
Identification of Quantitative Trait Loci for Node Number, Pod Number, and Seed Number in Soybean

 ,
,  , and
, and 
Abstract
1. Introduction
2. Results
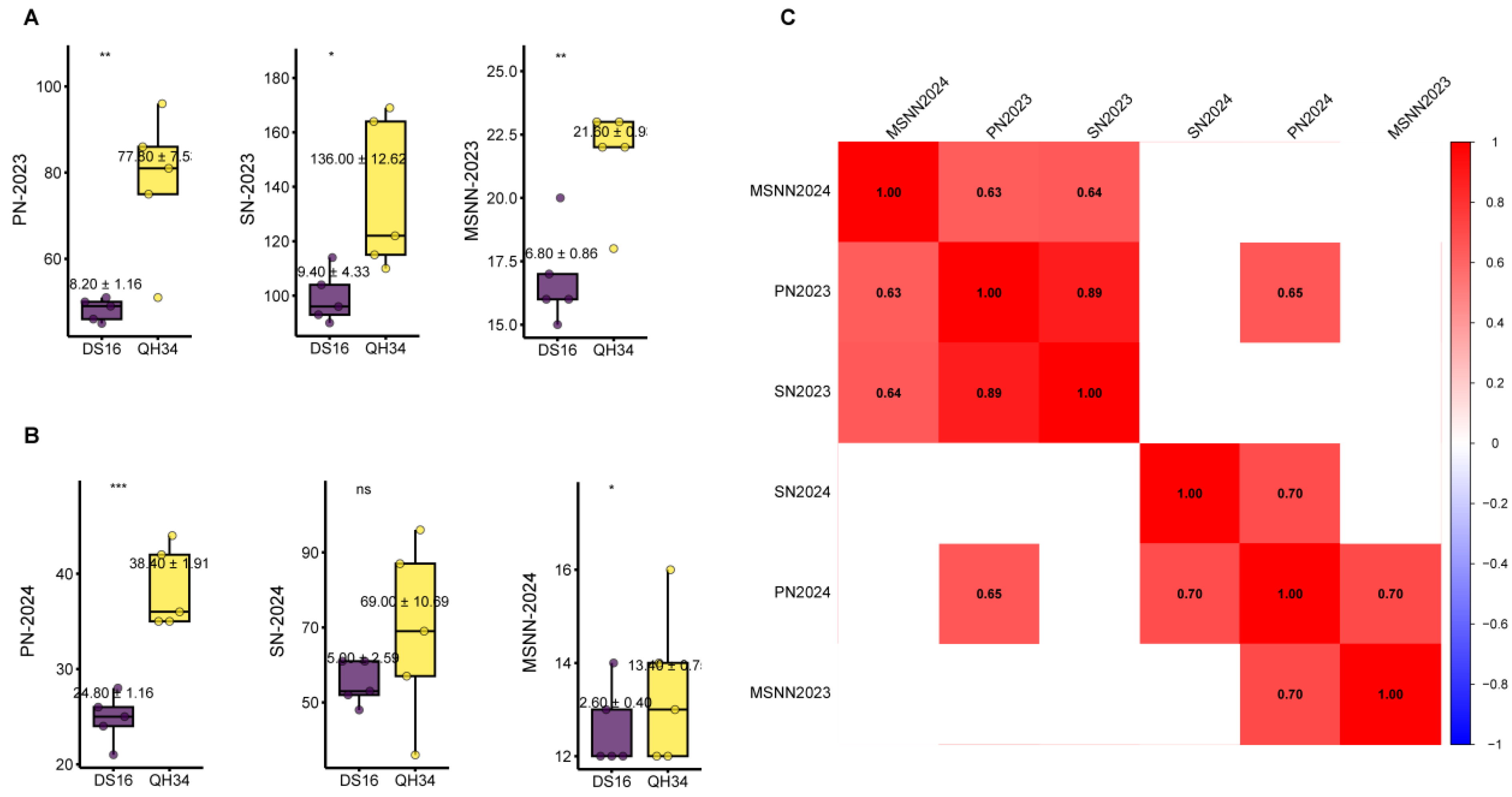
2.1. Phenotypic Characteristics of Parents and RILs
2.2. Construction of the High-Density Genetic Map
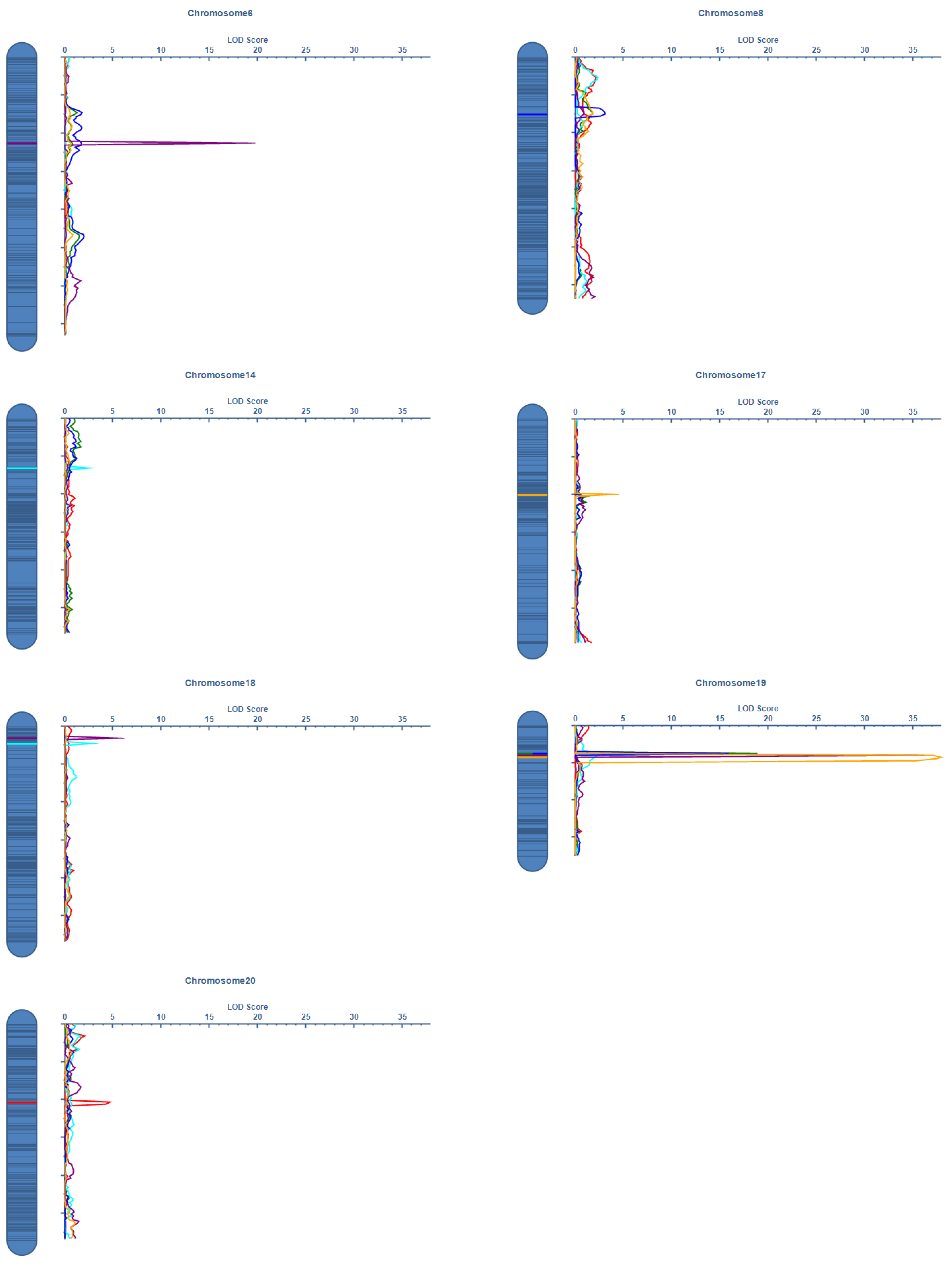
2.3. QTL Mapping and Genetic Architecture
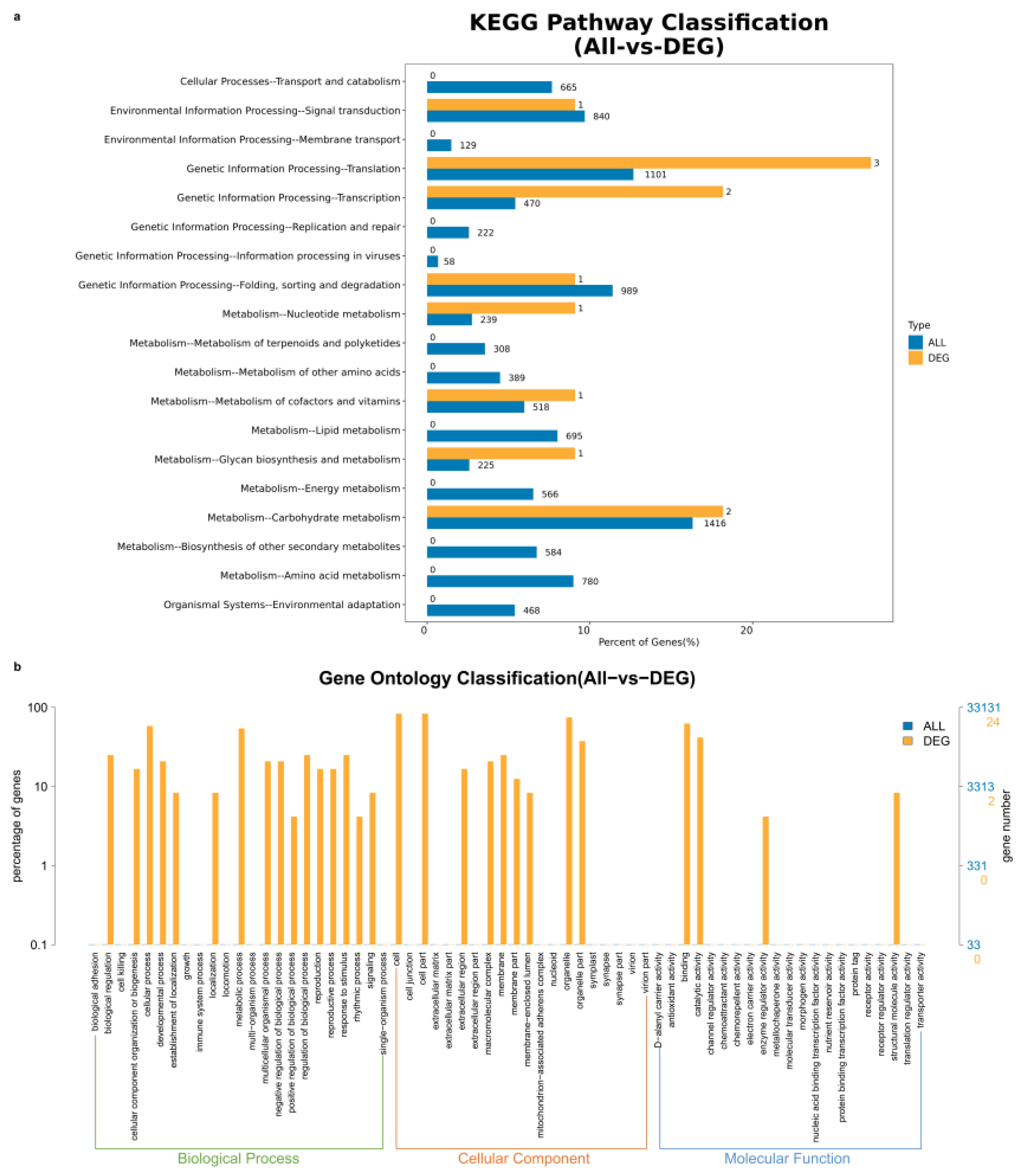
2.4. Gene GO Enrichment Analysis
2.5. Candidate Genes Mining
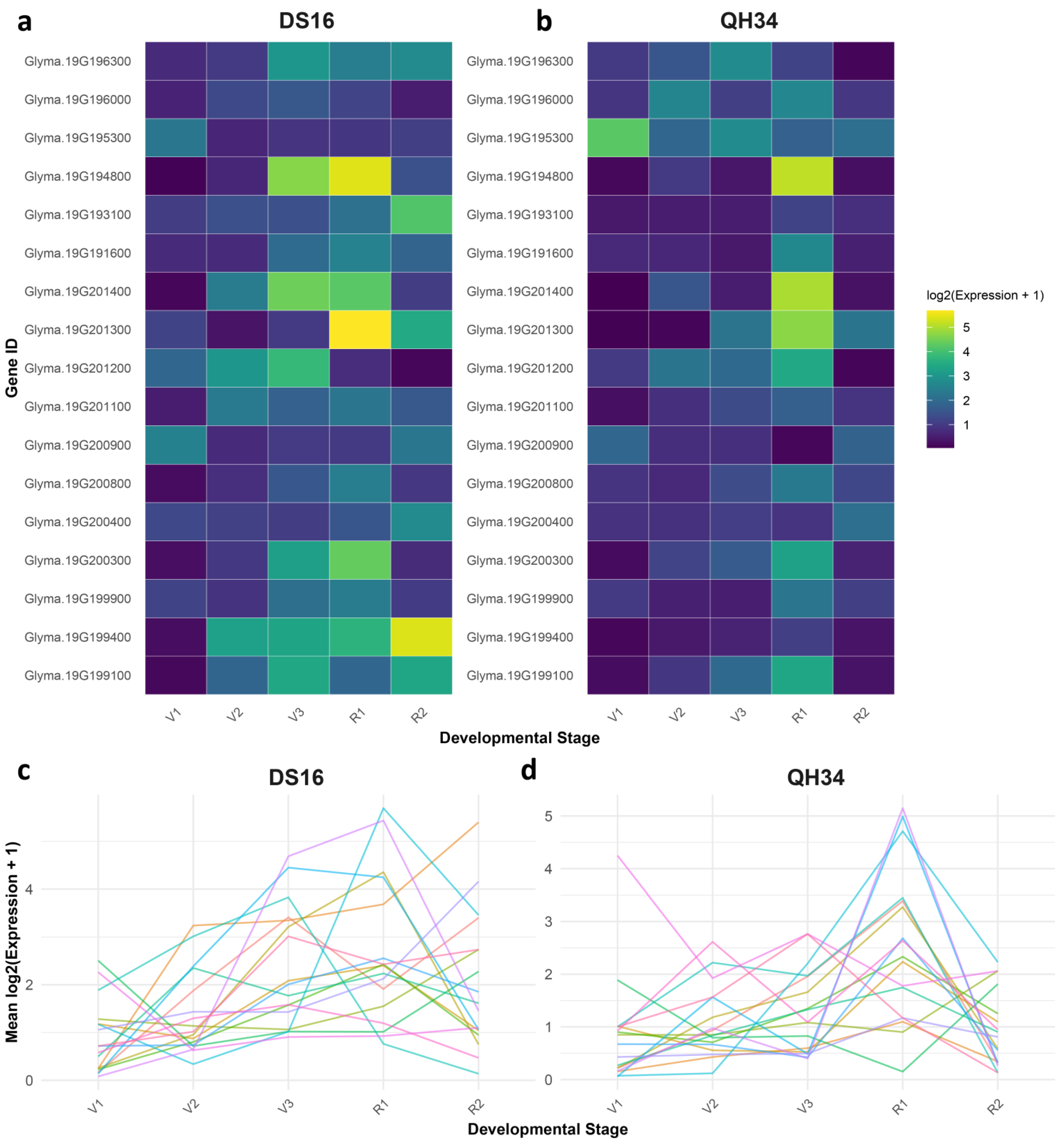
2.6. Differential Gene Expression Patterns Between QH34 and DS16 Lines
3. Discussion
4. Materials and Methods
4.1. Soybean Populations Assessments
4.2. Phenotypic Statistics and Analysis
4.3. DNA Extraction, Construction and Genotyping of SLAF Libraries
4.4. SLAF-Sequence and Genotype Parental Lines and RILs
4.5. Polymorphic SLAF Tag Encoding and Screening of Mapping SLAF Tags
4.6. QTL Mapping Analysis of RIL Population
4.7. Candidate Gene Prediction
4.8. Quantitative Real-Time PCR (qRT-PCR) Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MSNN | Main Stem Node Number |
| QTL | Quantitative Trait Loci |
| RILs | Recombinant inbred lines |
| PN | Pod number |
| SN | Seed number |
| SLAF | Specific locus-amplified fragment sequencing |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
References
- Chowdhury, S.; Fuller, R.A.; Dingle, H.; Chapman, J.W.; Zalucki, M.P. Migration in butterflies: A global overview. Biol. Rev. 2021, 96, 1462–1483. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, M.; Li, M.; Jiang, X.; Jiao, W.; Song, Q. A telomere-to-telomere gap-free assembly of soybean genome. Mol. Plant 2023, 16, 1711–1714. [Google Scholar] [CrossRef] [PubMed]
- Sedivy, E.J.; Wu, F.; Hanzawa, Y. Soybean domestication: The origin, genetic architecture and molecular bases. New Phytol. 2017, 214, 539–553. [Google Scholar] [CrossRef]
- Yu, H.; Bhat, J.A.; Li, C.; Zhao, B.; Guo, T.; Feng, X. Genome-wide survey identified superior and rare haplotypes for plant height in the north-eastern soybean germplasm of China. Mol. Breed. 2023, 43, 22. [Google Scholar] [CrossRef]
- Bhat, J.A.; Yu, D. High-throughput NGS-based genotyping and phenotyping: Role in genomics-assisted breeding for soybean improvement. Legume Sci. 2021, 3, e81. [Google Scholar] [CrossRef]
- Wang, B.; Smith, S.M.; Li, J. Genetic regulation of shoot architecture. Annu. Rev. Plant Biol. 2018, 69, 437–468. [Google Scholar] [CrossRef]
