
Identification and Functional Analysis of Key microRNAs in the Early Extrauterine Environmental Adaptation of Piglets

 , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Data Summary
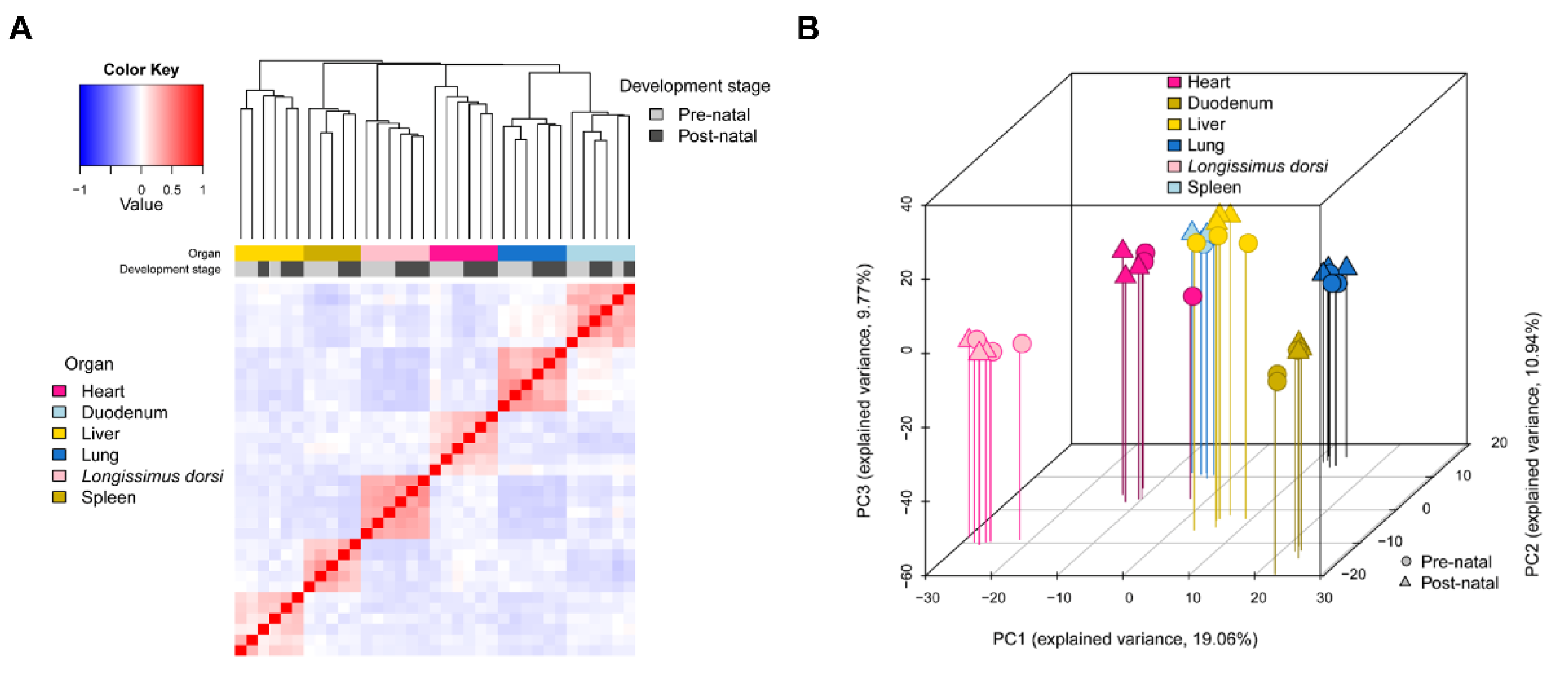
2.2. miRNA Expression Profiling
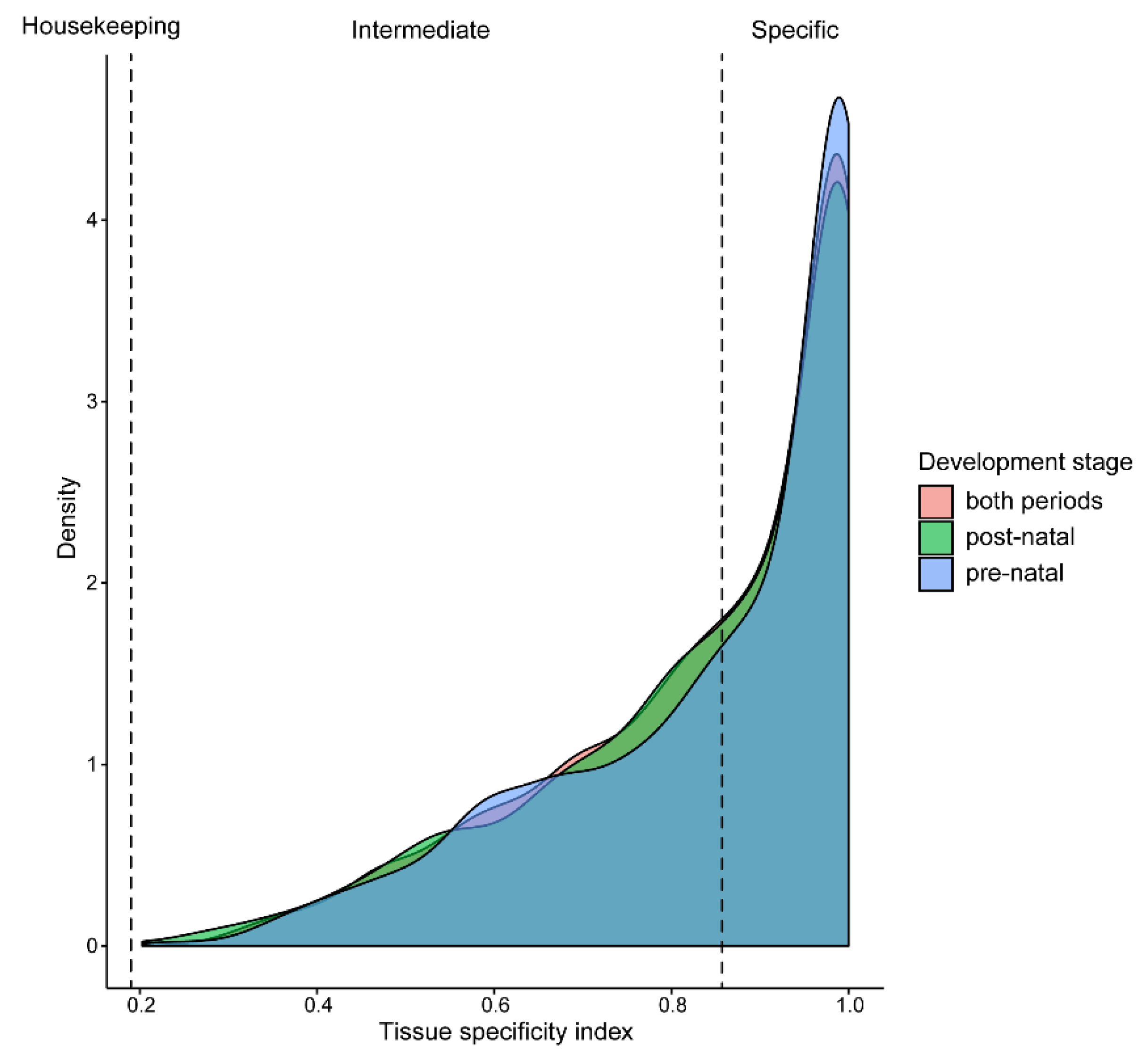
2.3. Tissue-Specific Analysis
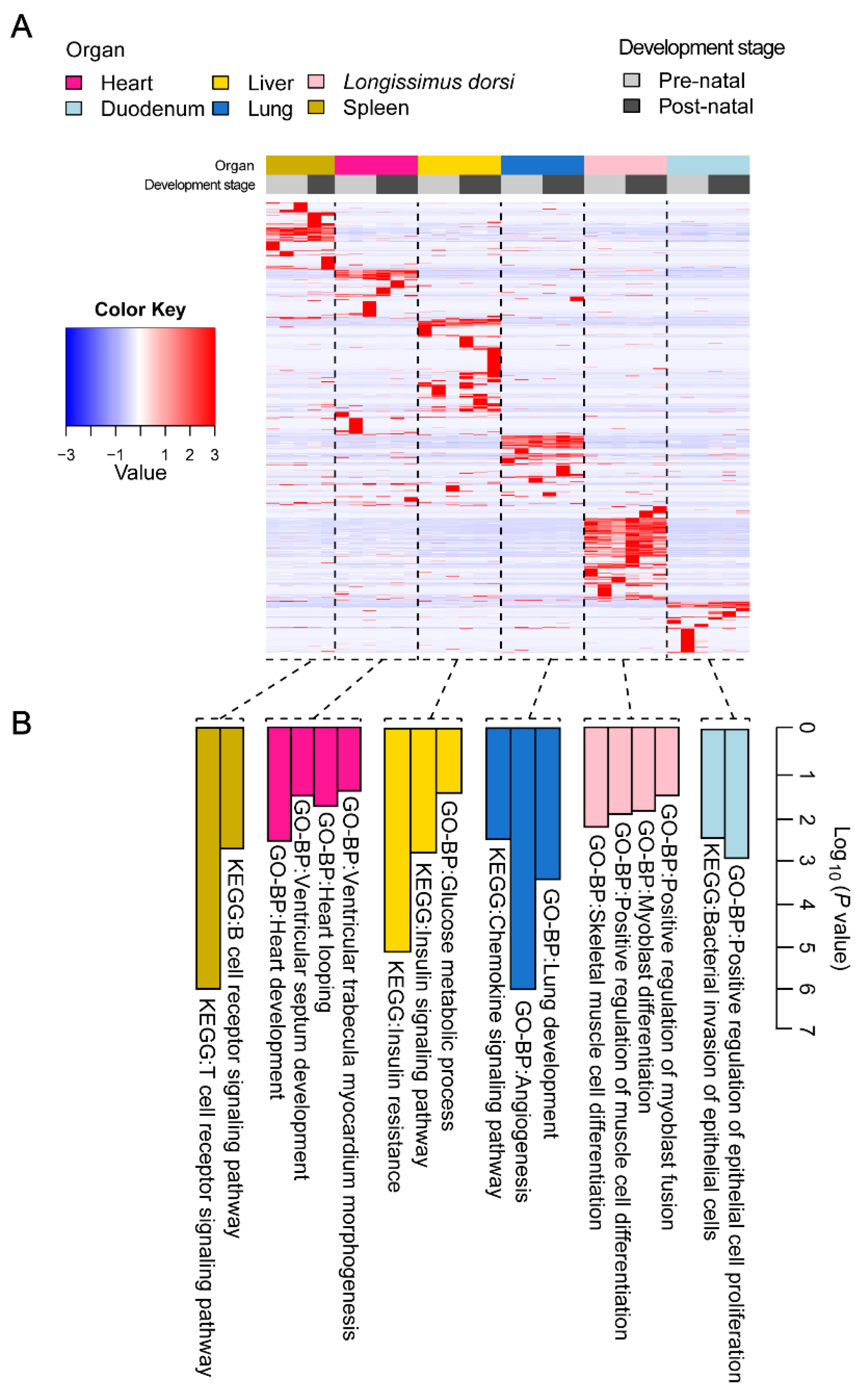
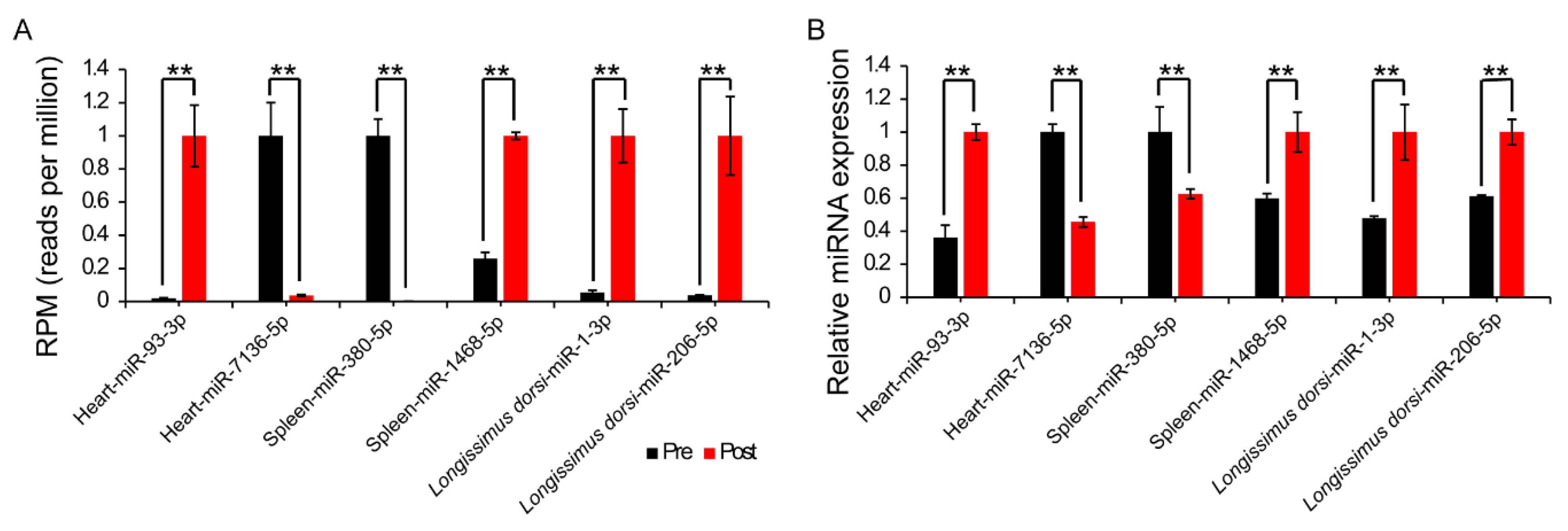
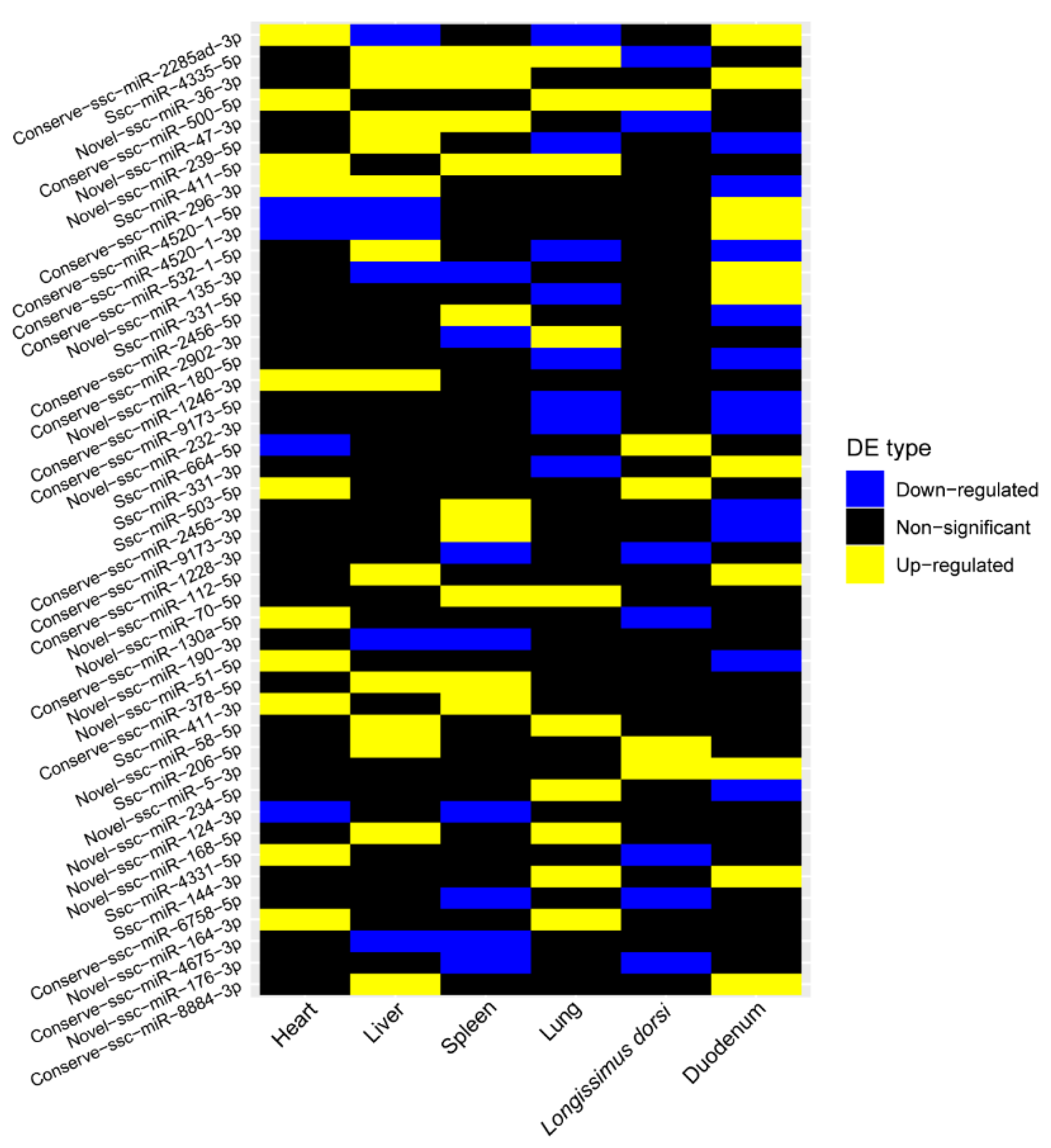
2.4. Differential Expression Analysis of miRNAs
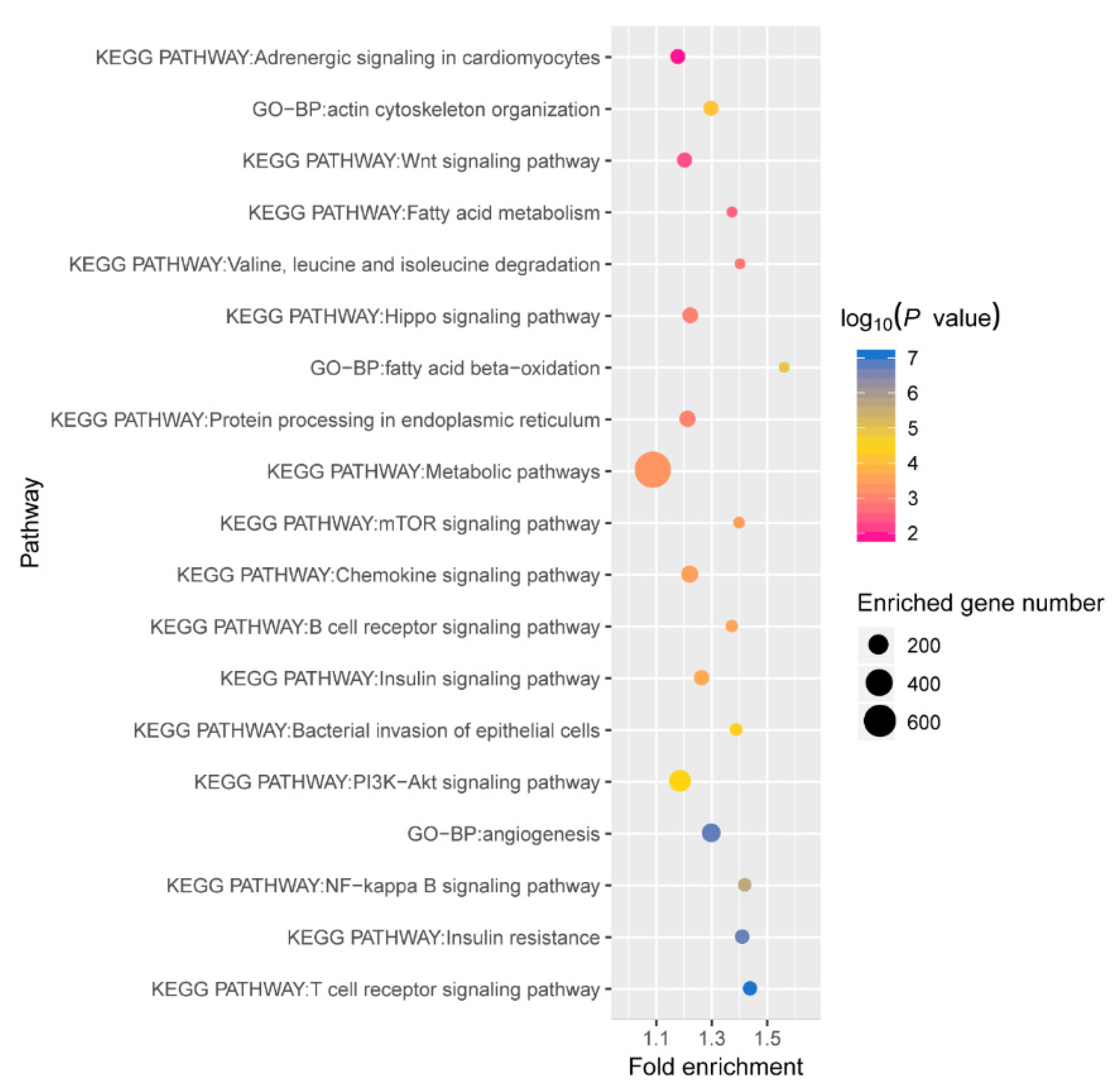
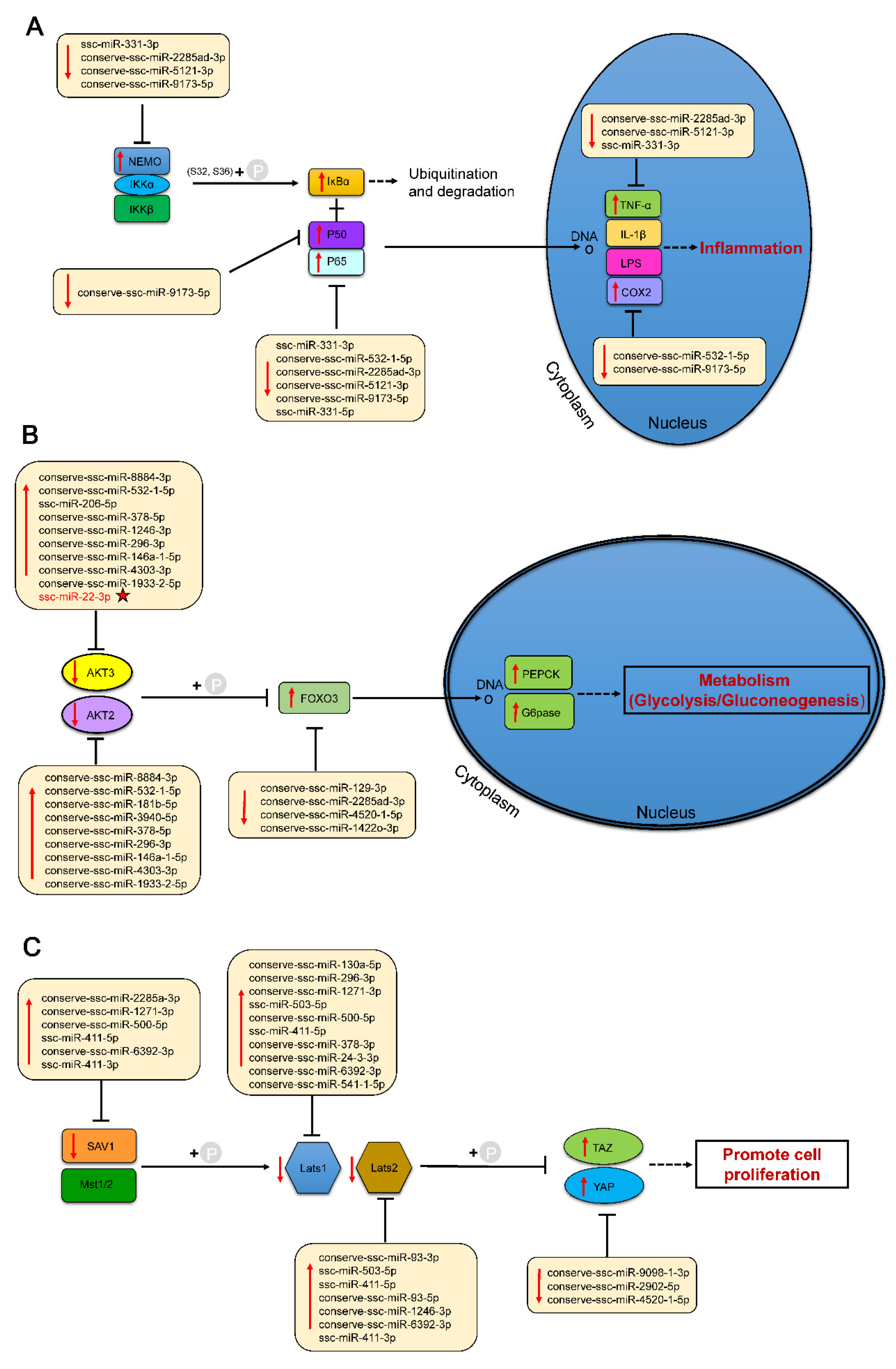
2.5. Regulatory Network of Differentially Expressed miRNAs in Six Tissues
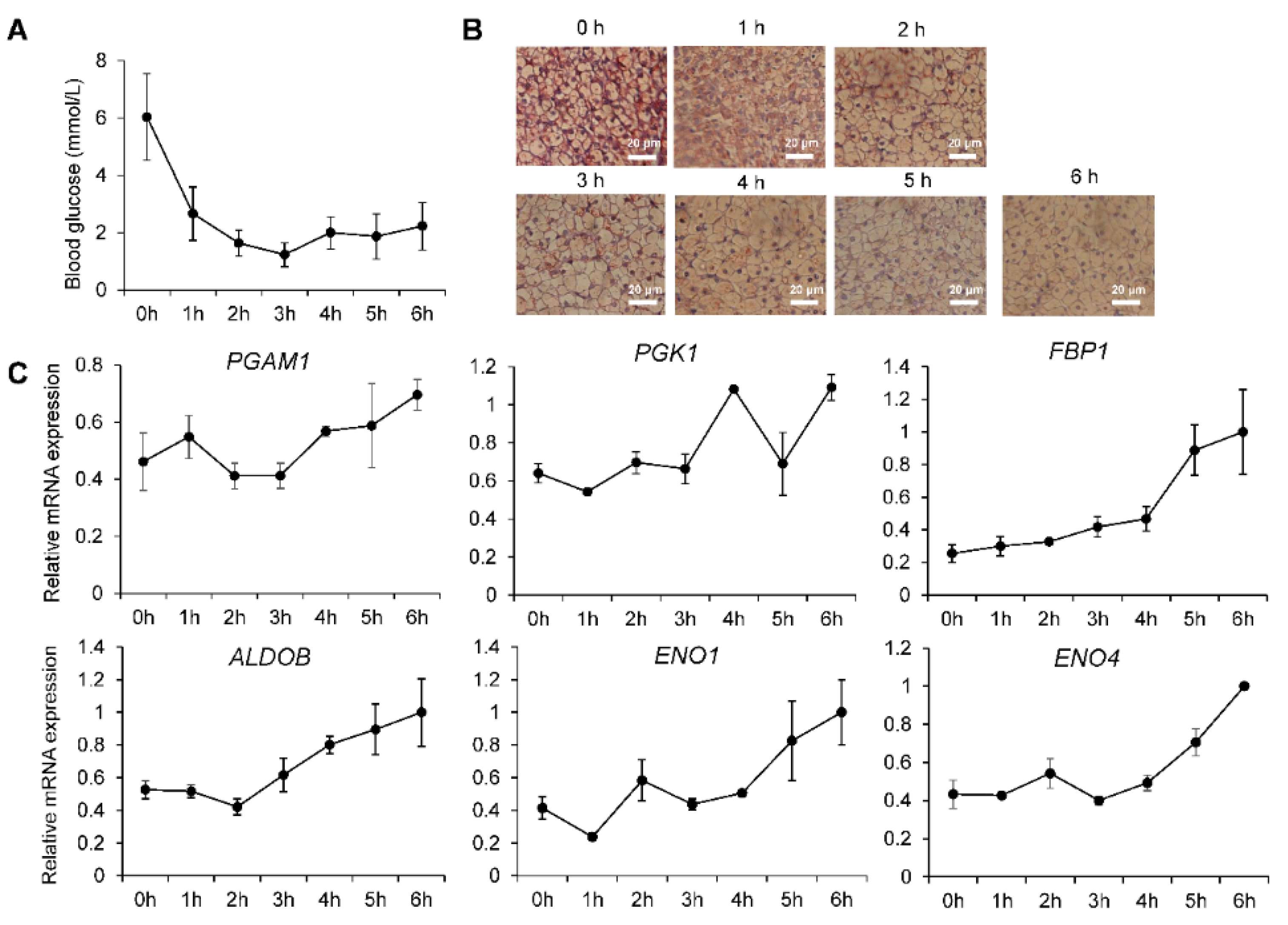
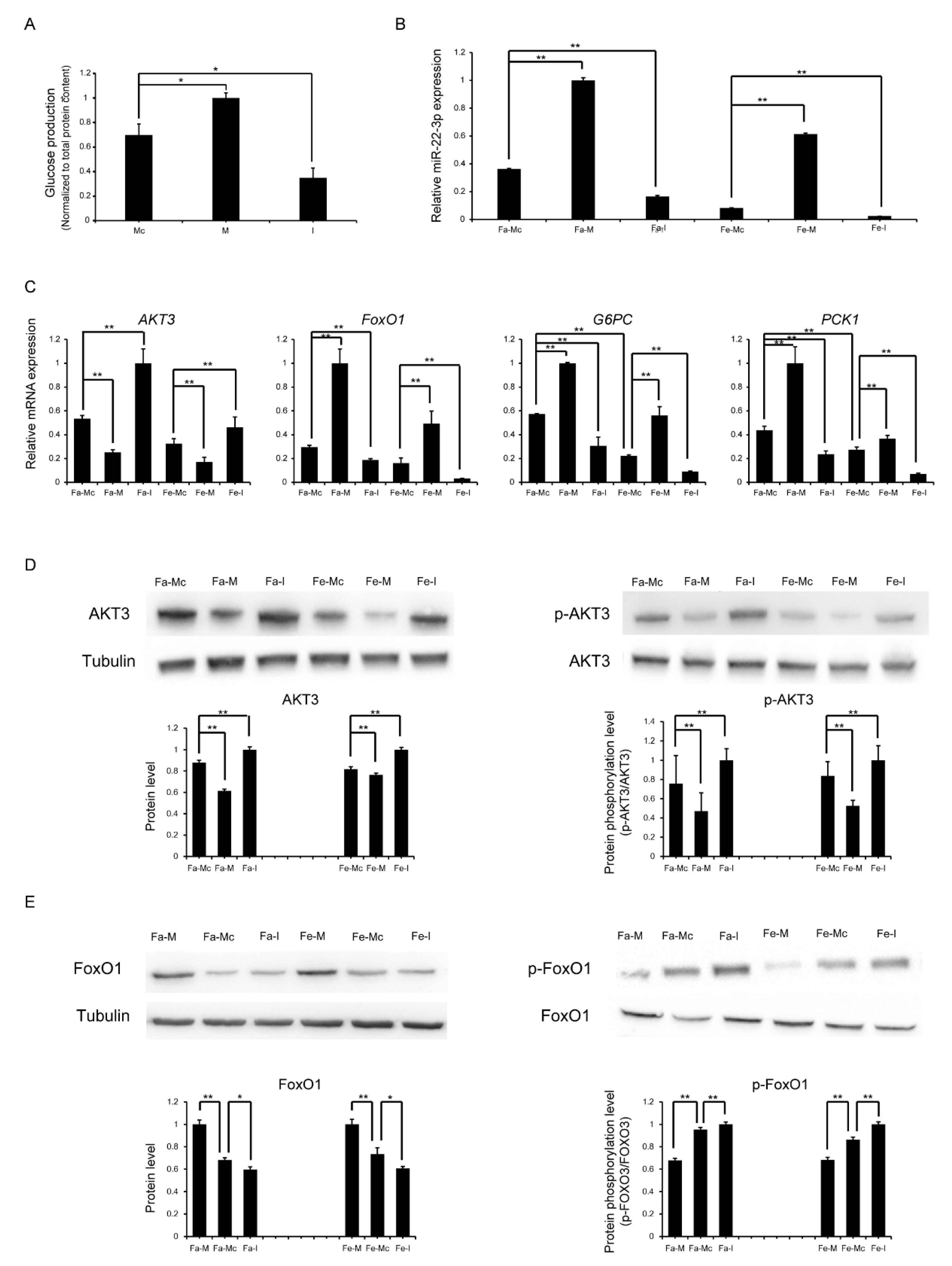
2.6. Re-Establishment of Glucose Homeostasis in Neonatal Piglets
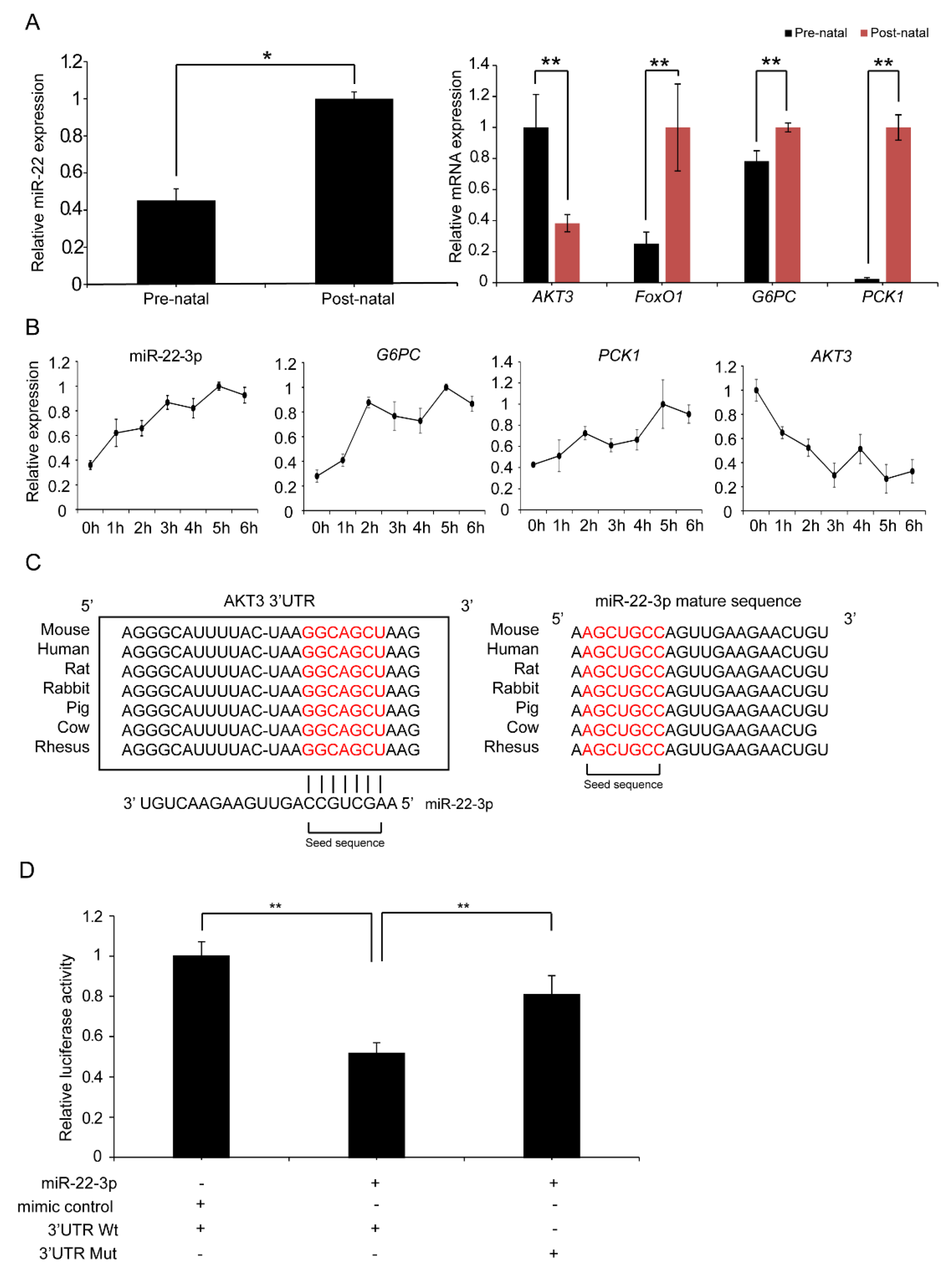
2.7. Validation of the Targeting Relationship Between AKT3 and miR-22-3p in the Liver
2.8. miR-22-3p Promotes Gluconeogenesis by Repressing AKT3 to Regulate the PI3K/AKT Signalling Pathway
