
Predicting Motif-Mediated Interactions Based on Viral Genomic Composition

 , ,
, ,
Abstract
1. Introduction
2. Results
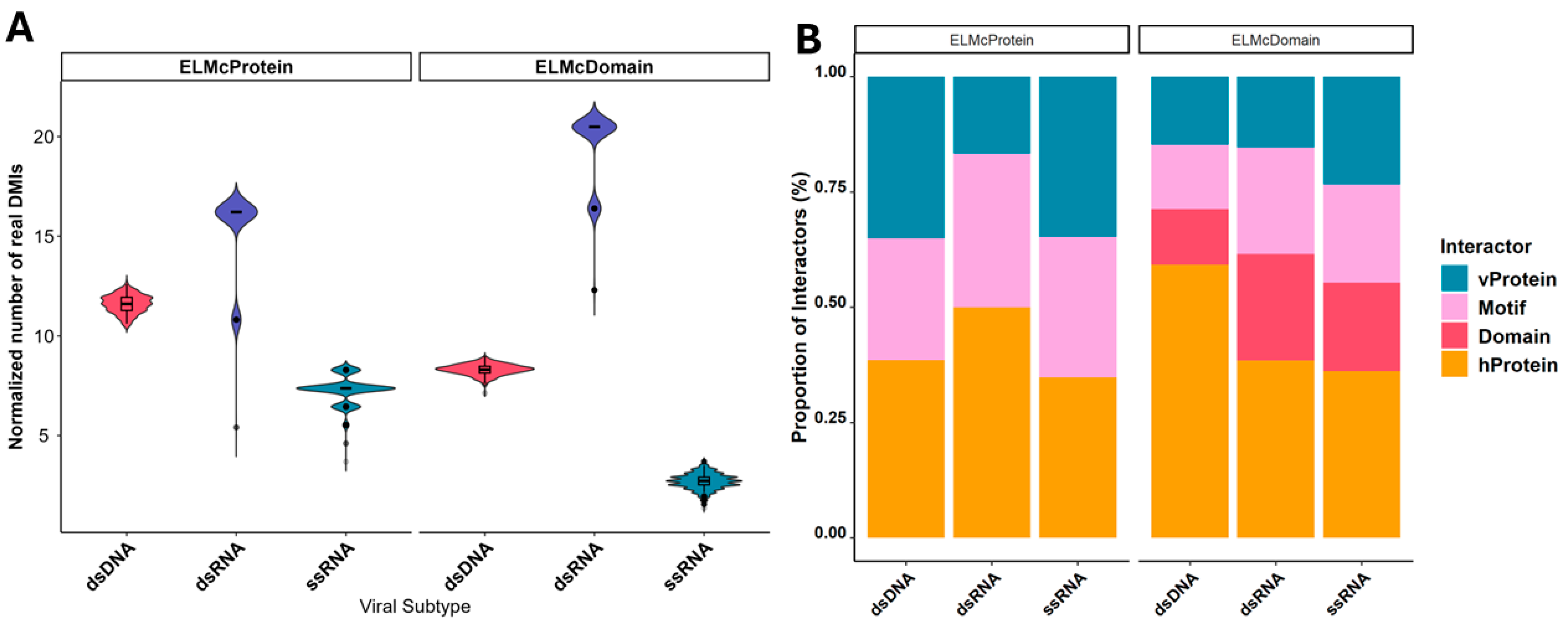
2.1. Enrichment of DMIs Based on Viral Genomic Composition
2.2. DMI Prediction Using Known Viral ELMs
2.3. Expanding the DMI Network Through Incorporating Domain Information
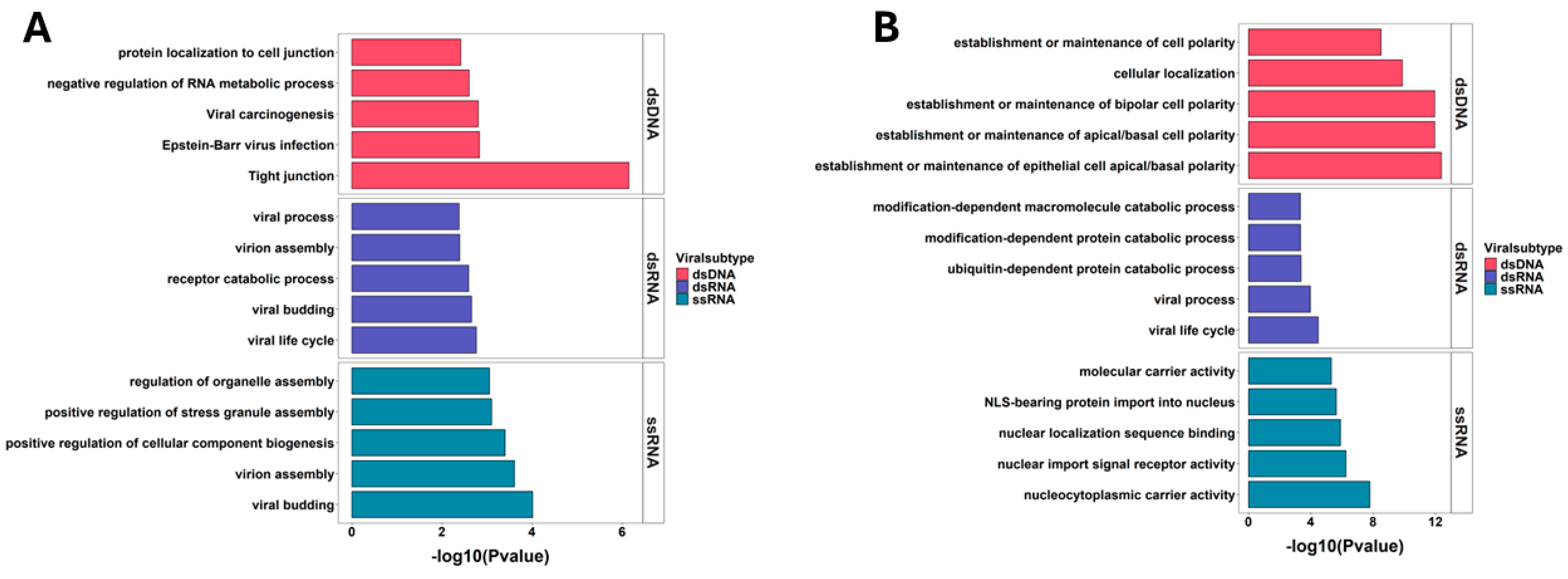
2.4. Host Proteins Targeted by Different Viral Genomic Categories
2.5. Pathways Hijacked by Different Viral Groups
2.6. Cross-Validation of Predictions Using ELM Known Interactions
3. Discussion
4. Materials and Methods
4.1. Data Retrieval and Processing
4.2. DMI Enrichment in Different Viral Groups
4.3. DMI Prediction in Different Viral Groups
4.4. Host-Hijacked Proteins and Pathway Analysis
- Unique to a single viral group: proteins targeted only by dsRNA, dsDNA, or ssRNA viruses.
- Shared between two viral groups: proteins targeted by combinations of two viral types (dsRNA + dsDNA, dsRNA + ssRNA, or dsDNA + ssRNA).
- Shared across all three viral groups: proteins targeted by dsRNA, dsDNA, and ssRNA viruses.
