
Mechanisms of Adipose Tissue Metabolism in Naturally Grazing Sheep at Different Growth Stages: Insights from mRNA and miRNA Profiles

 , , and
, , and
Abstract
1. Introduction
2. Results
2.1. mRNA Sequencing Analysis
2.1.1. Overview of mRNA Sequencing Data
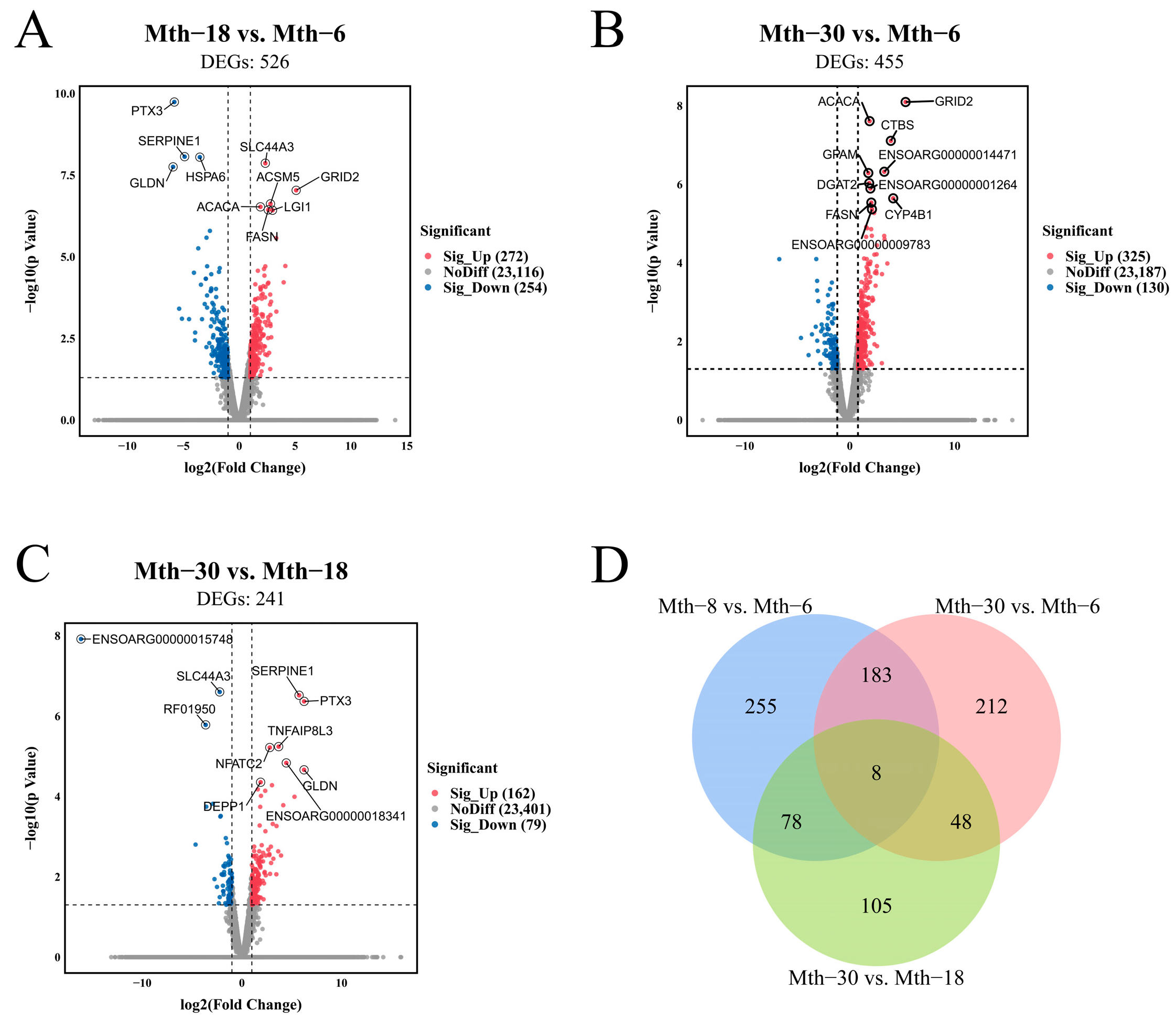
2.1.2. Differential Expression Analysis of Genes
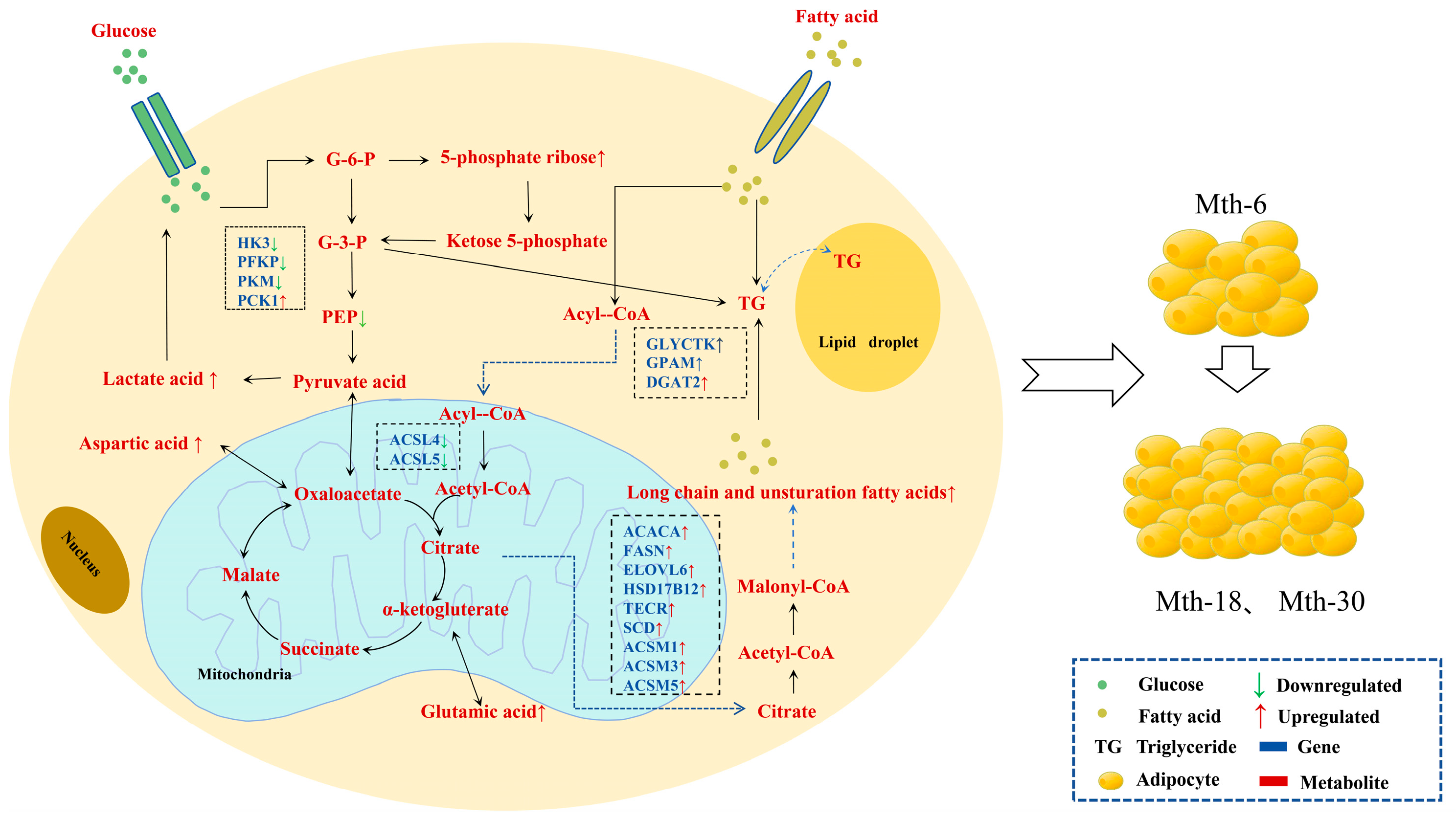
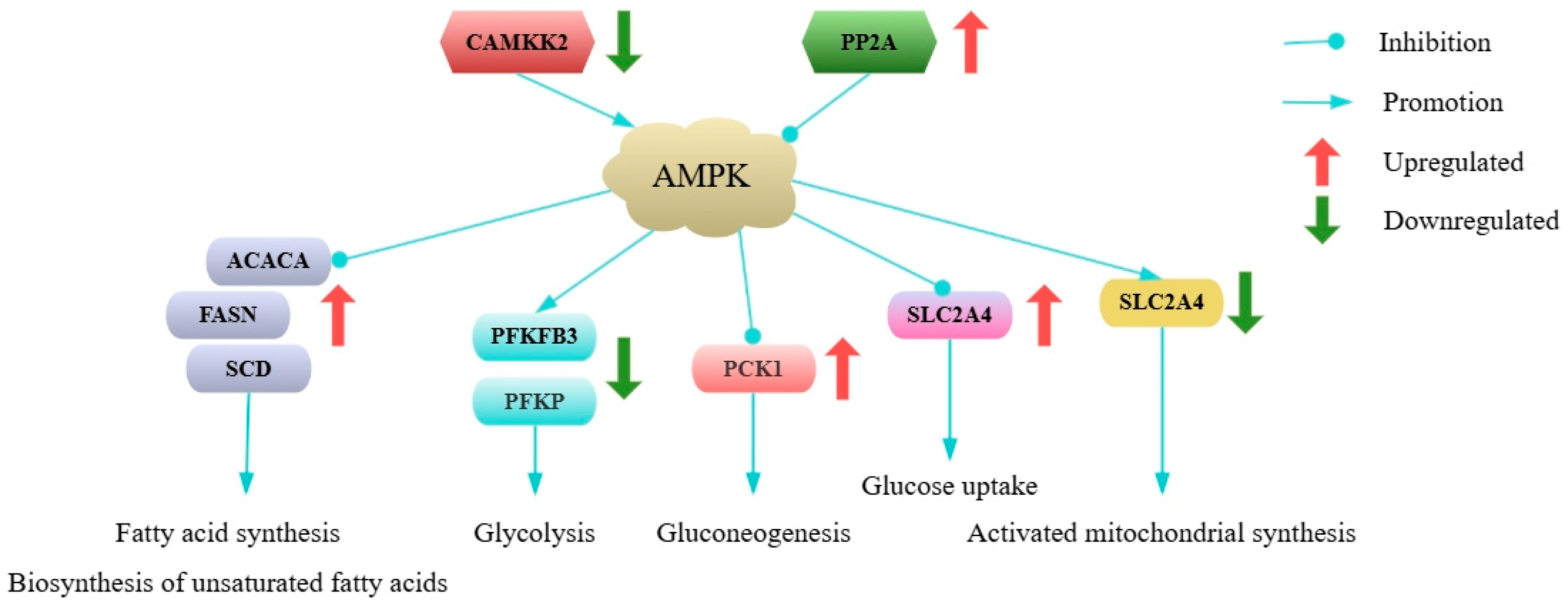
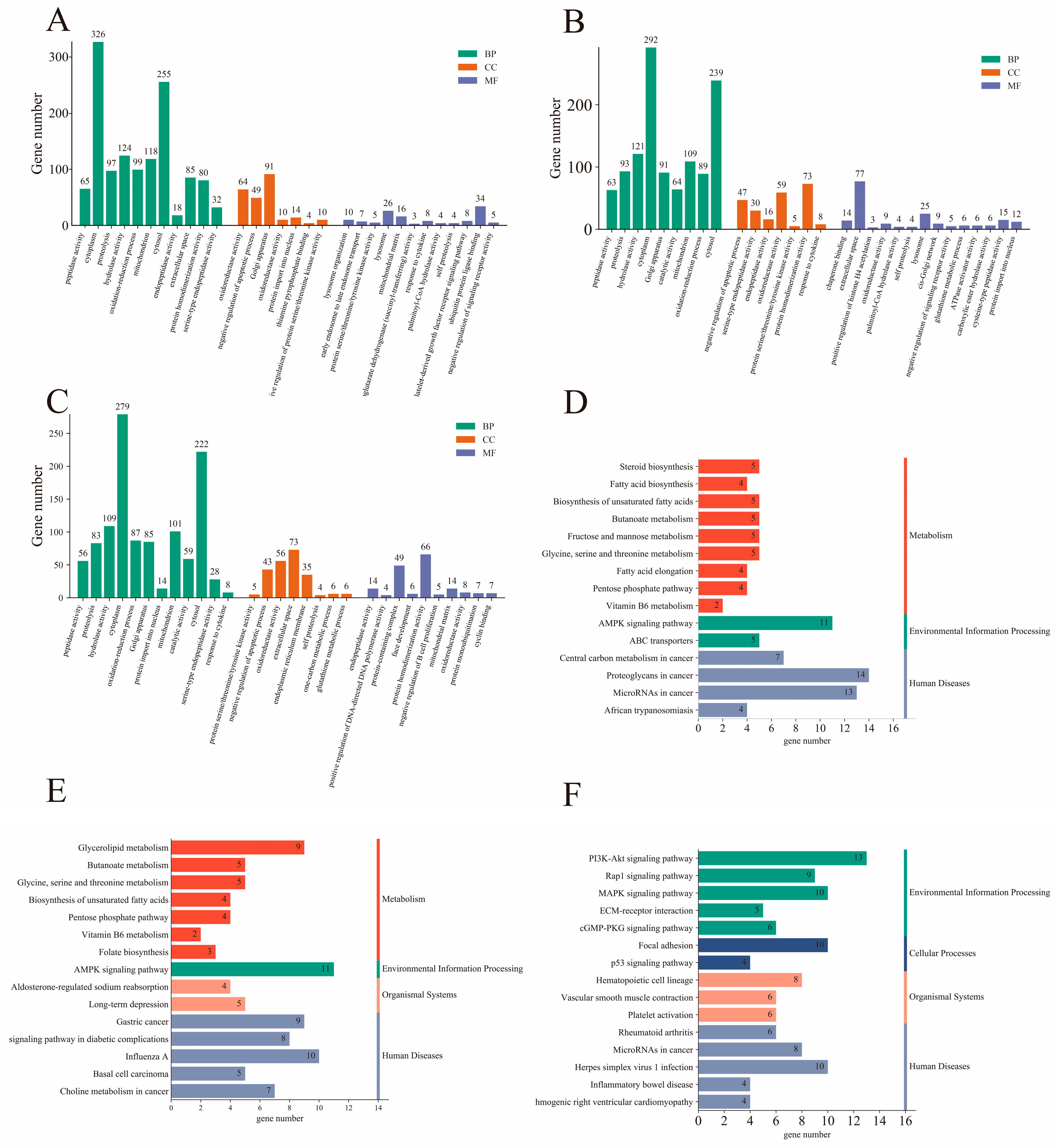
2.1.3. Functional Annotation and Enrichment Analysis of DEGs
2.2. miRNA Sequencing Analysis
2.2.1. Overview of miRNA Sequencing Data
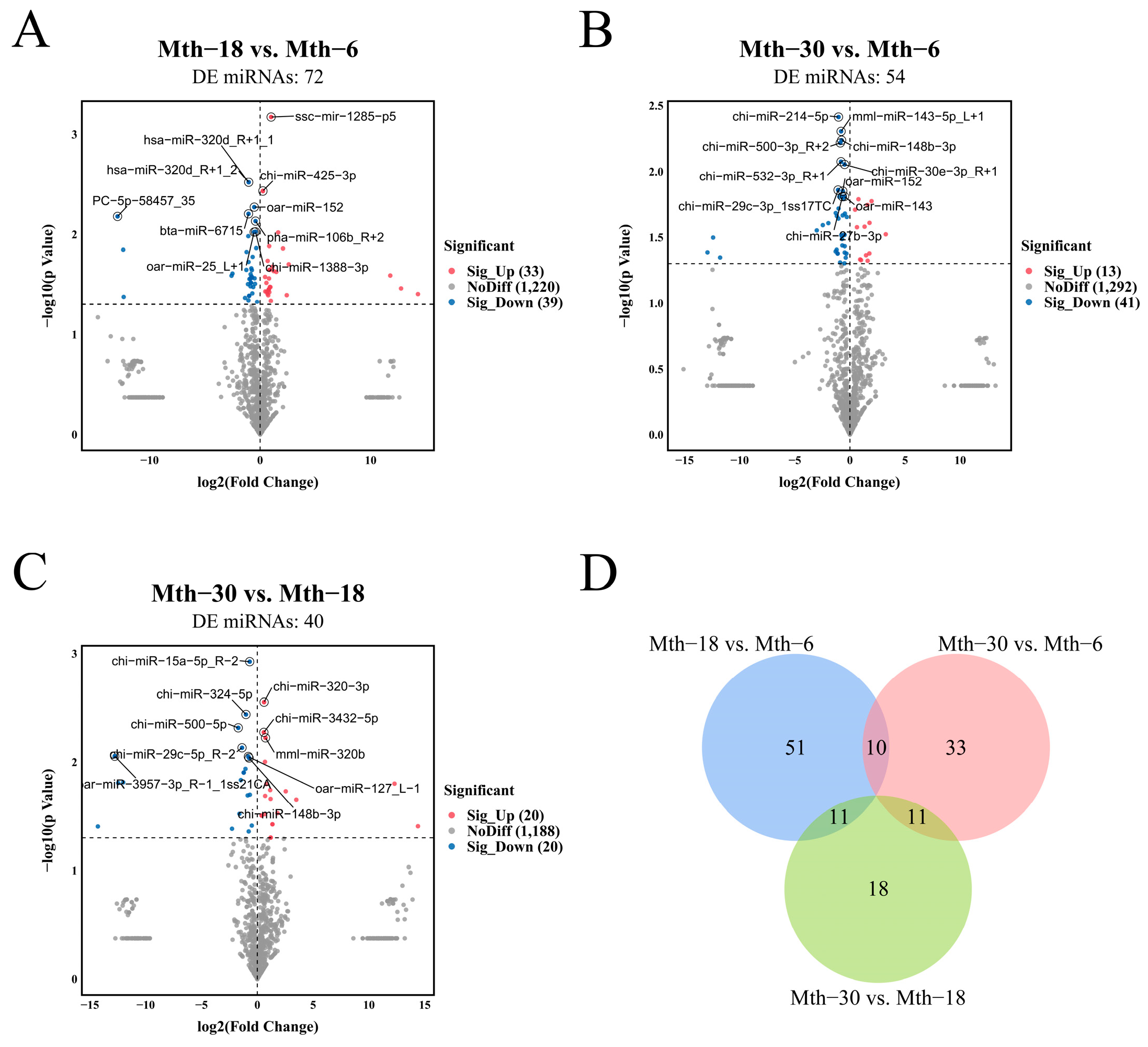
2.2.2. Differential Expression Analysis of miRNAs
2.2.3. Enrichment Analysis of Target Genes for DE miRNAs
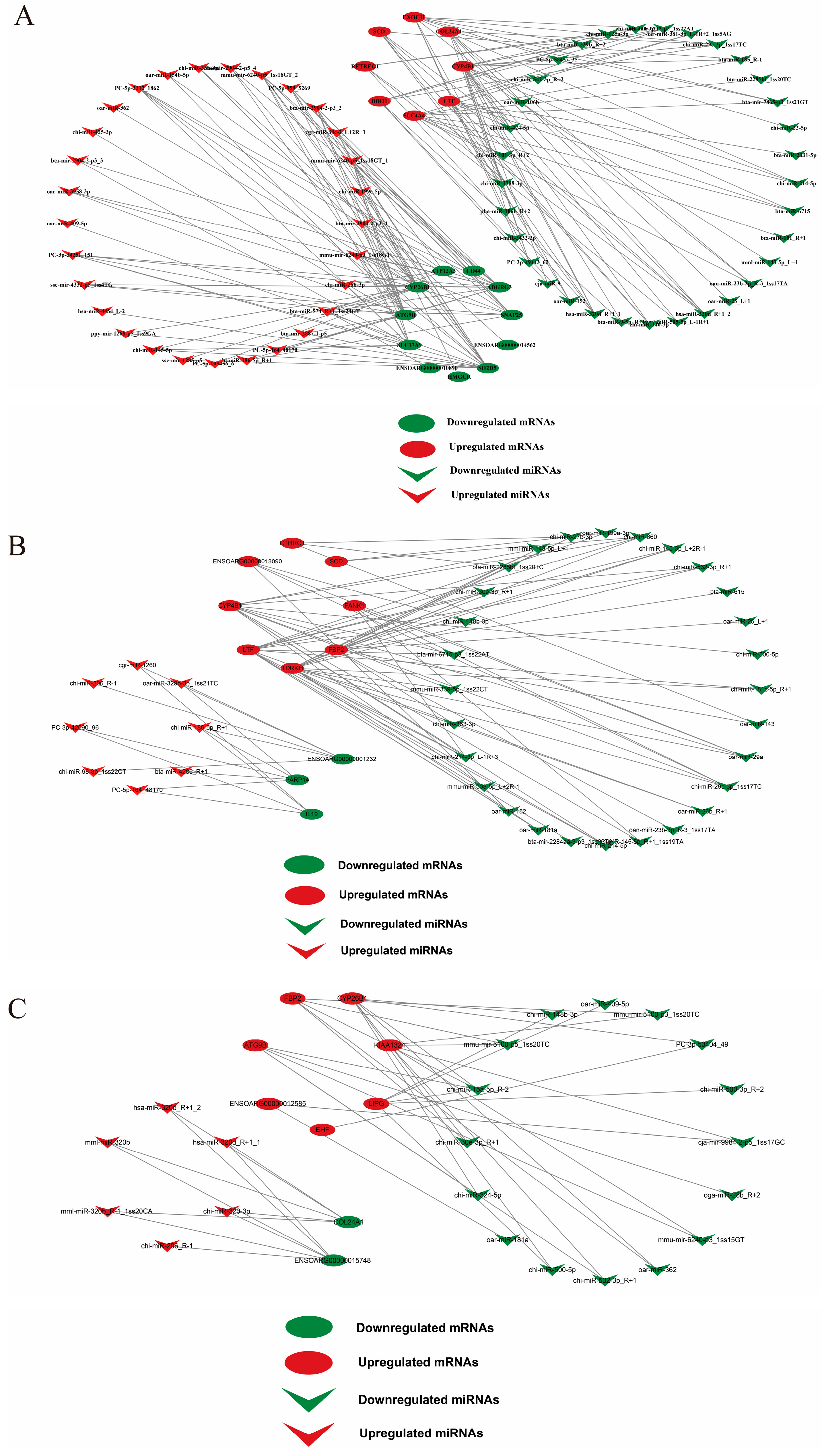
2.3. Integrated miRNA–mRNA Interaction Analysis
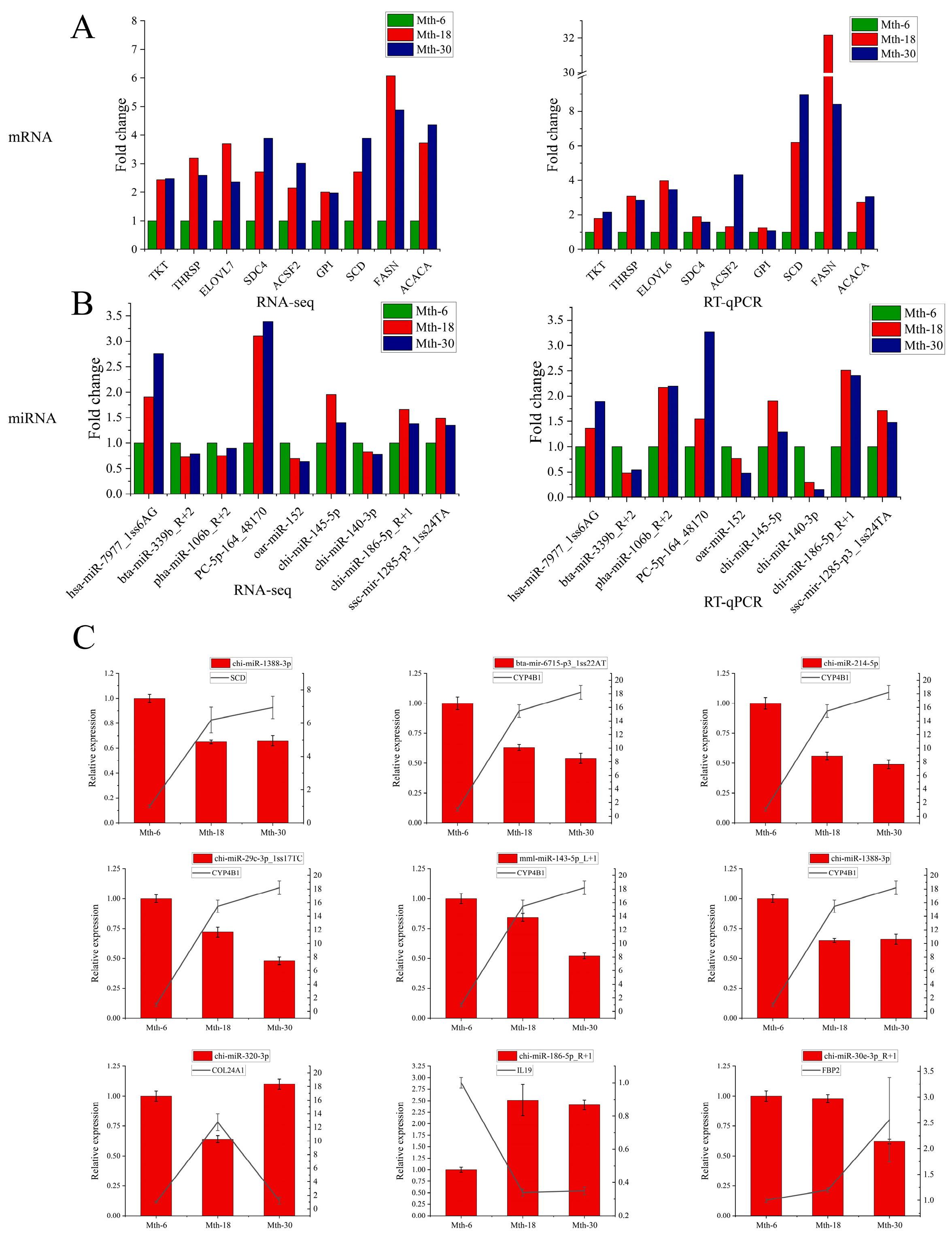
2.4. Validation Using RT-qPCR
3. Discussion
3.1. Effects of Gene Expression Changes on Adipose Tissue Metabolism
3.2. Effects of miRNA Expression Changes on Adipose Tissue Metabolism
4. Materials and Methods
4.1. Sample Collection
4.2. RNA Extraction
4.3. mRNA Sequencing and Data Analysis
4.4. miRNA Sequencing and Data Analysis
4.5. mRNA and miRNA Data Validation Using qRT-PCR
4.6. miRNA–mRNA Interaction Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Y.; Erdene, K.; Zhao, Y.B.; Li, C.; Wang, L.; Tian, F.; Ao, C.; Jin, H. Role of white adipose tissue browning in cold seasonal acclimation in grazing Mongolian sheep (Ovis aries). J. Therm. Biol. 2022, 109, 103333. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.X.; Wang, B.; Jing, J.N.; Ma, R.; Luo, Y.H.; Li, X.; Yan, Z.; Liu, Y.J.; Gao, L.; Ren, Y.L.; et al. Whole-body adipose tissue multi-omic analyses in sheep reveal molecular mechanisms underlying local adaptation to extreme environments. Commun. Biol. 2023, 6, 159. [Google Scholar] [CrossRef]
