
From Skeletal Muscle to Myocardium: Molecular Mechanisms of Exercise-Induced Irisin Regulation of Cardiac Fibrosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
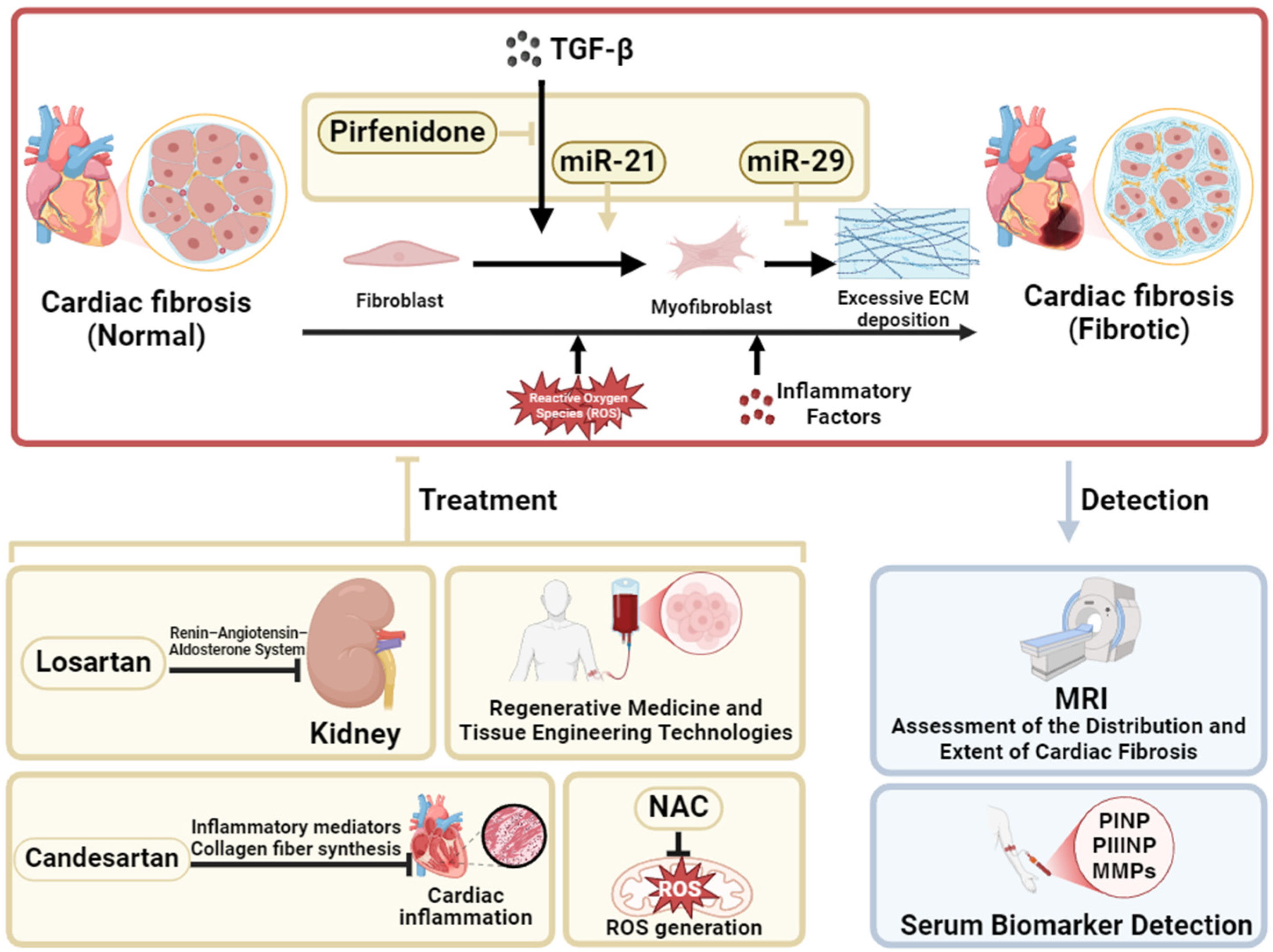
2. Current Status of the Treatment of Cardiac Fibrosis
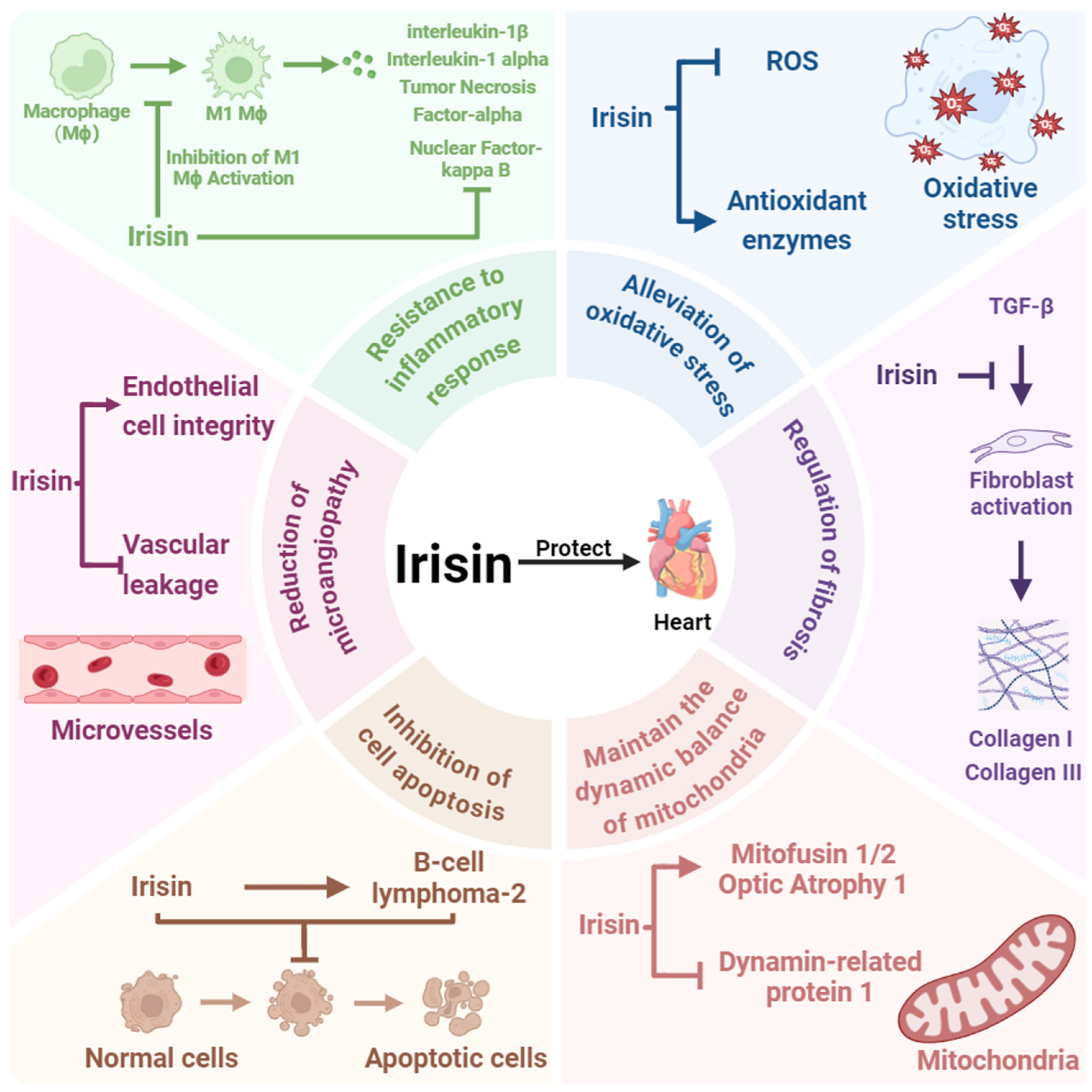
3. Irisin Is a Key Factor in Cardioprotection
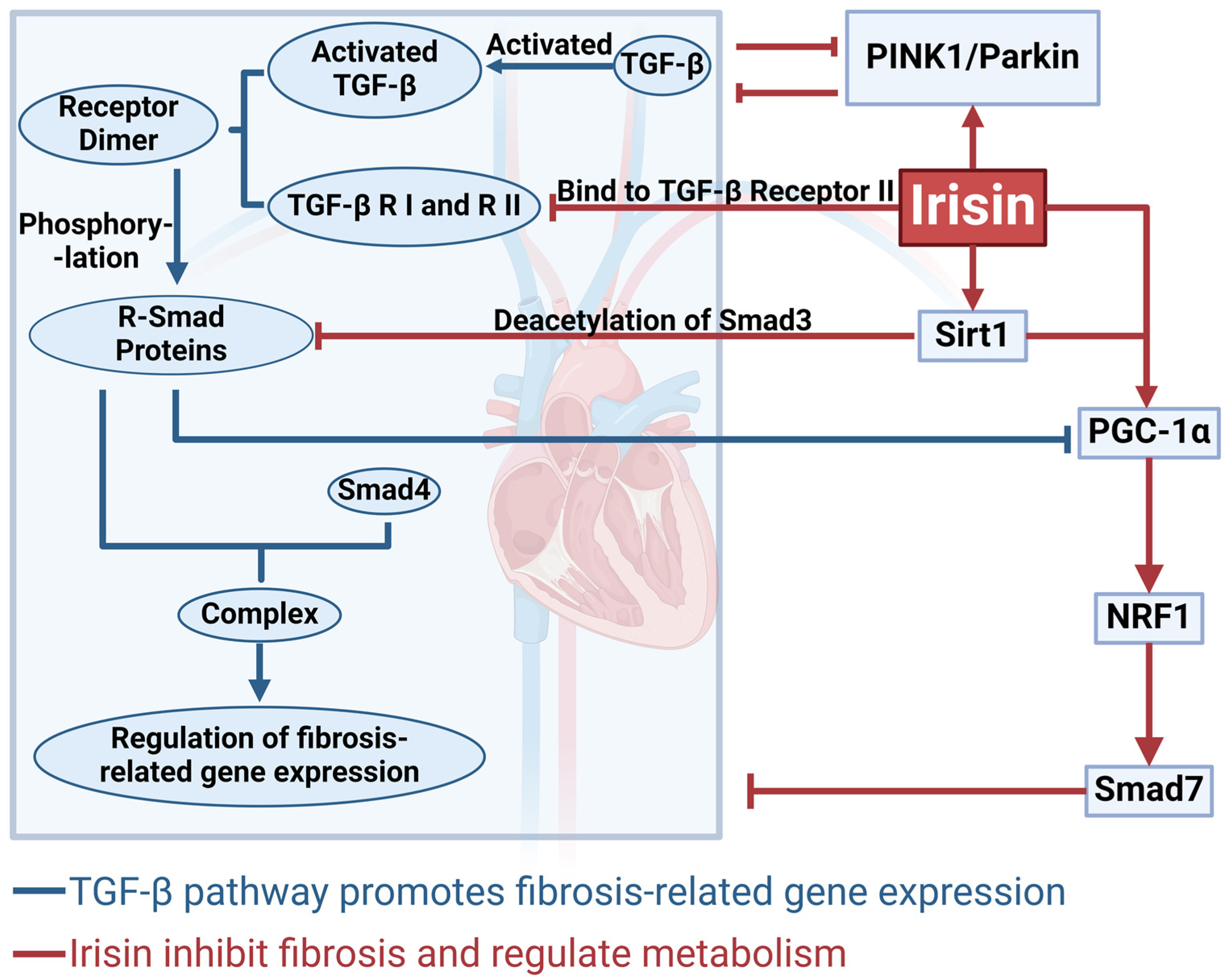
4. The Mechanism by Which Irisin Inhibits Cardiac Fibrosis via the TGF-β/Smad Signaling Pathway
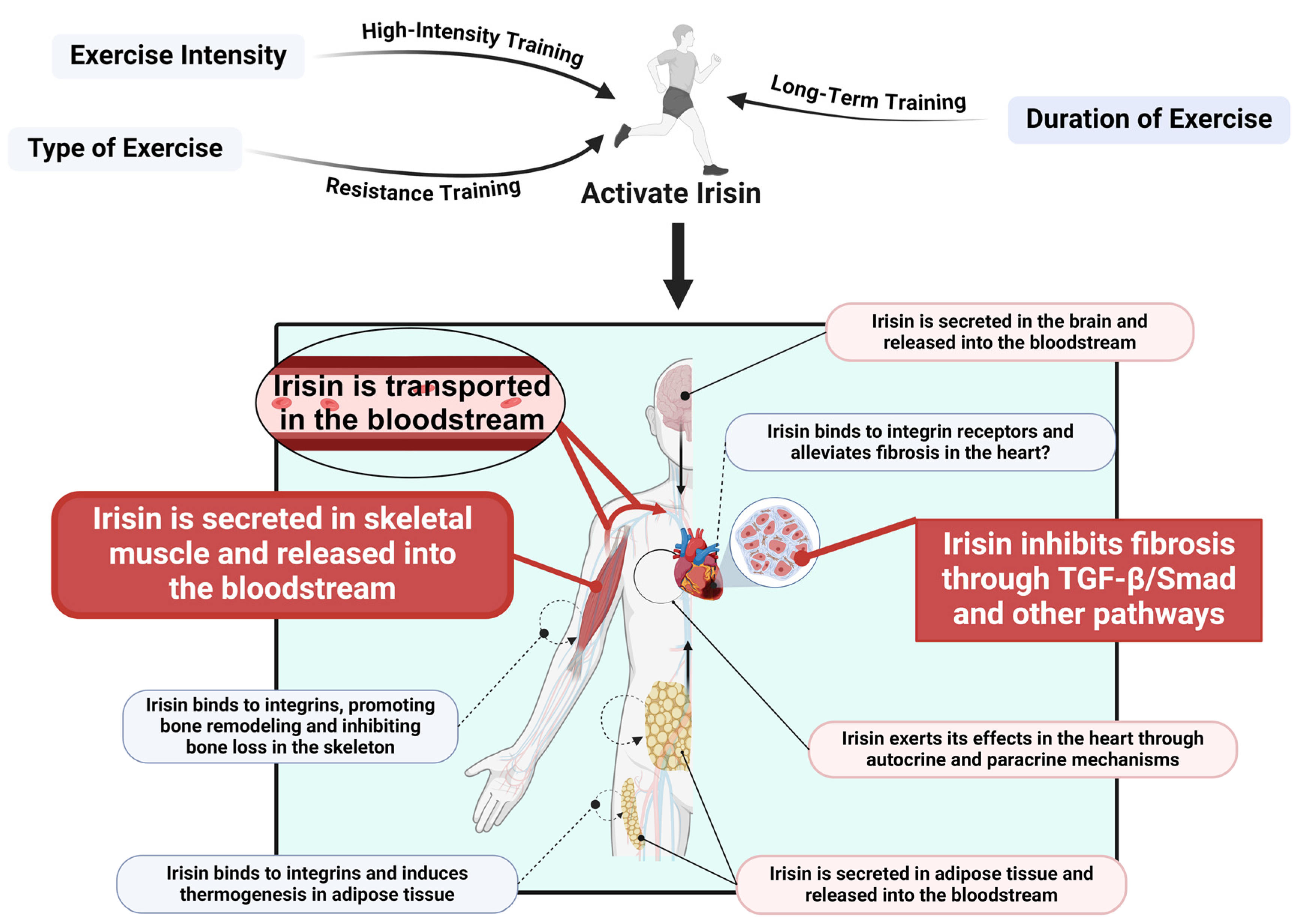
5. Exercise Promotes Irisin Release to Regulate Cardiac Fibrosis
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Naghavi, M.; Abajobir, A.A.; Abbafati, C.; Abbas, K.M.; Abd-Allah, F.; Abera, S.F.; Aboyans, V.; Adetokunboh, O.; Afshin, A.; Agrawal, A.; et al. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1151–1210. [Google Scholar] [CrossRef] [PubMed]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef]
- McMurray, J.J.; Pfeffer, M.A. Heart failure. Lancet 2005, 365, 1877–1889. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, W.; Ren, C.; Wang, Y.; Zhang, G.; Liu, J.; Wang, W. Cellular Phenotypic Transformation in Heart Failure Caused by Coronary Heart Disease and Dilated Cardiomyopathy: Delineating at Single-Cell Level. Biomedicines 2022, 10, 402. [Google Scholar] [CrossRef]
- Khan, R.; Sheppard, R. Fibrosis in heart disease: Understanding the role of transforming growth factor-beta in cardiomyopathy, valvular disease and arrhythmia. Immunology 2006, 118, 10–24. [Google Scholar] [CrossRef]
- Feng, N.; Yu, H.; Wang, Y.; Zhang, Y.; Xiao, H.; Gao, W. Exercise training attenuates angiotensin II-induced cardiac fibrosis by reducing POU2F1 expression. J. Sport Health Sci. 2023, 12, 464–476. [Google Scholar] [CrossRef]
- Fu, G.; Wang, Z.; Hu, S. Exercise improves cardiac fibrosis by stimulating the release of endothelial progenitor cell-derived exosomes and upregulating miR-126 expression. Front. Cardiovasc. Med. 2024, 11, 1323329. [Google Scholar] [CrossRef]
- Hu, S.; Liu, H.; Hu, Z.; Li, L.; Yang, Y. Follistatin-like 1: A dual regulator that promotes cardiomyocyte proliferation and fibrosis. J. Cell Physiol. 2020, 235, 5893–5902. [Google Scholar] [CrossRef]
- Liu, C.; Wei, A.; Wang, T. Irisin, an Effective Treatment for Cardiovascular Diseases. J. Cardiovasc. Dev. Dis. 2022, 9, 305. [Google Scholar] [CrossRef]