- Egli, D. The relationship between the number of nodes and pods in soybean communities. Crop Sci. 2013, 53, 1668–1676. [Google Scholar] [CrossRef]
- Liu, W.; Kim, M.Y.; Van, K.; Lee, Y.-H.; Li, H.; Liu, X.; Lee, S.-H. QTL identification of yield-related traits and their association with flowering and maturity in soybean. J. Crop Sci. Biotechnol. 2011, 14, 65–70. [Google Scholar] [CrossRef]
- Song, J.; Sun, X.; Zhang, K.; Liu, S.; Wang, J.; Yang, C.; Jiang, S.; Siyal, M.; Li, X.; Qi, Z. Identification of QTL and genes for pod number in soybean by linkage analysis and genome-wide association studies. Mol. Breed. 2020, 40, 60. [Google Scholar] [CrossRef]
- Yao, D.; Liu, Z.; Zhang, J.; Liu, S.; Qu, J.; Guan, S.; Pan, L.; Wang, D.; Liu, J.; Wang, P. Analysis of quantitative trait loci for main plant traits in soybean. Genet. Mol. Res. 2015, 14, 6101–6109. [Google Scholar] [CrossRef]
- Wang, P.; Sun, X.; Zhang, K.; Fang, Y.; Wang, J.; Yang, C.; Li, W.-X.; Ning, H. Mapping QTL/QTN and mining candidate genes for plant height and its response to planting densities in soybean [Glycine max (L.) Merr.] through a FW-RIL population. Mol. Breed. 2021, 41, 12. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.; Guo, C.; Sun, F.; Zhang, J.; Wang, Z.; Kong, J.; He, Q.; Sharmin, R.A.; Zhao, T. Genome-wide association studies for dynamic plant height and number of nodes on the main stem in summer sowing soybeans. Front. Plant Sci. 2018, 9, 1184. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Jiang, H.; Zhang, Z.; Zhao, Y.; Xie, J.; Wang, Q.; Zheng, H.; Hou, L.; Xiong, X.; Xin, D. Combined linkage mapping and BSA to identify QTL and candidate genes for plant height and the number of nodes on the main stem in soybean. Int. J. Mol. Sci. 2019, 21, 42. [Google Scholar] [CrossRef]
- Assefa, T.; Otyama, P.I.; Brown, A.V.; Kalberer, S.R.; Kulkarni, R.S.; Cannon, S.B. Genome-wide associations and epistatic interactions for internode number, plant height, seed weight and seed yield in soybean. BMC Genom. 2019, 20, 527. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Wang, Y.; Ren, H.; Du, W.; Yang, X.; Wang, D.; Cheng, Y.; Zhao, J.; Gai, J. Exploring the QTL–allele constitution of main stem node number and its differentiation among maturity groups in a Northeast China soybean population. Crop Sci. 2020, 60, 1223–1238. [Google Scholar] [CrossRef]