- Junior, F.F.; de Freitas Pena, A.; Grandis, F.A.; Koritiaki, N.A.; de Almeida Rego, F.C.; dos Santos, R.M.; de Azambuja Ribeiro, E.L. Subcutaneous fat thickness at slaughter in castrated and non-castrated Santa Inês and Dorper lambs and its influence on meat and carcass quality. Livest. Sci. 2021, 253, 104694. [Google Scholar] [CrossRef]
- Silva-Vignato, B.; Coutinho, L.L.; Poleti, M.D.; Cesar, A.S.M.; Moncau, C.T.; Regitano, L.C.A.; Balieiro, J.C.C. Gene co-expression networks associated with carcass traits reveal new pathways for muscle and fat deposition in Nelore cattle. BMC Genom. 2019, 20, 32. [Google Scholar] [CrossRef]
- Lu, Z.; Li, J.; Yuan, C.; Xi, B.; Yang, B.; Meng, X.; Guo, T.; Yue, Y.; Gao, Y.; Liu, J.; et al. Evaluation of Mutton Quality Characteristics of Dongxiang Tribute Sheep Based on Membership Function and Gas Chromatography and Ion Mobility Spectrometry. Front. Nutr. 2022, 9, 852399. [Google Scholar] [CrossRef]
- Jiao, D.; Ji, K.; Liu, H.; Wang, W.; Wu, X.; Zhou, J.; Zhang, Y.; Zhou, H.; Hickford, J.G.H.; Degen, A.A.; et al. Transcriptome analysis reveals genes involved in thermogenesis in two cold-exposed sheep breeds. Genes 2021, 12, 375. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liang, C.; Manthari, R.K.; Yu, Y.; Zhang, J.; Wang, J.; Cao, J. Distribution characteristics and regulation of amino acids and fatty acids in muscle and adipose tissues of sheep grown in natural grazing environment. Anim. Sci. J. Nihon Chikusan Gakkaiho 2022, 93, e13769. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Qu, H.; Xiong, X.; Wang, Y.; Liu, X.; Zhang, L.; Liao, X.; Liao, Q.; Sun, Z.; Ouyang, Q. Deficiency of Mitochondrial Glycerol 3-Phosphate Dehydrogenase Contributes to Hepatic Steatosis. Hepatology 2019, 70, 84–97. [Google Scholar] [CrossRef]
- Xiong, L.; Pei, J.; Wu, X.; Kalwar, Q.; Yan, P.; Guo, X. Effect of gender to fat deposition in Yaks based on transcriptomic and metabolomics analysis. Front. Cell Dev. Biol. 2021, 9, 653188. [Google Scholar] [CrossRef]
- Sun, B.; Hayashi, M.; Kudo, M.; Wu, L.; Qin, L.; Gao, M.; Liu, T. Madecassoside Inhibits Body Weight Gain via Modulating SIRT1-AMPK Signaling Pathway and Activating Genes Related to Thermogenesis. Front. Endocrinol. 2021, 12, 627950. [Google Scholar] [CrossRef]