- Sun, B.; Wu, H.; Lu, J.; Zhang, R.; Shen, X.; Gu, Y.; Shi, C.; Zhang, Y.; Yuan, W. Irisin reduces bone fracture by facilitating osteogenesis and antagonizing TGF-β/Smad signaling in a growing mouse model of osteogenesis imperfecta. J. Orthop. Translat. 2023, 38, 175–189. [Google Scholar] [CrossRef]
- Tang, Y.J.; Zhang, Z.; Yan, T.; Chen, K.; Xu, G.F.; Xiong, S.Q.; Wu, D.Q.; Chen, J.; Jose, P.A.; Zeng, C.Y.; et al. Irisin attenuates type 1 diabetic cardiomyopathy by anti-ferroptosis via SIRT1-mediated deacetylation of p53. Cardiovasc. Diabetol. 2024, 23, 116. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, R.A.; Gannon, N.P.; Barberena, M.A.; Garcia-Smith, R.; Bisoffi, M.; Mermier, C.M.; Conn, C.A.; Trujillo, K.A. Characterization of the metabolic effects of irisin on skeletal muscle in vitro. Diabetes Obes. Metab. 2014, 16, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.G.; Yuan, Y.P.; Wu, H.M.; Zhang, X.; Tang, Q.Z. Cardiac fibrosis: New insights into the pathogenesis. Int. J. Biol. Sci. 2018, 14, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol. Ther. 2009, 123, 255–278. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Bujak, M.; Frangogiannis, N.G. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res. 2007, 74, 184–195. [Google Scholar] [CrossRef]
- Wu, M.; Peng, Z.; Zu, C.; Ma, J.; Lu, S.; Zhong, J.; Zhang, S. Losartan Attenuates Myocardial Endothelial-To-Mesenchymal Transition in Spontaneous Hypertensive Rats via Inhibiting TGF-β/Smad Signaling. PLoS ONE 2016, 11, e0155730. [Google Scholar] [CrossRef]
- Wei, X.; Jin, J.; Wu, J.; He, Y.; Guo, J.; Yang, Z.; Chen, L.; Hu, K.; Li, L.; Jia, M.; et al. Cardiac-specific BACH1 ablation attenuates pathological cardiac hypertrophy by inhibiting the Ang II type 1 receptor expression and the Ca2+/CaMKII pathway. Cardiovasc. Res. 2023, 119, 1842–1855. [Google Scholar] [CrossRef]
- Kawamura, M.; Ito, H.; Onuki, T.; Miyoshi, F.; Watanabe, N.; Asano, T.; Tanno, K.; Kobayashi, Y. Candesartan decreases type III procollagen-N-peptide levels and inflammatory marker levels and maintains sinus rhythm in patients with atrial fibrillation. J. Cardiovasc. Pharmacol. 2010, 55, 511–517. [Google Scholar] [CrossRef]
