansari, J.; Gavins, F.N.E. Ischemia-reperfusion injury in sickle cell disease: From basics to therapeutics. Am. J. Pathol. 2019, 189, 706–718. [Google Scholar] [CrossRef]
- Hebbel, R.P.; Belcher, J.D.; Vercellotti, G.M. The multifaceted role of ischemia/reperfusion in sickle cell anemia. J. Clin. Investig. 2020, 130, 1062–1072. [Google Scholar] [CrossRef] [PubMed]
- Khaibullina, A.; Almeida, L.E.F.; Kamimura, S.; Zerfas, P.M.; Smith, M.L.; Vogel, S.; Wakim, P.; Vasconcelos, O.M.; Quezado, M.M.; Horkayne-Szakaly, I.; et al. Sickle cell disease mice have cerebral oxidative stress and vascular and white matter abnormalities. Blood Cell. Mol. Dis. 2021, 86, 102493. [Google Scholar] [CrossRef]
- Ajibola, K.A.; Adedokun, K.A.; Oduola, T.; Oparinde, D.P.; Ayelagbe, O.G.; Ojokuku, H.O. Assessment of iron status and interplay between lipid peroxidation and antioxidant capacity in common hemoglobin variants in Osun State, southwestern Nigeria. Kaohsiung J. Med. Sci. 2019, 35, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Park, F.; Soni, H.; Pressly, J.D.; Adebiyi, A. Acute hydroxyurea treatment reduces tubular damage following bilateral ischemia-reperfusion injury in a mouse model of sickle cell disease. Biochem. Biophys. Res. Commun. 2019, 515, 72–76. [Google Scholar] [CrossRef]
- Antwi-Boasiako, C.; Dankwah, G.B.; Aryee, R.; Hayfron-Benjamin, C.; Aboagye, G.; Campbell, A.D. Correlation of lipid peroxidation and nitric oxide metabolites, trace elements, and antioxidant enzymes in patients with sickle cell disease. J. Clin. Lab. Anal. 2020, 34, e23294. [Google Scholar] [CrossRef] [PubMed]
- Engwa, G.A.; Okolie, A.; Chidili, J.P.C.; Okore, P.A.; Onu, P.C.; Ugwu, M.O.; Oko, D.E.; Ferdinand, P.U. Relationship of oxidative stress and antioxidant response with vaso-occlusive crisis in sickle cell anaemia. Afr. Health Sci. 2021, 21, 150–158. [Google Scholar] [CrossRef]
- Yan, H.F.; Tuo, Q.Z.; Yin, Q.Z.; Lei, P. The pathological role of ferroptosis in ischemia/reperfusion-related injury. Zool. Res. 2020, 41, 220–230. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Ferroptosis in infection, inflammation, and immunity. J. Exp. Med. 2021, 218, e20210518. [Google Scholar] [CrossRef]
- Schmidt, H.M.; Wood, K.C.; Lewis, S.E.; Hahn, S.A.; Williams, X.M.; McMahon, B.; Baust, J.J.; Yuan, S.; Bachman, T.N.; Wang, Y.; et al. Xanthine oxidase drives hemolysis and vascular malfunction in sickle cell disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fang, Z.M.; Yi, X.; Wei, X.; Jiang, D.S. The interaction between ferroptosis and inflammatory signaling pathways. Cell Death Dis. 2023, 14, 205. [Google Scholar] [CrossRef]
- Wang, H.R.; Li, M.Z.; Cui, J.G.; Zhang, H.; Zhao, Y.; Li, J.L. Lycopene prevents phthalate-induced cognitive impairment via modulating ferroptosis. J. Agric. Food Chem. 2023, 71, 16727–16738. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Gomperts, E.; Nguyen, J.; Chen, C.; Abdulla, F.; Kiser, Z.M.; Gallo, D.; Levy, H.; Otterbein, L.E.; Vercellotti, G.M. Oral carbon monoxide therapy in murine sickle cell disease: Beneficial effects on vaso-occlusion, inflammation and anemia. PLoS ONE 2018, 13, e0205194. [Google Scholar] [CrossRef]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef]
- Liu, R.; Zhang, X.; Nie, L.; Sun, S.; Liu, J.; Chen, H. Heme oxygenase 1 in erythropoiesis: An important regulator beyond catalyzing heme catabolism. Ann. Hematol. 2023, 102, 1323–1332. [Google Scholar] [CrossRef]
- Azevedo, J.T.C.; Malmegrim, K.C.R. Immune mechanisms involved in sickle cell disease pathogenesis: Current knowledge and perspectives. Immunol. Lett. 2020, 224, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.E.; Trottier, E.D.; Kirby-Allen, M.; Pastore, Y. Acute complications in children with sickle cell disease: Prevention and management. Paediatr. Child Health 2022, 27, 50–62. [Google Scholar] [CrossRef]
