
2-Phenylcyclopropylmethylamine (PCPMA) Derivatives as D3R-Selective Ligands for 3D-QSAR, Docking and Molecular Dynamics Simulation Studies


Abstract
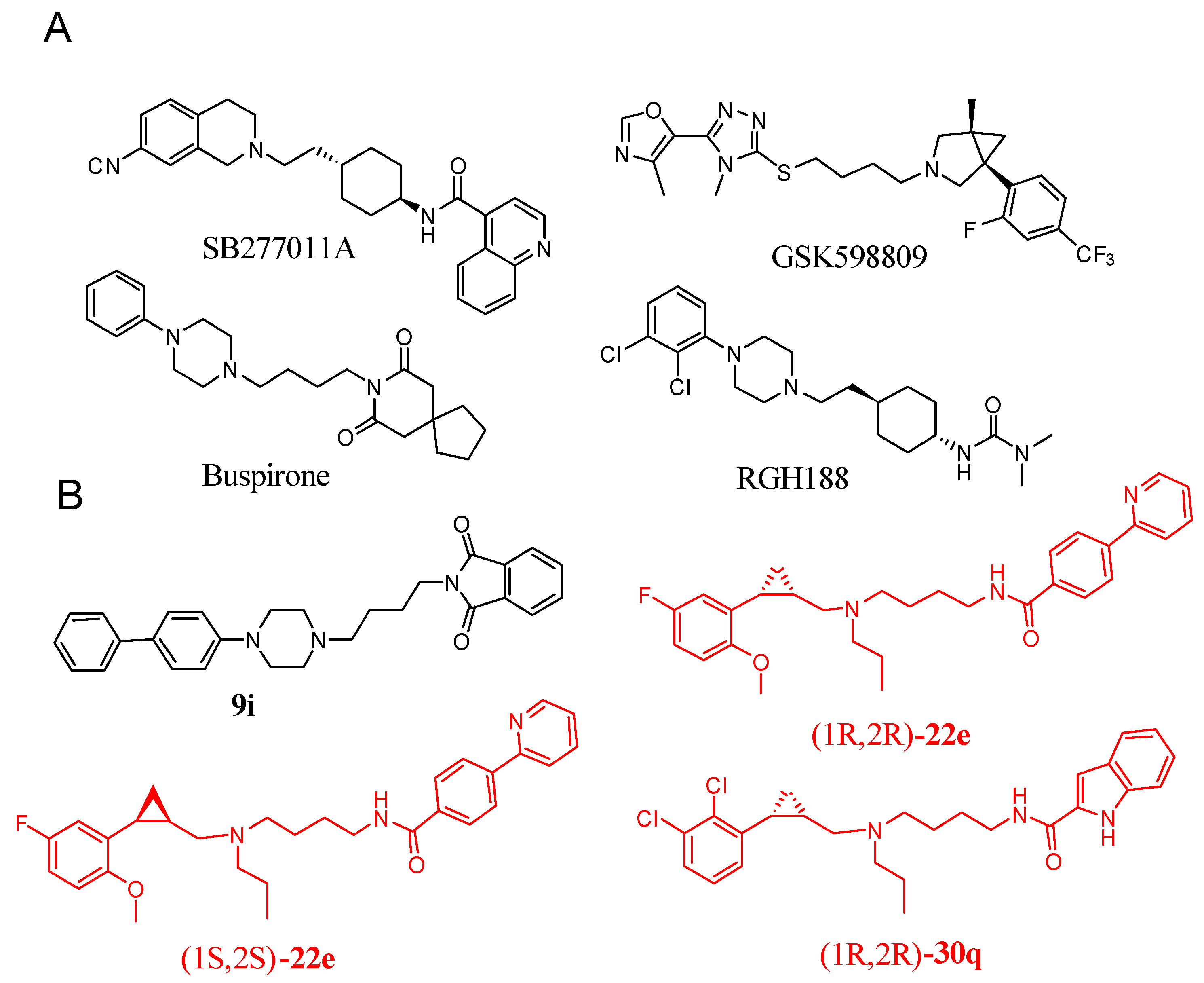
1. Introduction
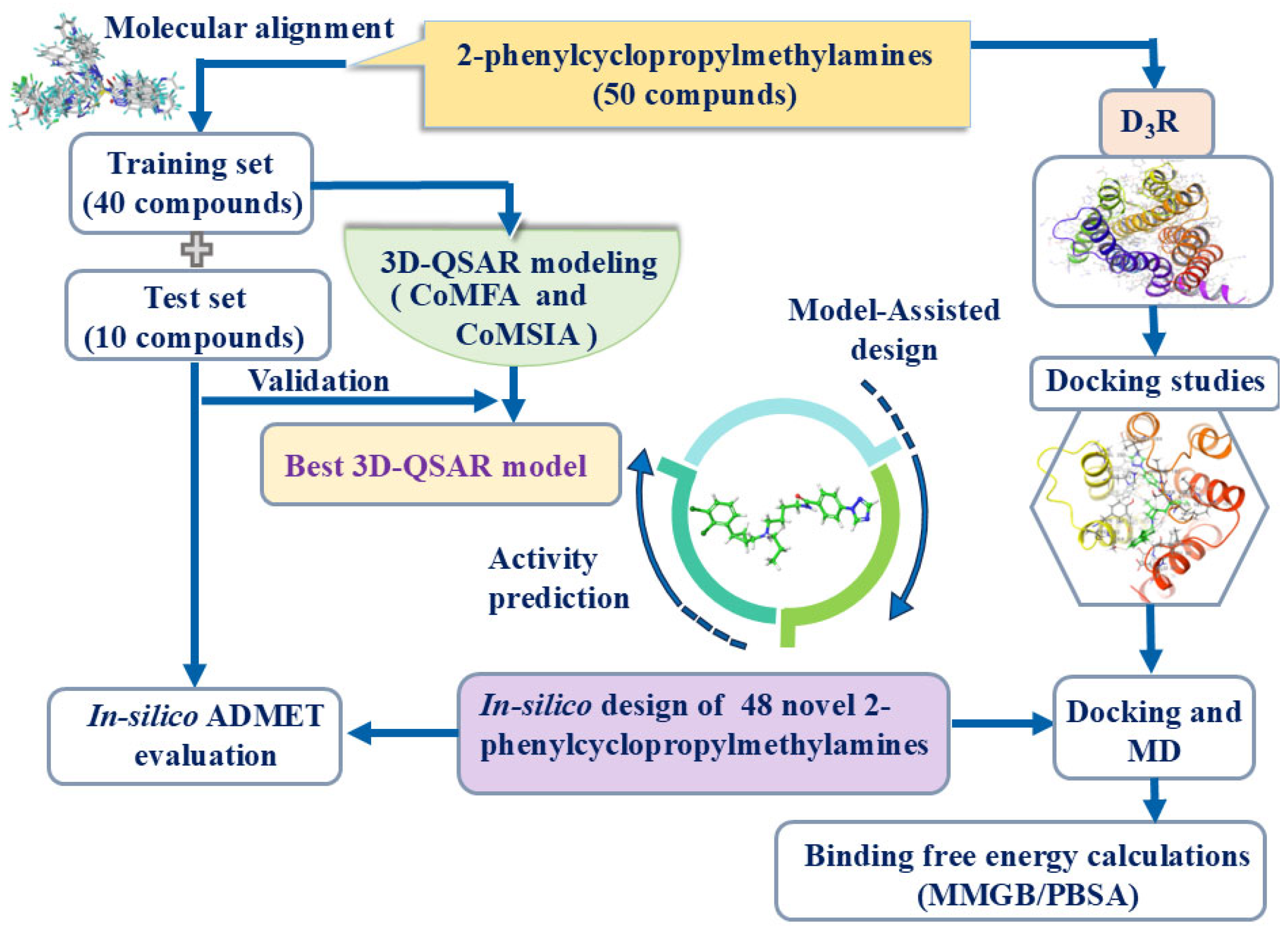
2. Results and Discussion
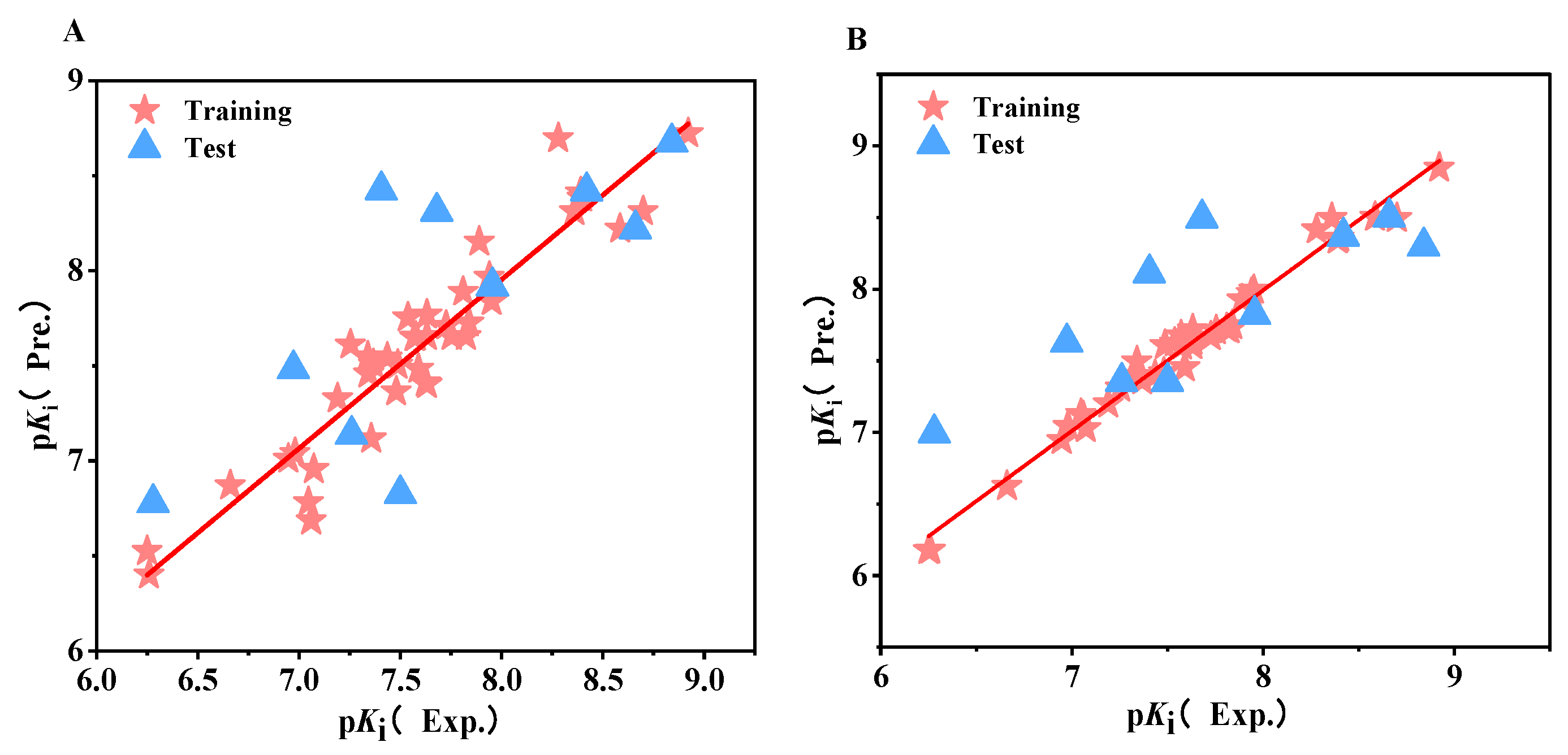
2.1. CoMFA and CoMSIA Statistical Results
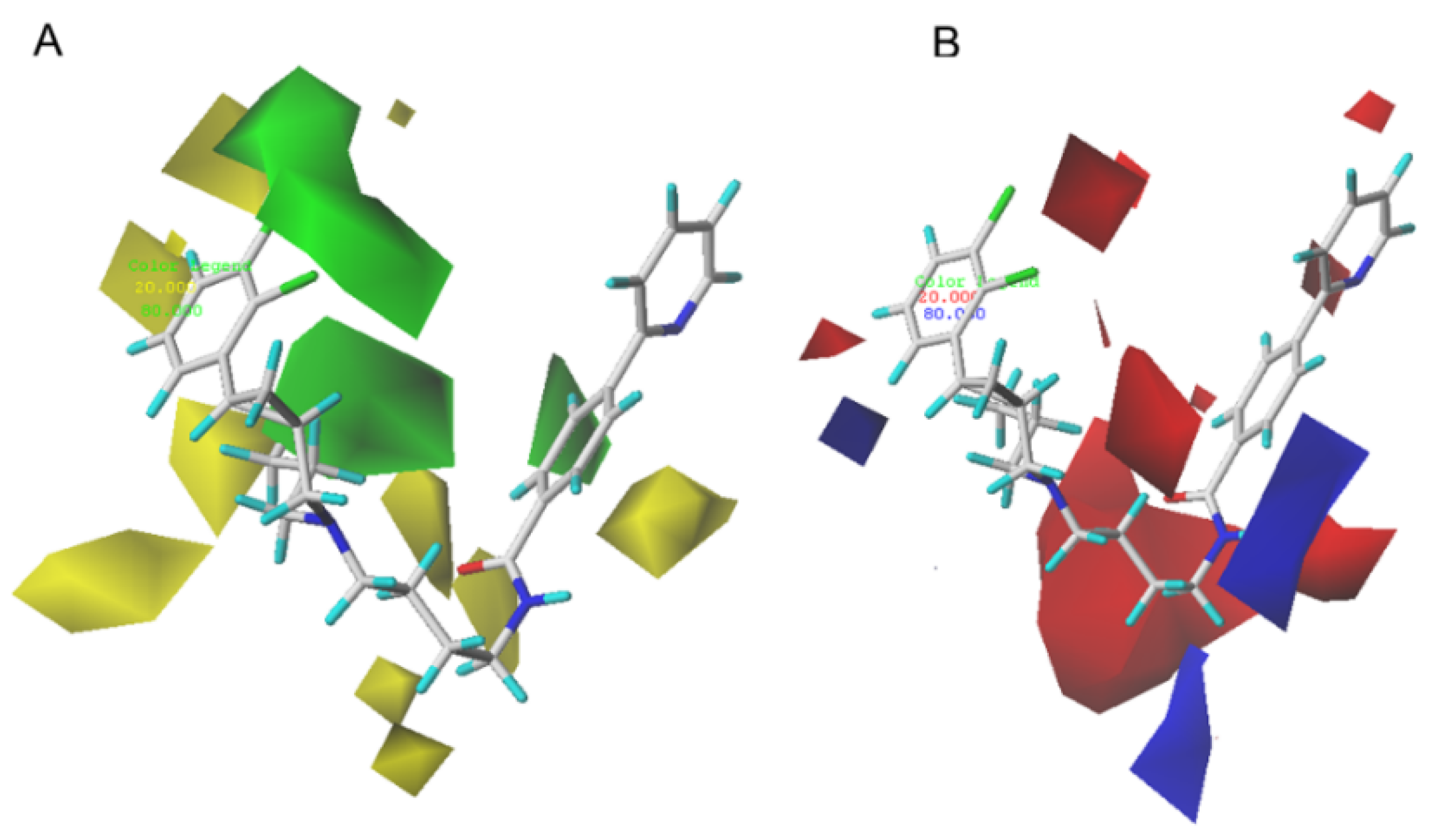
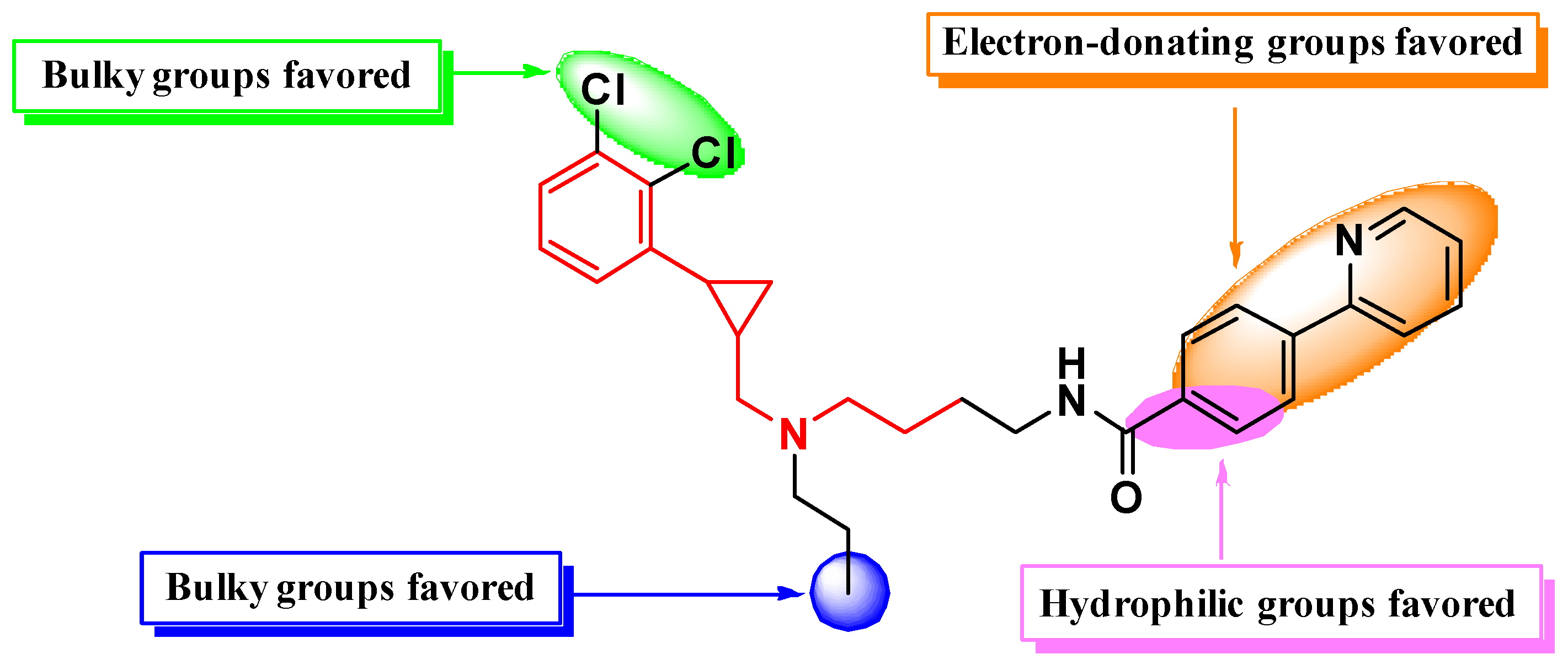
2.2. CoMFA and CoMSIA Contour Map Analysis
2.3. Design of Novel PCPMA-Like Compounds
2.4. ADMET and Drug Similarity Prediction Results
2.5. Molecular Docking
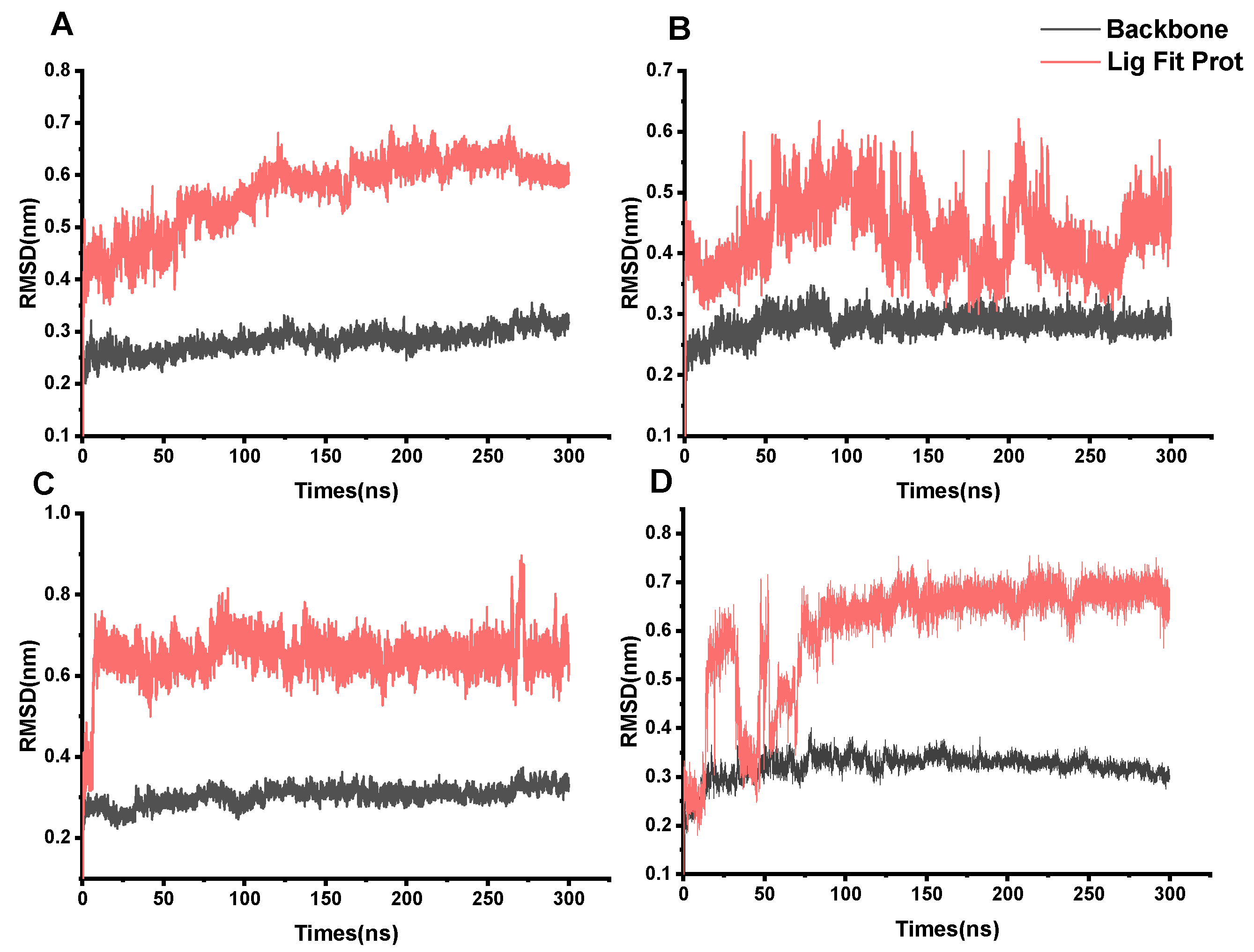
2.6. Molecular Dynamics (MD) Simulation
2.7. MMGB/PBSA Calculation of Binding Free Energy
3. Materials and Methods
3.1. 3D-QSAR Analysis Dataset
3.2. Compound Preparation and Restructuring
3.3. 3D-QSAR Models Production
3.4. Design of Novel PCPMA-Like D3R-Selective Ligands and Their Affinity Prediction with D3R
3.5. Pharmacokinetic ADMET and Drug Similarity Prediction
3.6. Molecular Docking
3.7. Molecular Dynamics Simulations (MD)
3.8. Combined Free Energy MMGB/PBSA Calculations and Per-Residue Free Energy Decomposition Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Levant, B. The D3 dopamine receptor: Neurobiology and potential clinical relevance. Pharmacol. Rev. 1997, 49, 231–252. [Google Scholar] [CrossRef] [PubMed]
- Löber, S.; Hübner, H.; Tschammer, N.; Gmeiner, P. Recent advances in the search for D3- and D4-selective drugs: Probes, models and candidates. Trends Pharmacol. Sci. 2011, 32, 148–157. [Google Scholar] [CrossRef]