- Fahim, A.M.; Liu, F.; He, J.; Wang, W.; Xing, G.; Gai, J. Evolutionary QTL-allele changes in main stem node number among geographic and seasonal subpopulations of Chinese cultivated soybeans. Mol. Genet. Genom. 2021, 296, 313–330. [Google Scholar] [CrossRef]
- Gai, J.; Wang, Y.; Wu, X.; Chen, S. A comparative study on segregation analysis and QTL mapping of quantitative traits in plants—With a case in soybean. Front. Agric. China 2007, 1, 1–7. [Google Scholar] [CrossRef]
- Wang, L.; Cheng, Y.; Ma, Q.; Mu, Y.; Huang, Z.; Xia, Q.; Zhang, G.; Nian, H. QTL fine-mapping of soybean (Glycine max L.) leaf type associated traits in two RILs populations. BMC Genom. 2019, 20, 260. [Google Scholar] [CrossRef]
- Zhang, W.-K.; Wang, Y.-J.; Luo, G.-Z.; Zhang, J.-S.; He, C.-Y.; Wu, X.-L.; Gai, J.-Y.; Chen, S.-Y. QTL mapping of ten agronomic traits on the soybean (Glycine max L. Merr.) genetic map and their association with EST markers. Theor. Appl. Genet. 2004, 108, 1131–1139. [Google Scholar] [CrossRef]
- Chen, S.; Sun, Y.; Shao, Z.; Li, W.; Du, H.; Kong, Y.; Li, X.; Zhang, C. Stable pleiotropic loci and candidate genes for fresh pod-and seed-related characteristics across multiple environments in soybean (Glycine max). Plant Breed. 2021, 140, 630–642. [Google Scholar] [CrossRef]
- Li, C.; Zou, J.; Jiang, H.; Yu, J.; Huang, S.; Wang, X.; Liu, C.; Guo, T.; Zhu, R.; Wu, X. Identification and validation of number of pod-and seed-related traits QTL s in soybean. Plant Breed. 2018, 137, 730–745. [Google Scholar] [CrossRef]
- Jeong, N.; Suh, S.J.; Kim, M.-H.; Lee, S.; Moon, J.-K.; Kim, H.S.; Jeong, S.-C. Ln is a key regulator of leaflet shape and number of seeds per pod in soybean. Plant Cell 2012, 24, 4807–4818. [Google Scholar] [CrossRef] [PubMed]
- Sayama, T.; Tanabata, T.; Saruta, M.; Yamada, T.; Anai, T.; Kaga, A.; Ishimoto, M. Confirmation of the pleiotropic control of leaflet shape and number of seeds per pod by the Ln gene in induced soybean mutants. Breed. Sci. 2017, 67, 363–369. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Zhang, H.; Sun, G.; Zhang, W.; Qiu, L. Evolution and association analysis of GmCYP78A10 gene with seed size/weight and pod number in soybean. Mol. Biol. Rep. 2015, 42, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Singh, M.; Srivastava, R.; Bajaj, D.; Saxena, M.S.; Rana, J.C.; Bansal, K.C.; Tyagi, A.K.; Parida, S.K. mQTL-seq delineates functionally relevant candidate gene harbouring a major QTL regulating pod number in chickpea. DNA Res. 2016, 23, 53–65. [Google Scholar] [CrossRef]