- Gbotosho, O.T.; Gollamudi, J.; Hyacinth, H.I. The role of inflammation in the cellular and molecular mechanisms of cardiopulmonary complications of sickle cell disease. Biomolecules 2023, 13, 381. [Google Scholar] [CrossRef]
- Sharma, R.; Antypiuk, A.; Vance, S.Z.; Manwani, D.; Pearce, Q.; Cox, J.E.; An, X.; Yazdanbakhsh, K.; Vinchi, F. Macrophage metabolic rewiring improves heme-suppressed efferocytosis and tissue damage in sickle cell disease. Blood 2023, 141, 3091–3108. [Google Scholar] [CrossRef]
- Kanias, T.; Lanteri, M.C.; Page, G.P.; Guo, Y.; Endres, S.M.; Stone, M.; Keating, S.; Mast, A.E.; Cable, R.G.; Triulzi, D.J.; et al. Ethnicity, sex, and age are determinants of red blood cell storage and stress hemolysis: Results of the REDS-III RBC-Omics study. Blood Adv. 2017, 1, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Van ’t Erve, T.J.; Wagner, B.A.; Martin, S.M.; Knudson, C.M.; Blendowski, R.; Keaton, M.; Holt, T.; Hess, J.R.; Buettner, G.R.; Ryckman, K.K.; et al. The heritability of hemolysis in stored human red blood cells. Transfusion 2015, 55, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Stolwijk, J.M.; Stefely, J.A.; Veling, M.T.; van ’t Erve, T.J.; Wagner, B.A.; Raife, T.J.; Buettner, G.R. Red blood cells contain enzymatically active GPx4 whose abundance anticorrelates with hemolysis during blood bank storage. Redox Biol. 2021, 46, 102073. [Google Scholar] [CrossRef]
- Yoshida, T.; Prudent, M.; D’Alessandro, A. Red blood cell storage lesion: Causes and potential clinical consequences. Blood Transfus. 2019, 17, 27–52. [Google Scholar]
- D’Alessandro, A.; Keele, G.R.; Hay, A.; Nemkov, T.; Earley, E.J.; Stephenson, D.; Vincent, M.; Deng, X.; Stone, M.; Dzieciatkowska, M.; et al. Ferroptosis regulates hemolysis in stored murine and human red blood cells. Blood 2025, 145, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Youssef, L.A.; Rebbaa, A.; Pampou, S.; Weisberg, S.P.; Stockwell, B.R.; Hod, E.A.; Spitalnik, S.L. Increased erythrophagocytosis induces ferroptosis in red pulp macrophages in a mouse model of transfusion. Blood 2018, 131, 2581–2593. [Google Scholar] [CrossRef]
- Soares, M.P.; Hamza, I. Macrophages and iron metabolism. Immunity 2016, 44, 492–504. [Google Scholar] [CrossRef]
- Telen, M.J. Beyond hydroxyurea: New and old drugs in the pipeline for sickle cell disease. Blood 2016, 127, 810–819. [Google Scholar] [CrossRef]
- Osunkwo, I.; Manwani, D.; Kanter, J. Current and novel therapies for the prevention of vaso-occlusive crisis in sickle cell disease. Ther. Adv. Hematol. 2020, 11, 2040620720955000. [Google Scholar] [CrossRef]
- Santana, S.S.; Pitanga, T.N.; de Santana, J.M.; Zanette, D.L.; de Vieira, J.J.; Yahouédéhou, S.C.M.A.; Adanho, C.S.A.; de Viana, S.M.; Luz, N.F.; Borges, V.M.; et al. Hydroxyurea scavenges free radicals and induces the expression of antioxidant genes in human cell cultures treated with hemin. Front. Immunol. 2020, 11, 1488. [Google Scholar] [CrossRef]
- Bruzzese, A.; Martino, E.A.; Mendicino, F.; Lucia, E.; Olivito, V.; Bova, C.; Filippelli, G.; Capodanno, I.; Neri, A.; Morabito, F.; et al. Iron chelation therapy. Eur. J. Haematol. 2023, 110, 490–497. [Google Scholar] [CrossRef]
- Hamdy, M.; El-Beshlawy, A.; Veríssimo, M.P.A.; Kanter, J.; Inusa, B.; Williams, S.; Lee, D.; Temin, N.T.; Fradette, C.; Tricta, F.; et al. Deferiprone versus deferoxamine for transfusional iron overload in sickle cell disease and other anemias: Pediatric subgroup analysis of the randomized, open-label FIRST study. Pediatr. Blood Cancer 2023, 71, e30711. [Google Scholar] [CrossRef] [PubMed]
- Scarpellini, C.; Klejborowska, G.; Lanthier, C.; Hassannia, B.; Vanden Berghe, T.; Augustyns, K. Beyond ferrostatin-1: A comprehensive review of ferroptosis inhibitors. Trends Pharmacol. Sci. 2023, 44, 902–916. [Google Scholar] [CrossRef]