- Liu, C.; Lu, X.Z.; Shen, M.Z.; Xing, C.Y.; Ma, J.; Duan, Y.Y.; Yuan, L.J. N-Acetyl Cysteine improves the diabetic cardiac function: Possible role of fibrosis inhibition. BMC Cardiovasc. Disord. 2015, 15, 84. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, X.R.; Wei, L.H.; Chung, A.C.; Yu, C.M.; Lan, H.Y. miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-β/Smad3 signaling. Mol. Ther. 2014, 22, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Ramanujam, D.; Schön, A.P.; Beck, C.; Vaccarello, P.; Felician, G.; Dueck, A.; Esfandyari, D.; Meister, G.; Meitinger, T.; Schulz, C.; et al. MicroRNA-21-Dependent Macrophage-to-Fibroblast Signaling Determines the Cardiac Response to Pressure Overload. Circulation 2021, 143, 1513–1525. [Google Scholar] [CrossRef]
- Schauerte, C.; Hübner, A.; Rong, S.; Wang, S.; Shushakova, N.; Mengel, M.; Dettling, A.; Bang, C.; Scherf, K.; Koelling, M.; et al. Antagonism of profibrotic microRNA-21 improves outcome of murine chronic renal allograft dysfunction. Kidney Int. 2017, 92, 646–656. [Google Scholar] [CrossRef]
- Everett, R.J.; Stirrat, C.G.; Semple, S.I.; Newby, D.E.; Dweck, M.R.; Mirsadraee, S. Assessment of myocardial fibrosis with T1 mapping MRI. Clin. Radiol. 2016, 71, 768–778. [Google Scholar] [CrossRef]
- Lijnen, P.J.; Maharani, T.; Finahari, N.; Prihadi, J.S. Serum collagen markers and heart failure. Cardiovasc. Hematol. Disord. Drug Targets 2012, 12, 51–55. [Google Scholar] [CrossRef]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, M.; Zhao, Y.; Dong, M. Irisin Protects Against LPS-Stressed Cardiac Damage Through Inhibiting Inflammation, Apoptosis, and Pyroptosis. Shock 2021, 56, 1009–1018. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, K.; Han, Y.; Zhu, H.; Zhou, X.; Tan, T.; Zeng, J.; Zhang, J.; Liu, Y.; Li, Y.; et al. Irisin Protects Heart Against Ischemia-Reperfusion Injury Through a SOD2-Dependent Mitochondria Mechanism. J. Cardiovasc. Pharmacol. 2018, 72, 259–269. [Google Scholar] [CrossRef]
- Tan, Y.; Ouyang, H.; Xiao, X.; Zhong, J.; Dong, M. Irisin ameliorates septic cardiomyopathy via inhibiting DRP1-related mitochondrial fission and normalizing the JNK-LATS2 signaling pathway. Cell Stress Chaperones 2019, 24, 595–608. [Google Scholar] [CrossRef]