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Experimental Animal Samples
5.2. Total RNA Extraction from Tissues and Cells
5.3. Total RNA Quality Control
- (1)
- Purity assessment of total RNA: A NanoDrop ND-2000 spectrophotometer was used to measure the absorbance of the total RNA sample at 260 nm and 280 nm and calculate the A260/A280 ratio. A ratio close to 2.0 (with a generally acceptable range between 1.9 and 2.1) indicated a qualified sample.
- (2)
- Integrity assessment of total RNA: The total RNA sample was assessed by 1.2% denaturing agarose gel electrophoresis, with an intensity ratio of the 28S band to the 18S band of approximately 2 and the absence of smears and trailing indicating a qualified sample.
5.4. Small RNA Library Construction and Sequencing
5.5. miRNA Data Analysis
5.6. Tissue-Specific Analysis
5.7. Differential miRNA Identification
5.8. miRNA Target Gene Prediction and Functional Enrichment
5.9. Dual-Luciferase Assay to Verify Target Relationships
5.10. HepG2 Cell Culture and Transfection
5.11. Reverse Transcription and Quantitative Real-Time PCR (qRT-PCR)
5.12. Western Blot
5.13. Glucose Production Assay
5.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van Woudenberg, C.D.; Wills, C.A.; Rubarth, L.B. Newborn transition to extrauterine life. Neonatal Netw. 2012, 31, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Askin, D.F. Fetal-to-neonatal transition—What is normal and what is not? Neonatal Netw. 2009, 28, e33–e40. [Google Scholar] [CrossRef]
- Van Hoover, C. Maternal, Fetal, & Neonatal Physiology: A Clinical Perspective, 2nd edition. J. Midwifery Women’s Health 2004, 49, 164–165. [Google Scholar] [CrossRef]
- Steiniger, B.; Ulfig, N.; Riße, M.; Barth, P.J. Fetal and early post-natal development of the human spleen: From primordial arterial B cell lobules to a non-segmented organ. Histochem. Cell Biol. 2007, 128, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Tchirikov, M.; Rybakowski, C.; Hüneke, B.; Schröder, H.J. Blood