- Sokoloff, P.; Giros, B.; Martres, M.-P.; Bouthenet, M.-L.; Schwartz, J.-C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 1990, 347, 146–151. [Google Scholar] [CrossRef]
- Chien, E.Y.T.; Liu, W.; Zhao, Q.; Katritch, V.; Han, G.W.; Hanson, M.A.; Shi, L.; Newman, A.H.; Javitch, J.A.; Cherezov, V.; et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [Google Scholar] [CrossRef]
- Sokoloff, P.; Giros, B.; Martres, M.P.; Andrieux, M.; Besancon, R.; Pilon, C.; Bouthenet, M.L.; Souil, E.; Schwartz, J.C. Localization and function of the D3 dopamine receptor. Arzneimittelforschung 1992, 42, 224–230. [Google Scholar]
- Zingales, V.; Torrisi, S.A.; Leggio, G.M.; Bucolo, C.; Drago, F.; Salomone, S. Pharmacological and Genetic Evidence of Dopamine Receptor 3-Mediated Vasoconstriction in Isolated Mouse Aorta. Biomolecules 2021, 11, 418. [Google Scholar] [CrossRef]
- Reilly, S.W.; Riad, A.A.; Hsieh, C.-J.; Sahlholm, K.; Jacome, D.A.; Griffin, S.; Taylor, M.; Weng, C.-C.; Xu, K.; Kirschner, N.; et al. Leveraging a Low-Affinity Diazaspiro Orthosteric Fragment to Reduce Dopamine D3 Receptor (D3R) Ligand Promiscuity across Highly Conserved Aminergic G-Protein-Coupled Receptors (GPCRs). J. Med. Chem. 2019, 62, 5132–5147. [Google Scholar] [CrossRef]
- Keck, T.M.; John, W.S.; Czoty, P.W.; Nader, M.A.; Newman, A.H. Identifying Medication Targets for Psychostimulant Addiction: Unraveling the Dopamine D3 Receptor Hypothesis. J. Med. Chem. 2015, 58, 5361–5380. [Google Scholar] [CrossRef]