- Wei, W.; Xiao, J.; Huang, N.; Xing, C.; Wang, J.; He, X.; Xu, J.; Wang, H.; Guo, X.; Jiang, R. Identification of central regulators related to abdominal fat deposition in chickens based on weighted gene co-expression network analysis. Poult. Sci. 2024, 103, 103436. [Google Scholar] [PubMed]
- Fan, Y.; Gan, M.; Tan, Y.; Chen, L.; Zhu, L.J.M. Mir-152 regulates 3T3-L1 preadipocyte proliferation and differentiation. Molecules 2019, 24, 3379. [Google Scholar] [CrossRef]
- Wang, Q.; Cao, H.; Su, X.; Liu, W. Identification of key miRNAs regulating fat metabolism based on RNA-seq from fat-tailed sheep and F2 of wild Argali. Gene 2022, 834, 146660. [Google Scholar] [PubMed]
- Bae, I.S.; Park, P.J.; Lee, J.H.; Cho, E.G.; Lee, T.R.; Kim, S.H. PPARγ-mediated G-protein coupled receptor 120 signaling pathway promotes transcriptional activation of miR-143 in adipocytes. Gene 2017, 626, 64–69. [Google Scholar]
- Wu, W.; Liu, K.; You, Z.; Zhang, J. MiR-196b-3p and miR-450b-3p are key regulators of adipogenesis in porcine intramuscular and subcutaneous adipocytes. BMC Genom. 2023, 24, 360. [Google Scholar]
- Yuan, Z.; Xiang, R.; Li, W.; Li, F.; Yue, X. Transcriptomic analyses revealed common tailed and perirenal adipose differentially expressed genes in four Chinese indigenous sheep breeds. Livest. Sci. 2019, 230, 103832. [Google Scholar]
- He, X.; Wu, R.; Yun, Y.; Qin, X.; Huang, Y.; Chen, L.; Han, Y.; Wu, J.; Sha, L.; Borjigin, G. MicroRNA and circular RNA profiling in the deposited fat tissue of Sunite sheep. Front. Vet. Sci. 2022, 9, 954882. [Google Scholar]
- Han, Y.; He, X.; Yun, Y.; Chen, L.; Huang, Y.; Wu, Q.; Qin, X.; Wu, H.; Wu, J.; Sha, R.; et al. The characterization of subcutaneous adipose tissue in Sunit sheep at different growth stages: A comprehensive analysis of the morphology, fatty acid profile, and metabolite profile. Foods 2024, 13, 544. [Google Scholar] [CrossRef]