- Liu, J.F.; Su, G.; Chen, L.X.; Zhou, J.P.; Gao, J.; Zhang, J.J.; Wu, Q.H.; Chen, W.; Chen, D.Y.; Zhang, Z.C. Irisin Attenuates Apoptosis Following Ischemia-Reperfusion Injury Through Improved Mitochondria Dynamics and ROS Suppression Mediated Through the PI3K/Akt/mTOR Axis. Mol. Neurobiol. 2023, 60, 4261–4272. [Google Scholar] [CrossRef]
- Pan, J.A.; Zhang, H.; Lin, H.; Gao, L.; Zhang, H.L.; Zhang, J.F.; Wang, C.Q.; Gu, J. Irisin ameliorates doxorubicin-induced cardiac perivascular fibrosis through inhibiting endothelial-to-mesenchymal transition by regulating ROS accumulation and autophagy disorder in endothelial cells. Redox Biol. 2021, 46, 102120. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Hwang, S.M.; Go, E.J.; Kim, Y.H.; Park, C.K. Irisin alleviates CFA-induced inflammatory pain by modulating macrophage polarization and spinal glial cell activation. Biomed. Pharmacother. 2024, 178, 117157. [Google Scholar] [CrossRef]
- Buechler, M.B.; Fu, W.; Turley, S.J. Fibroblast-macrophage reciprocal interactions in health, fibrosis, and cancer. Immunity 2021, 54, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Yang, M.; Huang, S.; Zhong, S.; Zhang, Q.; Ding, H.; Xiong, X.; Hu, Z.; Yang, Y. Different Roles of Resident and Non-resident Macrophages in Cardiac Fibrosis. Front. Cardiovasc. Med. 2022, 9, 818188. [Google Scholar] [CrossRef]
- Chen, R.R.; Fan, X.H.; Chen, G.; Zeng, G.W.; Xue, Y.G.; Liu, X.T.; Wang, C.Y. Irisin attenuates angiotensin II-induced cardiac fibrosis via Nrf2 mediated inhibition of ROS/ TGFβ1/Smad2/3 signaling axis. Chem.-Biol. Interact. 2019, 302, 11–21. [Google Scholar] [CrossRef]
- Hu, S.Y.; Zhou, Y.; Zhong, S.J.; Yang, M.; Huang, S.M.; Li, L.; Li, X.C.; Hu, Z.X. Shenmai Injection Improves Hypertensive Heart Failure by Inhibiting Myocardial Fibrosis via TGF-β 1/Smad Pathway Regulation. Chin. J. Integr. Med. 2023, 29, 119–126. [Google Scholar] [CrossRef]
- Zhou, B.; Ling, L.; Zhang, F.; Liu, T.Y.; Zhou, H.; Qi, X.H.; Chen, Q.; Li, Y.H.; Kang, Y.M.; Zhu, G.Q. Fibronectin Type III Domain-Containing 5 Attenuates Liver Fibrosis Via Inhibition of Hepatic Stellate Cell Activation. Cell. Physiol. Biochem. 2018, 48, 227–236. [Google Scholar] [CrossRef]
- Bi, J.; Zhang, J.; Ren, Y.; Du, Z.; Zhang, Y.; Liu, C.; Wang, Y.; Zhang, L.; Shi, Z.; Wu, Z.; et al. Exercise hormone irisin mitigates endothelial barrier dysfunction and microvascular leakage-related diseases. JCI Insight 2020, 5, e136277. [Google Scholar] [CrossRef]