- Nemli, S.; Asciogul, T.K.; Kaya, H.B.; Kahraman, A.; Eşiyok, D.; Tanyolac, B. Association mapping for five agronomic traits in the common bean (Phaseolus vulgaris L.). J. Sci. Food Agric. 2014, 94, 3141–3151. [Google Scholar] [CrossRef]
- Liu, Z.; Yao, D.; Zhang, J.; Li, Z.; Ma, J.; Liu, S.; Qu, J.; Guan, S.; Wang, D.; Pan, L. Identification of genes associated with the increased number of four-seed pods in soybean (Glycine max L.) using transcriptome analysis. Genet. Mol. Res. 2015, 14, 18895–18912. [Google Scholar] [CrossRef]
- Ren, H.; Zhao, K.; Zhang, C.; Lamlom, S.F.; Liu, X.; Wang, X.; Zhang, F.; Yuan, R.; Gao, Y.; Cao, B. Genetic analysis and QTL mapping of seed hardness trait in a soybean (Glycine max) recombinant inbred line (RIL) population. Gene 2024, 905, 148238. [Google Scholar] [CrossRef]
- Zhang, B.; Zhao, K.; Ren, H.; Lamlom, S.F.; Liu, X.; Wang, X.; Zhang, F.; Yuan, R.; Wang, J. Comparative Study of Isoflavone Synthesis Genes in Two Wild Soybean Varieties Using Transcriptomic Analysis. Agriculture 2023, 13, 1164. [Google Scholar] [CrossRef]
- Yang, Y.; Shi, J.; Wang, X.; Liu, G.; Wang, H. Genetic architecture and mechanism of seed number per pod in rapeseed: Elucidated through linkage and near-isogenic line analysis. Sci. Rep. 2016, 6, 24124. [Google Scholar] [CrossRef]
- Lamlom, S.F.; Zhang, Y.; Su, B.; Wu, H.; Zhang, X.; Fu, J.; Zhang, B.; Qiu, L.-J. Map-based cloning of a novel QTL qBN-1 influencing branch number in soybean [Glycine max (L.) Merr.]. Crop J. 2020, 8, 793–801. [Google Scholar] [CrossRef]
- Hyten, D.L.; Choi, I.Y.; Song, Q.; Specht, J.E.; Carter, T.E.; Shoemaker, R.C.; Hwang, E.Y.; Matukumalli, L.K.; Cregan, P.B. A high density integrated genetic linkage map of soybean and the development of a 1536 universal soy linkage panel for quantitative trait locus mapping. Crop Sci. 2010, 50, 960–968. [Google Scholar] [CrossRef]
- Jun, T.H.; Freewalt, K.; Michel, A.P.; Mian, R. Identification of novel QTL for leaf traits in soybean. Plant Breed. 2014, 133, 61–66. [Google Scholar] [CrossRef]
- Lee, S.; Freewalt, K.R.; McHale, L.K.; Song, Q.; Jun, T.-H.; Michel, A.P.; Dorrance, A.E.; Mian, M.R. A high-resolution genetic linkage map of soybean based on 357 recombinant inbred lines genotyped with BARCSoySNP6K. Mol. Breed. 2015, 35, 58. [Google Scholar] [CrossRef]