- Stemp, G.; Ashmeade, T.; Branch, C.L.; Hadley, M.S.; Hunter, A.J.; Johnson, C.N.; Nash, D.J.; Thewlis, K.M.; Vong, A.K.K.; Austin, N.E.; et al. Design and synthesis of trans-N-4-2-(6-cyano-1,2,3,4-tetrahydroisoquinolin-2-yl) ethyl cyclohexyl -4-quinolinecarboxamide (SB-277011): A potent and selective dopamine D3 receptor antagonist with high oral bioavailability and CNS penetration in the rat. J. Med. Chem. 2000, 43, 1878–1885. [Google Scholar] [CrossRef]
- Appel, N.M.; Li, S.-H.; Holmes, T.H.; Acri, J.B. Dopamine D3 Receptor Antagonist (GSK598809) Potentiates the Hypertensive Effects of Cocaine in Conscious, Freely-Moving Dogs. J. Pharmacol. Exp. Ther. 2015, 354, 484–492. [Google Scholar] [CrossRef]
- Nielsen, S.; Gowing, L.; Sabioni, P.; Le Foll, B. Pharmacotherapies for cannabis dependence. Cochrane Database Syst. Rev. 2019, 1, CD008940. [Google Scholar] [CrossRef] [PubMed]
- Gyertyán, I.; Kiss, B.; Sághy, K.; Laszy, J.; Szabó, G.; Szabados, T.; Gémesi, L.I.; Pásztor, G.; Zájer-Balázs, M.; Kapás, M.; et al. Cariprazine (RGH-188), a potent D3/D2 dopamine receptor partial agonist, binds to dopamine D3 receptors in vivo and shows antipsychotic-like and procognitive effects in rodents. Neurochem. Int. 2011, 59, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Shonberg, J.; Lopez, L.; Scammells, P.J.; Christopoulos, A.; Capuano, B.; Lane, J.R. Biased agonism at G protein-coupled receptors: The promise and the challenges—A medicinal chemistry perspective. Med. Res. Rev. 2014, 34, 1286–1330. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.R.; Sexton, P.M.; Christopoulos, A. Bridging the gap: Bitopic ligands of G-protein-coupled receptors. Trends Pharmacol. Sci. 2013, 34, 59–66. [Google Scholar] [CrossRef]
- Cao, Y.; Sun, N.; Zhang, J.; Liu, Z.; Tang, Y.-Z.; Wu, Z.; Kim, K.-M.; Cheon, S.H. Design, synthesis, and evaluation of bitopic arylpiperazine-phthalimides as selective dopamine D3 receptor agonists. Medchemcomm 2018, 9, 1457–1465. [Google Scholar] [CrossRef]
- Shaik, A.B.; Boateng, C.A.; Battiti, F.O.; Bonifazi, A.; Cao, J.; Chen, L.; Chitsazi, R.; Ravi, S.; Lee, K.H.; Shi, L.; et al. Structure Activity Relationships for a Series of Eticlopride-Based Dopamine D2/D3 Receptor Bitopic Ligands. J. Med. Chem. 2021, 64, 15313–15333. [Google Scholar] [CrossRef]
- Arroyo-Urea, S.; Nazarova, A.L.; Carrión-Antolí, Á.; Bonifazi, A.; Battiti, F.O.; Lam, J.H.; Newman, A.H.; Katritch, V.; García-Nafría, J. A bitopic agonist bound to the dopamine 3 receptor reveals a selectivity site. Nat. Commun. 2024, 15, 7759. [Google Scholar] [CrossRef]
- Dalton King, H.; Denhart, D.J.; Deskus, J.A.; Ditta, J.L.; Epperson, J.R.; Higgins, M.A.; Kung, J.E.; Marcin, L.R.; Sloan, C.P.; Mattson, G.K.; et al. Conformationally restricted homotryptamines. Part 4: Heterocyclic and naphthyl analogs of a potent selective serotonin reuptake inhibitor. Bioorg. Med. Chem. Lett. 2007, 17, 5647–5651. [Google Scholar] [CrossRef]