4.5. Cross-Validation of Predictions Using ELM Database
4.6. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Glavina, J.; Palopoli, N.; Chemes, L.B. Evolution of SLiM-mediated hijack functions in intrinsically disordered viral proteins. Essays Biochem. 2022, 66, 945–958. [Google Scholar] [CrossRef] [PubMed]
- Tenthorey, J.L.; Emerman, M.; Malik, H.S. Evolutionary Landscapes of Host-Virus Arms Races. Annu. Rev. Immunol. 2022, 40, 271–294. [Google Scholar] [CrossRef] [PubMed]
- Hraber, P.; O’Maille, P.E.; Silberfarb, A.; Davis-Anderson, K.; Generous, N.; McMahon, B.H.; Fair, J.M. Resources to Discover and Use Short Linear Motifs in Viral Proteins. Trends Biotechnol. 2020, 38, 113–127. [Google Scholar] [CrossRef]
- Villanueva, R.A.; Rouille, Y.; Dubuisson, J. Interactions between virus proteins and host cell membranes during the viral life cycle. Int. Rev. Cytol. 2005, 245, 171–244. [Google Scholar] [CrossRef] [PubMed]
- Becerra, A.; Bucheli, V.A.; Moreno, P.A. Prediction of virus-host protein-protein interactions mediated by short linear motifs. BMC Bioinform. 2017, 18, 163. [Google Scholar] [CrossRef]
- Idrees, S. Predicting Motif Mimicry in Viruses; UNSW Sydney: Kensington, Australia, 2020. [Google Scholar]
- Idrees, S.; Paudel, K.R.; Sadaf, T.; Hansbro, P.M. Uncovering domain motif interactions using high-throughput protein-protein interaction detection methods. FEBS Lett. 2024, 598, 725–742. [Google Scholar] [CrossRef]
- Mihalič, F.; Benz, C.; Kassa, E.; Lindqvist, R.; Simonetti, L.; Inturi, R.; Aronsson, H.; Andersson, E.; Chi, C.N.; Davey, N.E.; et al. Identification of motif-based interactions between SARS-CoV-2 protein domains and human peptide ligands pinpoint antiviral targets. Nat. Commun. 2023, 14, 5636. [Google Scholar] [CrossRef]
- Elkhaligy, H.; Balbin, C.A.; Gonzalez, J.L.; Liberatore, T.; Siltberg-Liberles, J. Dynamic, but Not Necessarily Disordered, Human-Virus Interactions Mediated through SLiMs in Viral Proteins. Viruses 2021, 13, 2369. [Google Scholar] [CrossRef]
- Yang, C.W. A comparative study of short linear motif compositions of the influenza A virus ribonucleoproteins. PLoS ONE 2012, 7, e38637. [Google Scholar] [CrossRef]
- Idrees, S.; Paudel, K.R. Proteome-wide assessment of human interactome as a source of capturing domain–motif and domain-domain interactions. J. Cell Commun. Signal. 2023, 18, e12014. [Google Scholar] [CrossRef]
- Idrees, S.; Paudel, K.R.; Hansbro, P.M. Prediction of motif-mediated viral mimicry through the integration of host-pathogen interactions. Arch. Microbiol. 2024, 206, 94. [Google Scholar] [CrossRef]
- Soorajkumar, A.; Alakraf, E.; Uddin, M.; Du Plessis, S.S.; Alsheikh-Ali, A.; Kandasamy, R.K. Computational Analysis of Short Linear Motifs in the Spike Protein of SARS-CoV-2 Variants Provides Possible Clues into the Immune Hijack and Evasion Mechanisms of Omicron Variant. Int. J. Mol. Sci. 2022, 23, 8822. [Google Scholar] [CrossRef] [PubMed]
- Halehalli, R.R.; Nagarajaram, H.A. Molecular principles of human virus protein-protein interactions. Bioinformatics 2015, 31, 1025–1033. [Google Scholar] [CrossRef]