- Liu, J.; Ning, C.; Li, B.; Li, R.; Wu, W.; Liu, H. Transcriptome comparison between prenatal and postnatal Large White livers identifies differences in the expression level of genes related to metabolism and postnatal growth. Gene 2019, 686, 92–103. [Google Scholar]
- Xiao, C.; Sun, T.; Yang, Z.; Zou, L.; Deng, J.; Yang, X. Whole-transcriptome RNA sequencing reveals the global molecular responses and circRNA/lncRNA-miRNA-mRNA ceRNA regulatory network in chicken fat deposition. Poult. Sci. 2022, 101, 102121. [Google Scholar]
- Maa, Z.; Lia, H.; Zhenga, H.; Jianga, K.; Yana, F.; Tiana, Y.; Kanga, X.; Wanga, Y.; Liua, X. Hepatic ELOVL6 mRNA is regulated by the gga-miR-22-3p in egg-laying hen. Gene 2017, 623, 72–79. [Google Scholar]
- Angkawijaya, A.E.; Nguyen, V.C.; Gunawan, F.; Nakamura, Y. A pair of arabidopsis diacylglycerol kinases essential for gametogenesis and endoplasmic reticulum phospholipid metabolism in leaves and flowers. Plant Cell 2020, 32, 2602–2620. [Google Scholar]
- Yu, H.; Zhao, Z.; Yu, X.; Li, J.; Lu, C.; Yang, R. Bovine lipid metabolism related gene GPAM: Molecular characterization, function identification, and association analysis with fat deposition traits. Gene 2017, 609, 9–18. [Google Scholar]
- Zhang, Z.; Shu, G.; Zhu, X.; Guo, J.; Cai, H.; Wang, S.; Wang, L.; Gao, P.; Xi, Q.; Zhang, Y. Effect of diacylglycerol acyltransferase 2 overexpression in 3T3-L1 is associated to an increase in mono-unsaturated fatty acid accumulation. J. Anim. Sci. Biotechnol. 2014, 5, 406–413. [Google Scholar]
- Riedel, E.O.; Hinrichs, A.; Kemter, E.; Dahlhoff, M.; Backman, M.; Rathkolb, B.; Prehn, C.; Adamski, J.; Renner, S.; Blutke, A.; et al. Functional changes of the liver in the absence of growth hormone (GH) action—Proteomic and metabolomic insights from a GH receptor deficient pig model. Mol. Metab. 2020, 36, 100978. [Google Scholar] [PubMed]
- Liu, T.; Feng, H.; Yousuf, S.; Xie, L.; Miao, X. Differential regulation of mRNAs and lncRNAs related to lipid metabolism in Duolang and Small Tail Han sheep. Sci. Rep. 2022, 12, 11157. [Google Scholar]