- Holm, L.; Kääriäinen, S.; Rosenström, P.; Schenkel, A. Searching protein structure databases with DaliLite v.3. Bioinformatics 2008, 24, 2780–2781. [Google Scholar] [CrossRef]
- Arnò, B.; Galli, F.; Roostalu, U.; Aldeiri, B.M.; Miyake, T.; Albertini, A.; Bragg, L.; Prehar, S.; McDermott, J.C.; Cartwright, E.J.; et al. TNAP limits TGF-β-dependent cardiac and skeletal muscle fibrosis by inactivating the SMAD2/3 transcription factors. J. Cell Sci. 2019, 132, jcs234948. [Google Scholar] [CrossRef]
- Peng, H.; Wang, Q.; Lou, T.; Qin, J.; Jung, S.; Shetty, V.; Li, F.; Wang, Y.; Feng, X.H.; Mitch, W.E.; et al. Myokine mediated muscle-kidney crosstalk suppresses metabolic reprogramming and fibrosis in damaged kidneys. Nat. Commun. 2017, 8, 1493. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Qin, S.; Tang, J.; Wang, T.; Ren, W.; Di, L.; Bo, W.; Ma, Y.; Wu, F.; Xu, Z.; et al. Resistance exercise upregulates Irisin expression and suppresses myocardial fibrosis following myocardial infarction via activating AMPK-Sirt1 and inactivating TGFβ1-Smad2/3. Acta Physiol. 2024, 240, e14163. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.Z.; Wen, D.; Zhang, M.; Xie, Q.; Ma, L.; Guan, Y.; Ren, Y.; Chen, J.; Hao, C.M. Sirt1 activation ameliorates renal fibrosis by inhibiting the TGF-β/Smad3 pathway. J. Cell Biochem. 2014, 115, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Han, S.; Zhu, J.; Cheng, F. MiR-132-3p activation aggravates renal ischemia-reperfusion injury by targeting Sirt1/PGC1alpha axis. Cell. Signal. 2023, 110, 110801. [Google Scholar] [CrossRef]
- Simic, P.; Williams, E.O.; Bell, E.L.; Gong, J.J.; Bonkowski, M.; Guarente, L. SIRT1 suppresses the epithelial-to-mesenchymal transition in cancer metastasis and organ fibrosis. Cell Rep. 2013, 3, 1175–1186. [Google Scholar] [CrossRef]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007, 100, 1512–1521. [Google Scholar] [CrossRef]
- Potente, M.; Ghaeni, L.; Baldessari, D.; Mostoslavsky, R.; Rossig, L.; Dequiedt, F.; Haendeler, J.; Mione, M.; Dejana, E.; Alt, F.W.; et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007, 21, 2644–2658. [Google Scholar] [CrossRef]