- Ren, H.; Qu, X.; Hong, H.; Sun, L.; Lamlom, S.F.; Liu, Z.; Lu, W. Multi-environment QTL mapping identifies major genetic loci influencing soybean main stem node architecture. PeerJ 2024, 12, e18539. [Google Scholar] [CrossRef]
- Ren, H.; Zhang, F.; Zhu, X.; Lamlom, S.F.; Liu, X.; Wang, X.; Zhao, K.; Wang, J.; Sun, M.; Yuan, M. Cultivation model and deficit irrigation strategy for reducing leakage of bundle sheath cells to CO2, improve 13C carbon isotope, photosynthesis and soybean yield in semi-arid areas. J. Plant Physiol. 2023, 285, 153979. [Google Scholar] [CrossRef]
- Ott, J.; Kamatani, Y.; Lathrop, M. Family-based designs for genome-wide association studies. Nat. Rev. Genet. 2011, 12, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Gong, J.; Xiao, X.; Zhang, Z.; Li, J.; Liu, A.; Lu, Q.; Shang, H.; Shi, Y.; Ge, Q. GWAS analysis and QTL identification of fiber quality traits and yield components in upland cotton using enriched high-density SNP markers. Front. Plant Sci. 2018, 9, 1067. [Google Scholar] [CrossRef]
- Fang, Y.; Liu, S.; Dong, Q.; Zhang, K.; Tian, Z.; Li, X.; Li, W.; Qi, Z.; Wang, Y.; Tian, X. Linkage analysis and multi-locus genome-wide association studies identify QTNs controlling soybean plant height. Front. Plant Sci. 2020, 11, 9. [Google Scholar] [CrossRef]
- Zhang, S.; Meng, L.; Wang, J.; Zhang, L. Background controlled QTL mapping in pure-line genetic populations derived from four-way crosses. Heredity 2017, 119, 256–264. [Google Scholar] [CrossRef]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Li, N.; Li, N.; Xu, Y.; Ma, S.; Wang, J. Construction of a high-density genetic map for watermelon (Citrullus lanatus L.) based on large-scale SNP discovery by specific length amplified fragment sequencing (SLAF-seq). Sci. Hortic. 2016, 203, 38–46. [Google Scholar] [CrossRef]
- Gong, D.; Huang, L.; Xu, X.; Wang, C.; Ren, M.; Wang, C.; Chen, M. Construction of a high-density SNP genetic map in flue-cured tobacco based on SLAF-seq. Mol. Breed. 2016, 36, 100. [Google Scholar] [CrossRef]
- Wang, L.; Li, X.; Wang, L.; Xue, H.; Wu, J.; Yin, H.; Zhang, S. Construction of a high-density genetic linkage map in pear (Pyrus communis × Pyrus pyrifolia nakai) using SSRs and SNPs developed by SLAF-seq. Sci. Hortic. 2017, 218, 198–204. [Google Scholar] [CrossRef]