- Liu, J.; Clough, S.J.; Hutchinson, A.J.; Adamah-Biassi, E.B.; Popovska-Gorevski, M.; Dubocovich, M.L. MT1 and MT2 Melatonin Receptors: A Therapeutic Perspective. Annu. Rev. Pharmacool. Toxicol. 2016, 56, 361–383. [Google Scholar] [CrossRef]
- Cho, S.J.; Jensen, N.H.; Kurome, T.; Kadari, S.; Manzano, M.L.; Malberg, J.E.; Caldarone, B.; Roth, B.L.; Kozikowski, A.P. Selective 5-Hydroxytryptamine 2C Receptor Agonists Derived from the Lead Compound Tranylcypromine: Identification of Drugs with Antidepressant-Like Action. J. Med. Chem. 2009, 52, 1885–1902. [Google Scholar] [CrossRef]
- Carpenter, J.; Wang, Y.; Wu, G.; Feng, J.; Ye, X.-Y.; Morales, C.L.; Broekema, M.; Rossi, K.A.; Miller, K.J.; Murphy, B.J.; et al. Utilization of an Active Site Mutant Receptor for the Identification of Potent and Selective Atypical 5-HT 2C Receptor Agonists. J. Med. Chem. 2017, 60, 6166–6190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Cheng, J.; McCorvy, J.D.; Lorello, P.J.; Caldarone, B.J.; Roth, B.L.; Kozikowski, A.P. Discovery of N-Substituted (2-Phenylcyclopropyl)methylamines as Functionally Selective Serotonin 2C Receptor Agonists for Potential Use as Antipsychotic Medications. J. Med. Chem. 2017, 60, 6273–6288. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Zhou, Q.; Yan, W.; Sun, J.; Kozikowski, A.P.; Zhao, S.; Huang, X.-P.; Cheng, J. Design and Synthesis of Bitopic 2-Phenylcyclopropylmethylamine (PCPMA) Derivatives as Selective Dopamine D3 Receptor Ligands. J. Med. Chem. 2020, 63, 4579–4602. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, L.; Lei, M. 3D-QSAR and docking studies on 2-arylbenzoxazole and linker-Y transthyretin amyloidogenesis inhibitors. Sci. China Chem. 2013, 56, 1550–1563. [Google Scholar] [CrossRef]
- Damale, M.G.; Harke, S.N.; Kalam Khan, F.A.; Shinde, D.B.; Sangshetti, J.N. Recent advances in multidimensional QSAR (4D-6D): A critical review. Mini Rev. Med. Chem. 2014, 14, 35–55. [Google Scholar] [CrossRef]
- Lambrinidis, G.; Vallianatou, T.; Tsantili-Kakoulidou, A. In vitro, in silico and integrated strategies for the estimation of plasma protein binding. A review. Adv. Drug Deliv. Rev. 2015, 86, 27–45. [Google Scholar] [CrossRef]
- Kim, K.H. Comparative molecular field analysis (CoMFA). In Molecular Similarity in Drug Design; Springer: Dordrecht, The Netherlands, 1995; pp. 291–331. [Google Scholar]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef]
- Völkner, M.; Wagner, F.; Steinheuer, L.M.; Carido, M.; Kurth, T.; Yazbeck, A.; Schor, J.; Wieneke, S.; Ebner, L.J.A.; Del Toro Runzer, C.; et al. HBEGF-TNF induce a complex outer retinal pathology with photoreceptor cell extrusion in human organoids. Nat. Commun. 2022, 13, 6183. [Google Scholar] [CrossRef]
- Katritch, V.; Cherezov, V.; Stevens, R.C. Structure-Function of the G Protein–Coupled Receptor Superfamily. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 531–556. [Google Scholar] [CrossRef]