- Tiano, J.P.; Springer, D.A.; Rane, S.G. SMAD3 negatively regulates serum irisin and skeletal muscle FNDC5 and peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) during exercise. J. Biol. Chem. 2015, 290, 7671–7684. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, H.; Li, A.M.; Liu, Y.T.; Liu, Y.; Zhang, W.; Yang, C.; Song, N.; Zhan, M.; Yang, S. VDR regulates mitochondrial function as a protective mechanism against renal tubular cell injury in diabetic rats. Redox Biol. 2024, 70, 103062. [Google Scholar] [CrossRef]
- Bi, J.; Zhang, J.; Ren, Y.; Du, Z.; Li, Q.; Wang, Y.; Wei, S.; Yang, L.; Zhang, J.; Liu, C.; et al. Irisin alleviates liver ischemia-reperfusion injury by inhibiting excessive mitochondrial fission, promoting mitochondrial biogenesis and decreasing oxidative stress. Redox Biol. 2019, 20, 296–306. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Wang, J.; Zhang, D.; Wu, H.; Li, W.; Wei, H.; Ta, N.; Fan, Y.; Liu, Y.; et al. Mitophagy receptor FUNDC1 is regulated by PGC-1α/NRF1 to fine tune mitochondrial homeostasis. EMBO Rep. 2021, 22, e50629. [Google Scholar] [CrossRef] [PubMed]
- Suliman, H.B.; Healy, Z.; Zobi, F.; Kraft, B.D.; Welty-Wolf, K.; Smith, J.; Barkauskas, C.; Piantadosi, C.A. Nuclear respiratory factor-1 negatively regulates TGF-β1 and attenuates pulmonary fibrosis. iScience 2022, 25, 103535. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Qin, S.; Liang, Q.; Xi, Y.; Bo, W.; Cai, M.; Tian, Z. Exercise Training Enhances Myocardial Mitophagy and Improves Cardiac Function via Irisin/FNDC5-PINK1/Parkin Pathway in MI Mice. Biomedicines 2021, 9, 701. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, S.; Markwald, R.R.; Hascall, V.C.; Dowling, W.; Lottes, R.G.; Baatz, J.E.; Beeson, G.; Beeson, C.C.; Perrella, M.A.; Thannickal, V.J.; et al. Transforming growth factor β1 (TGFβ1) regulates CD44V6 expression and activity through extracellular signal-regulated kinase (ERK)-induced EGR1 in pulmonary fibrogenic fibroblasts. J. Biol. Chem. 2017, 292, 10465–10489. [Google Scholar] [CrossRef]
- Shen, S.; Wu, G.; Luo, W.; Li, W.; Li, X.; Dai, C.; Huang, W.; Liang, G. Leonurine attenuates angiotensin II-induced cardiac injury and dysfunction via inhibiting MAPK and NF-κB pathway. Phytomedicine 2023, 108, 154519. [Google Scholar] [CrossRef]