- Meszaros, B.; Samano-Sanchez, H.; Alvarado-Valverde, J.; Calyseva, J.; Martinez-Perez, E.; Alves, R.; Shields, D.C.; Kumar, M.; Rippmann, F.; Chemes, L.B.; et al. Short linear motif candidates in the cell entry system used by SARS-CoV-2 and their potential therapeutic implications. Sci. Signal 2021, 14, eabd0334. [Google Scholar] [CrossRef] [PubMed]
- Pushker, R.; Mooney, C.; Davey, N.E.; Jacque, J.M.; Shields, D.C. Marked variability in the extent of protein disorder within and between viral families. PLoS ONE 2013, 8, e60724. [Google Scholar] [CrossRef]
- Kumar, M.; Gouw, M.; Michael, S.; Samano-Sanchez, H.; Pancsa, R.; Glavina, J.; Diakogianni, A.; Valverde, J.A.; Bukirova, D.; Calyseva, J.; et al. ELM-the eukaryotic linear motif resource in 2020. Nucleic Acids Res. 2020, 48, D296–D306. [Google Scholar] [CrossRef]
- Kumar, N.; Kaushik, R.; Tennakoon, C.; Uversky, V.N.; Longhi, S.; Zhang, K.Y.J.; Bhatia, S. Comprehensive Intrinsic Disorder Analysis of 6108 Viral Proteomes: From the Extent of Intrinsic Disorder Penetrance to Functional Annotation of Disordered Viral Proteins. J. Proteome Res. 2021, 20, 2704–2713. [Google Scholar] [CrossRef] [PubMed]
- Schuck, P.; Zhao, H. Diversity of short linear interaction motifs in SARS-CoV-2 nucleocapsid protein. mBio 2023, 14, e0238823. [Google Scholar] [CrossRef]
- Klink, G.V.; Kalinina, O.V.; Bazykin, G.A. Changing selection on amino acid substitutions in Gag protein between major HIV-1 subtypes. Virus Evol. 2024, 10, veae036. [Google Scholar] [CrossRef]
- Righetto, I.; Milani, A.; Cattoli, G.; Filippini, F. Comparative structural analysis of haemagglutinin proteins from type A influenza viruses: Conserved and variable features. BMC Bioinform. 2014, 15, 363. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Koonin, E.V.; Donchenko, A.P.; Blinov, V.M. Coronavirus genome: Prediction of putative functional domains in the non-structural polyprotein by comparative amino acid sequence analysis. Nucleic Acids Res. 1989, 17, 4847–4861. [Google Scholar] [CrossRef] [PubMed]
- Hou, D.; Zhang, L.; Deng, F.; Fang, W.; Wang, R.; Liu, X.; Guo, L.; Rayner, S.; Chen, X.; Wang, H.; et al. Comparative proteomics reveal fundamental structural and functional differences between the two progeny phenotypes of a baculovirus. J. Virol. 2013, 87, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Idrees, S.; Paudel, K.R.; Sadaf, T.; Hansbro, P.M. How different viruses perturb host cellular machinery via short linear motifs. EXCLI J. 2023, 22, 1113–1128. [Google Scholar]
- Idrees, S.; Perez-Bercoff, A.; Edwards, R.J. SLiMEnrich: Computational assessment of protein-protein interaction data as a source of domain-motif interactions. PeerJ 2018, 6, e5858. [Google Scholar] [CrossRef] [PubMed]
- Idrees, S.; Paudel, K.R. Bioinformatics prediction and screening of viral mimicry candidates through integrating known and predicted DMI data. Arch. Microbiol. 2023, 206, 30. [Google Scholar] [CrossRef]
- Banik, M.; Paudel, K.R.; Majumder, R.; Idrees, S. Prediction of virus-host interactions and identification of hot spot residues of DENV-2 and SH3 domain interactions. Arch. Microbiol. 2024, 206, 162. [Google Scholar] [CrossRef]
- Dyer, M.D.; Murali, T.M.; Sobral, B.W. The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathog. 2008, 4, e32. [Google Scholar] [CrossRef]