- Gulcin, İ. Antioxidants and antioxidant methods: An updated overview. Arch. Toxicol. 2020, 94, 651–715. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Zhang, Y.; Lou, H.; Ou, Z.; Liu, J.; Duan, W.; Wang, H.; Ge, Y.; Min, J.; Wang, F.; et al. GPX4 and vitamin E cooperatively protect hematopoietic stem and progenitor cells from lipid peroxidation and ferroptosis. Cell Death Dis. 2021, 12, 706. [Google Scholar] [CrossRef] [PubMed]
- Lan, D.; Yao, C.; Li, X.; Liu, H.; Wang, D.; Wang, Y.; Qi, S. Tocopherol attenuates the oxidative stress of BMSCs by inhibiting ferroptosis through the PI3k/AKT/mTOR pathway. Front. Bioeng. Biotechnol. 2022, 10, 938520. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef]
- Zhang, W.; Qian, S.; Kang, P.; Shi, C. Curcumin attenuates ferroptosis-induced myocardial injury in diabetic cardiomyopathy through the Nrf2 pathway. Cardiovasc. Ther. 2022, 2022, 3159717. [Google Scholar]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef]
- Patel, N.; Gonsalves, C.S.; Yang, M.; Malik, P.; Kalra, V.K. Placenta growth factor induces 5-lipoxygenase-activating protein to increase leukotriene formation in sickle cell disease. Blood 2009, 113, 1129–1138. [Google Scholar] [CrossRef]
- Haynes, J.; Obiako, B.; King, J.A.; Hester, R.B.; Ofori-Acquah, S. Activated neutrophil-mediated sickle red blood cell adhesion to lung vascular endothelium: Role of phosphatidylserine-exposed sickle red blood cells. Am. J. Phys. Heart Circ. Phys. 2006, 291, H1679–H1685. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Guan, Y.; Hu, W.; Xu, Z.; Ishfaq, M. An overview of pharmacological activities of baicalin and its aglycone baicalein: New insights into molecular mechanisms and signaling pathways. Iran. J. Basic Med. Sci. 2022, 25, 14–26. [Google Scholar] [PubMed]
- Nataraja, S.; Singh, M.; Demes, S.; Olson, L.; Stanwix, J.; Biddle, M.; Clarke, E.; Huang, Y.; Nguyen, J.; Chen, C.; et al. ML-0207/ASP8731: A novel BACH1 inhibitor that induces fetal hemoglobin in treatment of sickle cell disease. Blood 2021, 138, 854. [Google Scholar] [CrossRef]
- Belcher, J.D.; Nataraja, S.; Abdulla, F.; Zhang, P.; Chen, C.; Nguyen, J.; Ruan, C.; Singh, M.; Demes, S.; Olson, L.; et al. The BACH1 inhibitor ASP8731 inhibits inflammation and vaso-occlusion and induces fetal hemoglobin in sickle cell disease. Front. Med. 2023, 10, 1101501. [Google Scholar] [CrossRef]
- Nishizawa, H.; Matsumoto, M.; Shindo, T.; Saigusa, D.; Kato, H.; Suzuki, K.; Sato, M.; Ishii, Y.; Shimokawa, H.; Igarashi, K. Ferroptosis is controlled by the coordinated transcriptional regulation of glutathione and labile iron metabolism by the transcription factor BACH1. J. Biol. Chem. 2020, 295, 69–82. [Google Scholar] [CrossRef]
- Irikura, R.; Nishizawa, H.; Nakajima, K.; Yamanaka, M.; Chen, G.; Tanaka, K.; Onodera, M.; Matsumoto, M.; Igarashi, K. Ferroptosis model system by the re-expression of BACH1. J. Biochem. 2023, 174, 239–252. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, A.; Patanè, G.T.; Calderaro, A.; Barreca, D.; Tellone, E.; Putaggio, S. Crosstalk Between Sickle Cell Disease and Ferroptosis. Int. J. Mol. Sci. 2025, 26, 3675. https://doi.org/10.3390/ijms26083675
Russo A, Patanè GT, Calderaro A, Barreca D, Tellone E, Putaggio S. Crosstalk Between Sickle Cell Disease and Ferroptosis. International Journal of Molecular Sciences. 2025; 26(8):3675. https://doi.org/10.3390/ijms26083675
Chicago/Turabian StyleRusso, Annamaria, Giuseppe Tancredi Patanè, Antonella Calderaro, Davide Barreca, Ester Tellone, and Stefano Putaggio. 2025. "Crosstalk Between Sickle Cell Disease and Ferroptosis" International Journal of Molecular Sciences 26, no. 8: 3675. https://doi.org/10.3390/ijms26083675
APA StyleRusso, A., Patanè, G. T., Calderaro, A., Barreca, D., Tellone, E., & Putaggio, S. (2025). Crosstalk Between Sickle Cell Disease and Ferroptosis. International Journal of Molecular Sciences, 26(8), 3675. https://doi.org/10.3390/ijms26083675