- Mi, X.; Zhang, Z.; Cheng, J.; Xu, Z.; Zhu, K.; Ren, Y. Cardioprotective effects of Schisantherin A against isoproterenol-induced acute myocardial infarction through amelioration of oxidative stress and inflammation via modulation of PI3K-AKT/Nrf2/ARE and TLR4/MAPK/NF-κB pathways in rats. BMC Complement. Med. Ther. 2023, 23, 277. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, Z.; Wang, S.; Wang, X.; Mao, J. Targeting MAPK-ERK/JNK pathway: A potential intervention mechanism of myocardial fibrosis in heart failure. Biomed. Pharmacother. 2024, 173, 116413. [Google Scholar] [CrossRef]
- Liu, X.; Mujahid, H.; Rong, B.; Lu, Q.H.; Zhang, W.; Li, P.; Li, N.; Liang, E.S.; Wang, Q.; Tang, D.Q.; et al. Irisin inhibits high glucose-induced endothelial-to-mesenchymal transition and exerts a dose-dependent bidirectional effect on diabetic cardiomyopathy. J. Cell. Mol. Med. 2018, 22, 808–822. [Google Scholar] [CrossRef]
- Ma, Y.; Kuang, Y.; Bo, W.; Liang, Q.; Zhu, W.; Cai, M.; Tian, Z. Exercise Training Alleviates Cardiac Fibrosis through Increasing Fibroblast Growth Factor 21 and Regulating TGF-β1-Smad2/3-MMP2/9 Signaling in Mice with Myocardial Infarction. Int. J. Mol. Sci. 2021, 22, 12341. [Google Scholar] [CrossRef]
- Huh, J.Y.; Panagiotou, G.; Mougios, V.; Brinkoetter, M.; Vamvini, M.T.; Schneider, B.E.; Mantzoros, C.S. FNDC5 and irisin in humans: I. Predictors of circulating concentrations in serum and plasma and II. mRNA expression and circulating concentrations in response to weight loss and exercise. Metabolism 2012, 61, 1725–1738. [Google Scholar] [CrossRef]
- Roca-Rivada, A.; Castelao, C.; Senin, L.L.; Landrove, M.O.; Baltar, J.; Belén Crujeiras, A.; Seoane, L.M.; Casanueva, F.F.; Pardo, M. FNDC5/irisin is not only a myokine but also an adipokine. PLoS ONE 2013, 8, e60563. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Li, F.; Li, H.; Huang, Y.; Liu, Y.; Chen, Y. Effects and Molecular Mechanism of GST-Irisin on Lipolysis and Autocrine Function in 3T3-L1 Adipocytes. PLoS ONE 2016, 11, e0147480. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Wrann, C.D.; Jedrychowski, M.; Vidoni, S.; Kitase, Y.; Nagano, K.; Zhou, C.; Chou, J.; Parkman, V.A.; Novick, S.J.; et al. Irisin Mediates Effects on Bone and Fat via αV Integrin Receptors. Cell 2018, 175, 1756–1768.e17. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Ando, D.; Goto, K.; Kiuchi, M.; Yamakita, M.; Koyama, K. High-intensity exercise causes greater irisin response compared with low-intensity exercise under similar energy consumption. Tohoku J. Exp. Med. 2014, 233, 135–140. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Ando, D.; Takamatsu, K.; Goto, K. Resistance exercise induces a greater irisin response than endurance exercise. Metabolism 2015, 64, 1042–1050. [Google Scholar] [CrossRef]