flow through the ductus venosus in singleton and multifetal pregnancies and in fetuses with intrauterine growth retardation. Am. J. Obs. Gynecol. 1998, 178, 943–949. [Google Scholar] [CrossRef]
- Girard, J.; Ferré, P.; Pégorier, J.P.; Duée, P.H. Adaptations of glucose and fatty acid metabolism during perinatal period and suckling-weaning transition. Physiol. Rev. 1992, 72, 507–562. [Google Scholar] [CrossRef]
- Johnson, P.J. Neonatal pharmacology-pharmacokinetics. Neonatal Netw. 2011, 30, 54–61. [Google Scholar] [CrossRef]
- Clements, J.A. LUNG SURFACTANT: A Personal Perspective. Annu. Rev. Physiol. 1997, 59, 1–21. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Standart, N.; Jackson, R.J. MicroRNAs repress translation of m7Gppp-capped target mRNAs in vitro by inhibiting initiation and promoting deadenylation. Genes Dev. 2007, 21, 1975–1982. [Google Scholar] [CrossRef]
- Du, T.; Zamore, P.D. Beginning to understand microRNA function. Cell Res. 2007, 17, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.S.; Bhattacharyya, S.N.; Filipowicz, W. Repression of protein synthesis by miRNAs: How many mechanisms? Trends Cell Biol. 2007, 17, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; He, J.H.; Xiao, Z.D.; Zhang, Q.Q.; Chen, Y.Q.; Zhou, H.; Qu, L.H. Liver-enriched transcription factors regulate microRNA-122 that targets CUTL1 during liver development. Hepatology 2010, 52, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Kahn, D.; Gibson, W.S.; Round, J.L.; Scholz, R.L.; Chaudhuri, A.A.; Kahn, M.E.; Rao, D.S.; Baltimore, D. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 2010, 33, 607–619. [Google Scholar] [CrossRef]
- Kim, B.-M.; Woo, J.; Kanellopoulou, C.; Shivdasani, R.A. Regulation of mouse stomach development and Barx1 expression by specific microRNAs. Development 2011, 138, 1081–1086. [Google Scholar] [CrossRef]