- Berhanu, H.; Tesso, B.; Lule, D. Correlation and path coefficient analysis for seed yield and yield related traits in soybean (Glycine max (L.)) genotypes. Plant 2021, 9, 106–110. [Google Scholar] [CrossRef]
- Kumar, R.; Saini, M.; Taku, M.; Debbarma, P.; Mahto, R.K.; Ramlal, A.; Sharma, D.; Rajendran, A.; Pandey, R.; Gaikwad, K. Identification of quantitative trait loci (QTLs) and candidate genes for seed shape and 100-seed weight in soybean [Glycine max (L.) Merr.]. Front. Plant Sci. 2023, 13, 1074245. [Google Scholar] [CrossRef]
- Liu, C.; Chen, H.; Yu, Q.; Gu, H.; Li, Y.; Tu, B.; Zhang, H.; Zhang, Q.; Liu, X. Identification of quantitative trait loci and candidate genes for seed sucrose and soluble sugar concentrations in soybean. Crop Sci. 2023, 63, 2976–2992. [Google Scholar] [CrossRef]
- Guo, B.; Sleper, D.; Arelli, P.; Shannon, J.; Nguyen, H. Identification of QTLs associated with resistance to soybean cyst nematode races 2, 3 and 5 in soybean PI 90763. Theor. Appl. Genet. 2005, 111, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Masuda, R.; Tsubokura, Y.; Yasui, T.; Yamada, T.; Takahashi, K.; Nagaya, T.; Sayama, T.; Ishimoto, M.; Hajika, M. Identification of quantitative trait loci associated with boiled seed hardness in soybean. Breed. Sci. 2014, 64, 362–370. [Google Scholar] [CrossRef]
- Orazaly, M.; Chen, P.; Zhang, B.; Florez-Palacios, L.; Zeng, A. Confirmation of SSR markers and QTL for seed calcium content and hardness of soybean. J. Crop Improv. 2018, 32, 71–89. [Google Scholar] [CrossRef]
- Xu, X.; Xu, R.; Zhu, B.; Yu, T.; Qu, W.; Lu, L.; Xu, Q.; Qi, X.; Chen, X. A high-density genetic map of cucumber derived from Specific Length Amplified Fragment sequencing (SLAF-seq). Front. Plant Sci. 2015, 5, 768. [Google Scholar] [CrossRef]
- Bleckmann, A.; Alter, S.; Dresselhaus, T. The beginning of a seed: Regulatory mechanisms of double fertilization. Front. Plant Sci. 2014, 5, 452. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasulu, N.; Wobus, U. Seed-development programs: A systems biology–based comparison between dicots and monocots. Annu. Rev. Plant Biol. 2013, 64, 189–217. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Olalde, J.I.; Zuñiga-Mayo, V.M.; Montes, R.A.C.; Marsch-Martínez, N.; de Folter, S. Inside the gynoecium: At the carpel margin. Trends Plant Sci. 2013, 18, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Zhang, Q. Genetic and molecular bases of rice yield. Annu. Rev. Plant Biol. 2010, 61, 421–442. [Google Scholar] [CrossRef]