- Chen, L.; Lin, Y.; Yan, X.; Ni, H.; Chen, F.; He, F. 3D-QSAR studies on the structure–bitterness analysis of citrus flavonoids. Food Funct. 2023, 14, 4921–4930. [Google Scholar] [CrossRef]
- Salas, C.O.; Zarate, A.M.; Kryštof, V.; Mella, J.; Faundez, M.; Brea, J.; Loza, M.I.; Brito, I.; Hendrychová, D.; Jorda, R.; et al. Promising 2,6,9-Trisubstituted Purine Derivatives for Anticancer Compounds: Synthesis, 3D-QSAR, and Preliminary Biological Assays. Int. J. Mol. Sci. 2019, 21, 161. [Google Scholar] [CrossRef] [PubMed]
- Zięba, A.; Laitinen, T.; Patel, J.Z.; Poso, A.; Kaczor, A.A. Docking-Based 3D-QSAR Studies for 1,3,4-oxadiazol-2-one Derivatives as FAAH Inhibitors. Int. J. Mol. Sci. 2021, 22, 6108. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Shi, S.; Yi, J.; Wang, N.; He, Y.; Wu, Z.; Peng, J.; Deng, Y.; Wang, W.; Wu, C.; et al. ADMETlab 3.0: An updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Res. 2024, 52, W422–W431. [Google Scholar] [CrossRef]
- Smith, D.A.; Beaumont, K.; Maurer, T.S.; Di, L. Volume of Distribution in Drug Design. J. Med. Chem. 2015, 58, 5691–5698. [Google Scholar] [CrossRef]
- Hollenberg, P.F. Mechanisms of cytochrome P450 and peroxidase-catalyzed xenobiotic metabolism. FASEB J. 1992, 6, 686–694. [Google Scholar] [CrossRef]
- Lipinski, C. Interview by Peter Kirkpatrick. Nat. Rev. Drug Discov. 2012, 11, 900–901. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Plinski, E.F.; Plinska, S. Veber’s Rules in Terahertz Light. 2020. Available online: https://www.researchsquare.com/article/rs-12857/v1 (accessed on 4 April 2024).