- Hosseini, S.F.; Bakhtiarizadeh, M.R.; Salehi, A. Meta-analysis of RNA-Seq datasets highlights novel genes/pathways involved in fat deposition in fat-tail of sheep. Front. Vet. Sci. 2023, 10, 1159921. [Google Scholar]
- Martín-Bernabé, A.; Tarragó-Celada, J.; Cunin, V.; Michelland, S.; Cortés, R.; Poignant, J.; Boyault, C.; Rachidi, W.; Bourgoin-Voillard, S.; Cascante, M.; et al. Quantitative proteomic approach reveals altered metabolic pathways in response to the inhibition of lysine deacetylases in A549 cells under normoxia and hypoxia. Int. J. Mol. Sci. 2021, 22, 3378. [Google Scholar] [CrossRef]
- Kudryavtseva, A.V.; Fedorova, M.S.; Zhavoronkov, A.; Moskalev, A.A.; Zasedatelev, A.S.; Dmitriev, A.A.; Sadritdinova, A.F.; Karpova, I.Y.; Nyushko, K.M.; Kalinin, D.V.; et al. Effect of lentivirus-mediated shRNA inactivation of HK1, HK2, and HK3 genes in colorectal cancer and melanoma cells. BMC Genet. 2016, 17, 156. [Google Scholar]
- Wang, D.; Liu, F.; Yang, W.; Sun, Y.; Wang, X.; Sui, X.; Yang, J.; Wang, Q.; Song, W.; Zhang, M.; et al. Meldonium ameliorates hypoxia-induced lung injury and oxidative stress by regulating platelet-type phosphofructokinase-mediated glycolysis. Front. Pharmacol. 2022, 13, 863451. [Google Scholar]
- Lunt, S.Y.; Muralidhar, V.; Hosios, A.M.; Israelsen, W.J.; Gui, D.Y.; Newhouse, L.; Ogrodzinski, M.; Hecht, V.; Xu, K.; Acevedo, P.N.M.; et al. Pyruvate kinase isoform expression alters nucleotide synthesis to impact cell proliferation. Mol. Cell 2015, 57, 95–107. [Google Scholar] [PubMed]
- Latorre-Muro, P.; Baeza, J.; Hurtado-Guerrero, R.; Hicks, T.; Delso, I.; Hernández-Ruiz, C.; Velázquez-Campoy, A.; Lawton, A.J.; Angulo, J.; Denu, J.M.; et al. Self-acetylation at the active site of phosphoenolpyruvate carboxykinase (PCK1) controls enzyme activity. J. Biol. Chem. 2021, 296, 100205. [Google Scholar]
- Tang, Y.; Zhang, Y.; Wang, C.; Sun, Z.; Li, L.; Cheng, S.; Zhou, W. Overexpression of PCK1 Gene Antagonizes Hepatocellular Carcinoma Through the Activation of Gluconeogenesis and Suppression of Glycolysis Pathways. Cell. Physiol. Biochem. 2018, 47, 344–355. [Google Scholar] [PubMed]