- Saghai-Maroof, M.A.; Soliman, K.M.; Jorgensen, R.A.; Allard, R. Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc. Natl. Acad. Sci. USA 1984, 81, 8014–8018. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Jia, J.; Huan, W.; Cai, Z.-d.; Wei, R.-q.; Huang, J.-h.; Xia, Q.-j.; Xiao, X.-h.; Hai, N.; Cheng, Y.-b. Identification and validation of stable and novel quantitative trait loci for pod shattering in soybean [Glycinemax (L.) Merr.]. J. Integr. Agric. 2022, 21, 3169–3184. [Google Scholar] [CrossRef]
- Xian, P.; Cai, Z.; Jiang, B.; Xia, Q.; Cheng, Y.; Yang, Y.; Zhou, Q.; Lian, T.; Ma, Q.; Wang, Y. GmRmd1 encodes a TIR-NBS-BSP protein and confers resistance to powdery mildew in soybean. Plant Commun. 2022, 3, 100418. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Environment | Traits | Min | Max | Mean | SD | Variance | Skewness | Kurtosis | CV |
|---|---|---|---|---|---|---|---|---|---|
| Harbin | PN | 22.8 | 104 | 52.37 | 13.24 | 175.39 | 0.55 | 0.45 | 0.25 |
| SN | 35.6 | 239.4 | 111.43 | 34.22 | 1171.23 | 0.4 | 0.19 | 0.31 | |
| MSNN | 8 | 28.8 | 18.57 | 3.61 | 13.07 | −0.2031 | −0.0508 | 0.23 | |
| Sanya | PN | 9.8 | 82 | 27.25 | 9.89 | 97.76 | 1.96 | 6.96 | 0.36 |
| SN | 22 | 192 | 57.16 | 21.35 | 455.82 | 2.21 | 8.85 | 0.37 | |
| MSNN | 5.2 | 15.8 | 8.98 | 1.94 | 3.77 | 0.524 | −0.1229 | 0.22 |
| Linkage Group | Marker (No) | Map Distance (cM) | Average Map Distance (cM) | Gaps < 5 cM (%) | Max Gap (cM) |
|---|---|---|---|---|---|
| Gm01 | 348 | 145.08 | 0.42 | 100.00 | 4.54 |
| Gm02 | 372 | 156.70 | 0.42 | 99.46 | 5.56 |
| Gm03 | 365 | 167.92 | 0.46 | 99.18 | 7.46 |
| Gm04 | 309 | 140.07 | 0.45 | 99.03 | 7.28 |
| Gm05 | 183 | 160.41 | 0.88 | 98.35 | 5.38 |
| Gm06 | 211 | 145.65 | 0.69 | 99.05 | 8.32 |
| Gm07 | 371 | 161.22 | 0.44 | 99.46 | 9.49 |
| Gm08 | 255 | 125.88 | 0.50 | 100.00 | 4.00 |
| Gm09 | 357 | 133.83 | 0.38 | 99.72 | 5.50 |
| Gm10 | 455 | 123.76 | 0.27 | 99.56 | 7.94 |
| Gm11 | 131 | 122.39 | 0.94 | 96.92 | 10.95 |
| Gm12 | 133 | 128.10 | 0.97 | 97.73 | 14.34 |
| Gm13 | 243 | 125.73 | 0.52 | 99.17 | 10.69 |
| Gm14 | 224 | 148.46 | 0.67 | 99.55 | 11.70 |
| Gm15 | 625 | 167.77 | 0.27 | 99.36 | 7.90 |
| Gm16 | 390 | 192.55 | 0.49 | 99.23 | 7.58 |
| Gm17 | 345 | 148.67 | 0.43 | 98.84 | 6.76 |
| Gm18 | 283 | 142.60 | 0.51 | 100.00 | 4.43 |
| Gm19 | 484 | 152.16 | 0.32 | 99.79 | 6.31 |
| Gm20 | 213 | 156.31 | 0.74 | 99.06 | 5.82 |