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I. Selection criteria for drug-like compounds. Med. Res. Rev. 2003, 23, 302–321. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Lanka, G.; Begum, D.; Banerjee, S.; Adhikari, N.; P, Y.; Ghosh, B. Pharmacophore-based virtual screening, 3D QSAR, Docking, ADMET, and MD simulation studies: An in silico perspective for the identification of new potential HDAC3 inhibitors. Comput. Biol. Med. 2023, 166, 107481. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Lin, G.; Zhang, R.; Liang, Z.; Wu, L.; Wu, W. Studies on molecular mechanism between ACE and inhibitory peptides in different bioactivities by 3D-QSAR and MD simulations. J. Mol. Liq. 2020, 304, 112702. [Google Scholar] [CrossRef]
- Repasky, M.P.; Shelley, M.; Friesner, R.A. Flexible Ligand Docking with Glide. Curr. Protoc. Bioinform. 2007, 18, 8–12. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Chen, J.; Zeng, Q.; Wang, W.; Sun, H.; Hu, G. Decoding the Identification Mechanism of an SAM-III Riboswitch on Ligands through Multiple Independent Gaussian-Accelerated Molecular Dynamics Simulations. J. Chem. Inf. Model. 2022, 62, 6118–6132. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations w ith GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Chohan, T.A.; Chen, J.-J.; Qian, H.-Y.; Pan, Y.-L.; Chen, J.-Z. Molecular modeling studies to characterize N-phenylpyrimidin-2-amine selectivity for CDK2 and CDK4 through 3D-QSAR and molecular dynamics simulations. Mol. Biosyst. 2016, 12, 1250–1268. [Google Scholar] [CrossRef]
- Chohan, T.A.; Qian, H.-Y.; Pan, Y.-L.; Chen, J.-Z. Molecular simulation studies on the binding selectivity of 2-anilino-4 -(thiazol-5-yl)-pyrimidines in complexes with CDK2 and CDK7. Mol. Biosyst. 2016, 12, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Kubinyi, H. QSAR and 3D QSAR in drug design Part 1: Methodology. Drug Discovery Today 1997, 2, 457–467. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLS Statistic | CoMFA | CoMSIA |
|---|---|---|
| q2 | 0.607 | 0.643 |
| r2 | 0.981 | 0.889 |
| SEE | 0.094 | 0.207 |
| F | 149.222 | 69.912 |
| N | 10 | 4 |
| Steric | 0.393 | 0.375 |
| Electrostatic | 0.607 | - |
| Hydrophobic | - | 0.725 |
| SEP | 0.5 | 0.51 |
| RMSE | 0.28 | 0.35 |
| Compounds | Structures | pKi (Pre.) | |
|---|---|---|---|
| CoMFA | CoMSIA | ||
| 30r | 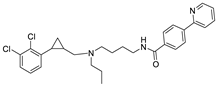 | 8.845 | 8.723 |
| D1 | 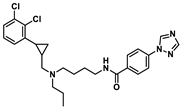 | 8.966 | 8.706 |
| D2 | 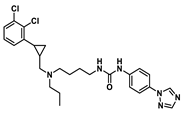 | 8.871 | 8.586 |
| D3 | 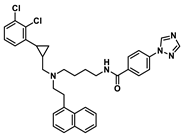 | 8.869 | 8.248 |
| D4 | 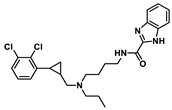 | 8.925 | 8.379 |
| 30r | D1 | D2 | D3 | D4 | |
|---|---|---|---|---|---|
| Molecular weight | 509.2 | 499.19 | 514.29 | 611.22 | 472.18 |
| nRot | 13 | 13 | 14 | 14 | 12 |
| Flexibility | 0.591 | 0.619 | 0.667 | 0.438 | 0.6 |
| LogP | 6.325 | 5.27 | 5.525 | 6.755 | 5.917 |
| TPSA 1/Å2 | 45.23 | 63.05 | 75.08 | 63.05 | 61.02 |
| SAscore 2 | <6 | <6 | <6 | <6 | <6 |
| nHA | 4 | 4 | 7 | 6 | 5 |
| nHD | 1 | 1 | 2 | 1 | 2 |
| System | 30r | D1 | D2 | D3 | D4 |
|---|---|---|---|---|---|
| ΔGvdW | −72.88 | −69.53 | −62.28 | −70.08 | −58.04 |
| ΔGele | −16.68 | −20.65 | −46.98 | −14.39 | −37.35 |
| ΔGgas | −89.56 | −90.18 | −109.26 | −84.47 | −95.39 |
| ΔGpolar(PB) | 51.86 | 48.41 | 69.15 | 50.53 | 61.19 |
| ΔGnonpolar | −6.28 | −5.58 | −6.27 | −6.63 | −5.56 |
| ΔGsol | 45.58 | 42.83 | 62.89 | 43.89 | 55.64 |
| ΔGtotal | −43.98 | −47.35 | −46.36 | −40.58 | −39.75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, L.; Gao, Y.; Zhang, S.; Zhao, L.; Zhao, R.; Sun, P.; Pan, X.; Zhang, W. 2-Phenylcyclopropylmethylamine (PCPMA) Derivatives as D3R-Selective Ligands for 3D-QSAR, Docking and Molecular Dynamics Simulation Studies. Int. J. Mol. Sci. 2025, 26, 3559. https://doi.org/10.3390/ijms26083559
Guo L, Gao Y, Zhang S, Zhao L, Zhao R, Sun P, Pan X, Zhang W. 2-Phenylcyclopropylmethylamine (PCPMA) Derivatives as D3R-Selective Ligands for 3D-QSAR, Docking and Molecular Dynamics Simulation Studies. International Journal of Molecular Sciences. 2025; 26(8):3559. https://doi.org/10.3390/ijms26083559
Chicago/Turabian StyleGuo, Li, Yuepeng Gao, Sujuan Zhang, Lingmi Zhao, Runxin Zhao, Pinghua Sun, Xinhui Pan, and Wei Zhang. 2025. "2-Phenylcyclopropylmethylamine (PCPMA) Derivatives as D3R-Selective Ligands for 3D-QSAR, Docking and Molecular Dynamics Simulation Studies" International Journal of Molecular Sciences 26, no. 8: 3559. https://doi.org/10.3390/ijms26083559
APA StyleGuo, L., Gao, Y., Zhang, S., Zhao, L., Zhao, R., Sun, P., Pan, X., & Zhang, W. (2025). 2-Phenylcyclopropylmethylamine (PCPMA) Derivatives as D3R-Selective Ligands for 3D-QSAR, Docking and Molecular Dynamics Simulation Studies. International Journal of Molecular Sciences, 26(8), 3559. https://doi.org/10.3390/ijms26083559