- Yi, D.; Yu, H.; Lu, K.; Ruan, C.; Ding, C.; Tong, L.; Zhao, X.; Chen, D. AMPK signaling in energy control, cartilage biology, and osteoarthritis. Front. Cell Dev. Biol. 2021, 9, 696602. [Google Scholar]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 2017, 66, 789–800. [Google Scholar]
- Li, C.; Hao, J.; Qiu, H.; Xin, H. CaMKK2 alleviates myocardial ischemia/reperfusion injury by inhibiting oxidative stress and inflammation via the action on the AMPK-AKT-GSK-3β/Nrf2 signaling cascade. Inflamm. Res. 2023, 72, 1409–1425. [Google Scholar]
- Hridya, H.; Amrita, A.; Sankari, M.; Doss, C.; Gopalakrishnan, M. Inhibitory effect of brazilein on tyrosinase and melanin synthesis: Kinetics and in silico approach. Int. J. Biol. Macromolec 2015, 81, 228–234. [Google Scholar]
- Yu, Z.; Jian, Z.; Shen, S.-H.; Purisima, E.; Wang, E. Global analysis of microRNA target gene expression reveals that miRNA targets are lower expressed in mature mouse and Drosophila tissues than in the embryos. Nucleic Acids Res. 2007, 35, 152–164. [Google Scholar] [PubMed]
- Yu, X.; Fang, X.; Gao, M.; Mi, J.; Zhang, X.; Xia, L.; Zhao, Z.; Albrecht, E.; Maak, S.; Yang, R. Isolation and identification of bovine preadipocytes and screening of MicroRNAs associated with adipogenesis. Animals 2020, 10, 818. [Google Scholar] [CrossRef]
- An, X.; Ma, K.; Zhang, Z.; Zhao, T.; Zhang, X.; Tang, B.; Li, Z. MiR-17, miR-21, and miR-143 enhance adipogenic differentiation from porcine bone marrow-derived mesenchymal stem cells. Dna Cell Biol. 2016, 35, 410–416. [Google Scholar]
- Li, W.; Song, Z.; Wenzhen, Z.; Gong, C.; Rajwali, K.; Zainaguli, J.; Shijun, L.; Linsen, Z. MiR-424 promotes bovine adipogenesis through an unconventional post-transcriptional regulation of STK11. Front. Genet. 2020, 11, 145. [Google Scholar]
- Bao, J.; Wang, Z.; Zhao, M.; Wang, Y.; Zhang, J.; Li, Y.; Gegen, T.; Jia, Y. Study on quality characteristics of natural pastures in different steppe types. Feed Res. 2022, 4, 75–78. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway id | Pathway Name | Upregulated Gene | Downregulated Gene |