| Total | 6297 | 2945.26 | 0.47 | 99.17 | 14.34 |
| Year | Traits | Chr. | Position | Left Marker | Right Marker | LOD | PVE(%) | Add | Physical Length |
|---|---|---|---|---|---|---|---|---|---|
| Harbin | PN | 20 | 41 | Marker2448322 | Marker2501150 | 4.6804 | 5.8529 | −3.3846 | 83,365 bp |
| SN | 14 | 26 | Marker5556237 | Marker5717653 | 2.5198 | 3.4514 | −6.1588 | 36.05 kbp | |
| SN | 18 | 9 | Marker4230716 | Marker4407522 | 2.8469 | 3.8975 | −6.5107 | 381 kbp | |
| MSNN | 6 | 45 | Marker777950 | Marker766908 | 7.8568 | 7.7379 | −1.2729 | 5.54 Mbp | |
| MSNN | 18 | 6 | Marker4165022 | Marker4228378 | 5.493 | 3.804 | −0.7547 | 64.58 kbp | |
| MSNN | 19 | 16 | Marker1939109 | Marker1978745 | 22.5907 | 24.5668 | 2.2155 | 101.90 kbp | |
| Sanya | PN | 19 | 15 | Marker1990755 | Marker1770324 | 18.1568 | 23.0188 | 4.6921 | 274,223 bp |
| SN | 19 | 15 | Marker1990755 | Marker1770324 | 15.2414 | 19.5888 | 9.4462 | 274,223 bp | |
| MSNN | 17 | 40 | Marker5907974 | Marker5935243 | 3.8931 | 3.4569 | −0.3523 | 2.84 Mbp | |
| MSNN | 19 | 17 | Marker1978745 | Marker1711920 | 37.9218 | 43.588 | 1.2453 | 1.12 Mbp |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Zha, B.; Yuan, R.; Zhao, K.; Sun, J.; Liu, X.; Wang, X.; Zhang, F.; Zhang, B.; Lamlom, S.F.; et al. Identification of Quantitative Trait Loci for Node Number, Pod Number, and Seed Number in Soybean. Int. J. Mol. Sci. 2025, 26, 2300. https://doi.org/10.3390/ijms26052300
Zhang C, Zha B, Yuan R, Zhao K, Sun J, Liu X, Wang X, Zhang F, Zhang B, Lamlom SF, et al. Identification of Quantitative Trait Loci for Node Number, Pod Number, and Seed Number in Soybean. International Journal of Molecular Sciences. 2025; 26(5):2300. https://doi.org/10.3390/ijms26052300
Chicago/Turabian StyleZhang, Chunlei, Bire Zha, Rongqiang Yuan, Kezhen Zhao, Jianqiang Sun, Xiulin Liu, Xueyang Wang, Fengyi Zhang, Bixian Zhang, Sobhi F. Lamlom, and et al. 2025. "Identification of Quantitative Trait Loci for Node Number, Pod Number, and Seed Number in Soybean" International Journal of Molecular Sciences 26, no. 5: 2300. https://doi.org/10.3390/ijms26052300
APA StyleZhang, C., Zha, B., Yuan, R., Zhao, K., Sun, J., Liu, X., Wang, X., Zhang, F., Zhang, B., Lamlom, S. F., Ren, H., & Qiu, L. (2025). Identification of Quantitative Trait Loci for Node Number, Pod Number, and Seed Number in Soybean. International Journal of Molecular Sciences, 26(5), 2300. https://doi.org/10.3390/ijms26052300