- Durmus Tekir, S.; Cakir, T.; Ulgen, K.O. Infection Strategies of Bacterial and Viral Pathogens through Pathogen-Human Protein-Protein Interactions. Front. Microbiol. 2012, 3, 46. [Google Scholar] [CrossRef]
- Idrees, S.; Chen, H.; Panth, N.; Paudel, K.R.; Hansbro, P.M. Exploring Viral-Host Protein Interactions as Antiviral Therapies: A Computational Perspective. Microorganisms 2024, 12, 630. [Google Scholar] [CrossRef]
- Pichlmair, A.; Kandasamy, K.; Alvisi, G.; Mulhern, O.; Sacco, R.; Habjan, M.; Binder, M.; Stefanovic, A.; Eberle, C.A.; Goncalves, A.; et al. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature 2012, 487, 486–490. [Google Scholar] [CrossRef]
- Durmus, S.; Ulgen, K.O. Comparative interactomics for virus-human protein-protein interactions: DNA viruses versus RNA viruses. FEBS Open Bio 2017, 7, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Garamszegi, S.; Franzosa, E.A.; Xia, Y. Signatures of pleiotropy, economy and convergent evolution in a domain-resolved map of human-virus protein-protein interaction networks. PLoS Pathog. 2013, 9, e1003778. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.J.; Palopoli, N. Computational prediction of short linear motifs from protein sequences. Methods Mol. Biol. 2015, 1268, 89–141. [Google Scholar] [CrossRef] [PubMed]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Adler, P.; Vilo, J.; Peterson, H. g:Profiler-interoperable web service for functional enrichment analysis and gene identifier mapping (2023 update). Nucleic Acids Res. 2023, 51, W207–W212. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viral Group | vhPPIs 1 | potDMI 2 | predDMI 3 | E-Score |
|---|---|---|---|---|
| dsRNA | 76 | 2 | 2 | 19 ** |
| ssRNA | 18,932 | 17 | 6 | 6.7 ** |
| dsDNA | 12,755 | 27 | 19 | 11.6 ** |
| ssDNA | 321 | 0 | 0 | NA |
| Group | Targeted Host Proteins |
|---|---|
| dsRNA | 1 |
| dsDNA | 61 |
| ssRNA | 10 |
| dsRNA + dsDNA | 3 |
| dsRNA + ssRNA | 2 |
| dsDNA + ssRNA | 6 |
| dsRNA + dsDNA + ssRNA | 1 |
| Viral Group | Known in ELM | Known (%) | Stringency |
|---|---|---|---|
| dsDNA | 31 | 79.4% * | ELMc-Protein |
| dsRNA | 2 | 66.6% * | ELMc-Protein |
| ssRNA | 6 | 66.7% * | ELMc-Protein |
| dsDNA | 21 | 16.5% * | ELMc-Domain |
| dsRNA | 2 | 40.0% * | ELMc-Domain |
| ssRNA | 6 | 31.5% * | ELMc-Domain |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Idrees, S.; Paudel, K.R.; Banik, M.; Suwal, N.; Thapa, R.; Bashyal, S. Predicting Motif-Mediated Interactions Based on Viral Genomic Composition. Int. J. Mol. Sci. 2025, 26, 3674. https://doi.org/10.3390/ijms26083674
Idrees S, Paudel KR, Banik M, Suwal N, Thapa R, Bashyal S. Predicting Motif-Mediated Interactions Based on Viral Genomic Composition. International Journal of Molecular Sciences. 2025; 26(8):3674. https://doi.org/10.3390/ijms26083674
Chicago/Turabian StyleIdrees, Sobia, Keshav Raj Paudel, Mithila Banik, Newton Suwal, Rajan Thapa, and Saroj Bashyal. 2025. "Predicting Motif-Mediated Interactions Based on Viral Genomic Composition" International Journal of Molecular Sciences 26, no. 8: 3674. https://doi.org/10.3390/ijms26083674
APA StyleIdrees, S., Paudel, K. R., Banik, M., Suwal, N., Thapa, R., & Bashyal, S. (2025). Predicting Motif-Mediated Interactions Based on Viral Genomic Composition. International Journal of Molecular Sciences, 26(8), 3674. https://doi.org/10.3390/ijms26083674