|---|---|---|---|
| ko00100 | Steroid biosynthesis | DHCR24, TM7SF2, CYP1B1, HSD17B12, STS | LIPA, SQLE, MSMO1 |
| ko00061 | Fatty acid biosynthesis | ACACA, FASN | ACSL4, ACSL5 |
| ko01040 | Biosynthesis of unsaturated fatty acids | ELOVL6, ENSOARG00000005352 (TECR), HSD17B12, SCD | ACOT7 |
| ko00650 | Butanoate metabolism | ACSM1, ACSM3, ACSM5, BDH1, HMGCLL1 | |
| ko04152 | AMPK signaling pathway | ACACA, FASN, PCK1, PFKFB4, PPARGC1A, PPP2R2B, SCD | HMGCR, PFKP, PIK3R3, PFKFB3 |
| ko00051 | Fructose and mannose metabolism | PFKFB4 | HK3, PFKFB3, PFKP, TIGAR |
| ko00260 | Glycine, serine, and threonine metabolism | ALAS2, GLYCTK | PSAT1 |
| ko00062 | Fatty acid elongation | ELOVL6, ENSOARG00000005352 (TECR), HSD17B12 | ACOT7 |
| ko00030 | Pentose phosphate pathway | GLYCTK, GPI, TKT | PFKP |
| ko04514 | Cell adhesion molecules (CAMs) | CNTN1, NFASC, NTNG1 | CD274, CD86, PVR, SELE |
| ko00561 | Glycerolipid metabolism | DGAT2, GLYCTK, GPAM |
| Pathway id | Pathway Name | Upregulated Gene | Downregulated Gene |
|---|---|---|---|
| ko00561 | Glycerolipid metabolism | DGAT2, DGKA, GLYCTK, GPAM, LPL | |
| ko04152 | AMPK signaling pathway | ACACA, FASN, FBP2, HNF4A, PCK1, PFKFB4, SCD, SLC2A4 | CAMKK2, FOXO3, PIK3R3 |
| ko00650 | Butanoate metabolism | ACSM1, ACSM3, ACSM5, BDH1, ECHS1 | |
| ko00260 | Glycine, serine, and threonine metabolism | ALAS2, GLYCTK, PHGDH | |
| ko01040 | Biosynthesis of unsaturated fatty acids | ACAA1, ELOVL6, HSD17B12, SCD | |
| ko00030 | Pentose phosphate pathway | FBP2, GLYCTK, RGN, TKT | |
| ko04010 | MAPK signaling pathway | CSF1, FGF1, FGF2, FGFR2, GADD45G, PRKCA, RPS6KA2, RRAS2 | DUSP10, JUN, KITLG, NR4A1, TAOK1, VEGFC |
| ko00062 | Fatty acid elongation | ECHS1, ELOVL6, HSD17B12 | |
| ko04151 | PI3K-Akt signaling pathway | COL1A1, COL1A2, CSF1, FGF1, FGF2, FGFR2, FOXO3, LAMB3, PCK1, PIK3R3, THBS1, VEGFC | KITLG, NR4A1, PRKCA, SGK2 |