- Huh, J.Y.; Siopi, A.; Mougios, V.; Park, K.H.; Mantzoros, C.S. Irisin in response to exercise in humans with and without metabolic syndrome. J. Clin. Endocrinol. Metab. 2015, 100, E453–E457. [Google Scholar] [CrossRef]
- Kraemer, R.R.; Shockett, P.; Webb, N.D.; Shah, U.; Castracane, V.D. A transient elevated irisin blood concentration in response to prolonged, moderate aerobic exercise in young men and women. Horm. Metab. Res. 2014, 46, 150–154. [Google Scholar] [CrossRef]
- Jedrychowski, M.P.; Wrann, C.D.; Paulo, J.A.; Gerber, K.K.; Szpyt, J.; Robinson, M.M.; Nair, K.S.; Gygi, S.P.; Spiegelman, B.M. Detection and Quantitation of Circulating Human Irisin by Tandem Mass Spectrometry. Cell Metab. 2015, 22, 734–740. [Google Scholar] [CrossRef]
- Ulupinar, S.; Ozbay, S.; Gencoglu, C.; Altinkaynak, K.; Sebin, E.; Oymak, B. Exercise in the cold causes greater irisin release but may not be enough for adropin. Chin. J. Physiol. 2021, 64, 129–134. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Li, L.; Yang, M.; Li, B.; Hu, S. From Skeletal Muscle to Myocardium: Molecular Mechanisms of Exercise-Induced Irisin Regulation of Cardiac Fibrosis. Int. J. Mol. Sci. 2025, 26, 3550. https://doi.org/10.3390/ijms26083550
Wang Z, Li L, Yang M, Li B, Hu S. From Skeletal Muscle to Myocardium: Molecular Mechanisms of Exercise-Induced Irisin Regulation of Cardiac Fibrosis. International Journal of Molecular Sciences. 2025; 26(8):3550. https://doi.org/10.3390/ijms26083550
Chicago/Turabian StyleWang, Zhao, Lin Li, Meng Yang, Biao Li, and Siyuan Hu. 2025. "From Skeletal Muscle to Myocardium: Molecular Mechanisms of Exercise-Induced Irisin Regulation of Cardiac Fibrosis" International Journal of Molecular Sciences 26, no. 8: 3550. https://doi.org/10.3390/ijms26083550
APA StyleWang, Z., Li, L., Yang, M., Li, B., & Hu, S. (2025). From Skeletal Muscle to Myocardium: Molecular Mechanisms of Exercise-Induced Irisin Regulation of Cardiac Fibrosis. International Journal of Molecular Sciences, 26(8), 3550. https://doi.org/10.3390/ijms26083550