| Pathway id | Pathway Name | Upregulated Gene | Downregulated Gene |
|---|---|---|---|
| ko04151 | PI3K-Akt signaling pathway | CREB3L1, CSF1, ENSOARG00000005037 (THBS2S), ENSOARG00000011855 (ITGB3, CD61), FLT4, IL4R, ITGA5, RELN | ANGPT1, BRCA1, KIT, VEGFC, PPP2R2B |
| ko04010 | MAPK signaling pathway | CSF1, DUSP4, FLT4, GADD45B, GADD45G | ANGPT1, KIT, VEGFC |
| ko04022 | cGMP-PKG signaling pathway | CREB3L1 | PDE3A, MYLK3 |
| ko00051 | Fructose and mannose metabolism | FBP2, HK3 | |
| ko04014 | Ras signaling pathway | CSF1, ENSOARG00000005737(RASSF5, RAPL), FLT4 | ANGPT1, KIT, PLD1, VEGFC |
| ko04923 | Regulation of lipolysis in adipocytes | PRKG1 | |
| ko0452 | ECM–receptor interaction | CD44, ENSOARG00000005037 (THBS2S), ENSOARG00000011855 (ITGB3, CD61), ITGA5, RELN |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, X.; Han, Y.; Chen, L.; Yun, Y.; Huang, Y.; Borjigin, G.; Nashun, B. Mechanisms of Adipose Tissue Metabolism in Naturally Grazing Sheep at Different Growth Stages: Insights from mRNA and miRNA Profiles. Int. J. Mol. Sci. 2025, 26, 3324. https://doi.org/10.3390/ijms26073324
He X, Han Y, Chen L, Yun Y, Huang Y, Borjigin G, Nashun B. Mechanisms of Adipose Tissue Metabolism in Naturally Grazing Sheep at Different Growth Stages: Insights from mRNA and miRNA Profiles. International Journal of Molecular Sciences. 2025; 26(7):3324. https://doi.org/10.3390/ijms26073324
Chicago/Turabian StyleHe, Xige, Yunfei Han, Lu Chen, Yueying Yun, Yajuan Huang, Gerelt Borjigin, and Buhe Nashun. 2025. "Mechanisms of Adipose Tissue Metabolism in Naturally Grazing Sheep at Different Growth Stages: Insights from mRNA and miRNA Profiles" International Journal of Molecular Sciences 26, no. 7: 3324. https://doi.org/10.3390/ijms26073324
APA StyleHe, X., Han, Y., Chen, L., Yun, Y., Huang, Y., Borjigin, G., & Nashun, B. (2025). Mechanisms of Adipose Tissue Metabolism in Naturally Grazing Sheep at Different Growth Stages: Insights from mRNA and miRNA Profiles. International Journal of Molecular Sciences, 26(7), 3324. https://doi.org/10.3390/ijms26073324