- Horak, M.; Novak, J.; Bienertova-Vasku, J. Muscle-specific microRNAs in skeletal muscle development. Dev. Biol. 2016, 410, 1–13. [Google Scholar] [CrossRef]
- White, M.F. Insulin signaling in health and disease. Science 2003, 302, 1710–1711. [Google Scholar] [CrossRef]
- Senese, R.; Cioffi, F.; Petito, G.; de Lange, P.; Russo, A.; Goglia, F.; Lanni, A.; Potenza, N. miR-22-3p is involved in gluconeogenic pathway modulated by 3,5-diiodo-L-thyronine (T2). Sci. Rep. 2019, 9, 16645. [Google Scholar] [CrossRef]
- Mendonca, A.; Thandapani, P.; Nagarajan, P.S.; Venkatesh, S.; Sundaresan, S. Role of microRNAs in regulation of insulin secretion and insulin signaling involved in type 2 diabetes mellitus. J. Biosci. 2022, 47, 58. [Google Scholar] [CrossRef]
- Arner, P.; Kulyté, A. MicroRNA regulatory networks in human adipose tissue and obesity. Nat. Rev. Endocrinol. 2015, 11, 276–288. [Google Scholar] [CrossRef]
- Sessa, R.; Hata, A. Role of microRNAs in lung development and pulmonary diseases. Pulm. Circ. 2013, 3, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Plasencia, M.; Castellano, J.J.; Díaz, T.; He, Y.; Marrades, R.M.; Navarro, A. Discovering genes and microRNAs involved in human lung development unveils IGFBP3/miR-34a dynamics and their relevance for alveolar differentiation. Stem Cell Res. Ther. 2024, 15, 263. [Google Scholar] [CrossRef] [PubMed]
- Harafuji, N.; Schneiderat, P.; Walter, M.C.; Chen, Y.W. miR-411 is up-regulated in FSHD myoblasts and suppresses myogenic factors. Orphanet J. Rare Dis. 2013, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ding, Y.; Yuan, Z.; Liu, J.; Sun, J.; Lei, F.; Wu, S.; Li, S.; Zhang, D. MicroRNA-500 sustains nuclear factor-κB activation and induces gastric cancer cell proliferation and resistance to apoptosis. Oncotarget 2015, 6, 2483–2495. [Google Scholar] [CrossRef] [PubMed]
- Forrest, A.R.; Kanamori-Katayama, M.; Tomaru, Y.; Lassmann, T.; Ninomiya, N.; Takahashi, Y.; de Hoon, M.J.; Kubosaki, A.; Kaiho, A.; Suzuki, M.; et al. Induction of microRNAs, mir-155, mir-222, mir-424 and mir-503, promotes monocytic differentiation through combinatorial regulation. Leukemia 2010, 24, 460–466. [Google Scholar] [CrossRef]
- Yan, B.; Zhao, J.L. miR-1228 prevents cellular apoptosis through targeting of MOAP1 protein. Apoptosis 2012, 17, 717–724. [Google Scholar] [CrossRef]
- Labialle, S.; Marty, V.; Bortolin-Cavaillé, M.L.; Hoareau-Osman, M.; Pradère, J.P.; Valet, P.; Martin, P.G.; Cavaillé, J. The miR-379/miR-410 cluster at the imprinted Dlk1-Dio3 domain controls neonatal metabolic adaptation. EMBO J. 2014, 33, 2216–2230. [Google Scholar] [CrossRef]
- Anderson, C.; Catoe, H.; Werner, R. MIR-206 regulates connexin43 expression during skeletal muscle development. Nucleic Acids Res. 2006, 34, 5863–5871. [Google Scholar] [CrossRef]
- Hassan, F.; Nuovo, G.J.; Crawford, M.; Boyaka, P.N.; Kirkby, S.; Nana-Sinkam, S.P.; Cormet-Boyaka, E. MiR-101 and miR-144 regulate the expression of the CFTR chloride channel in the lung. PLoS ONE 2012, 7, e50837. [Google Scholar] [CrossRef]
- Liao, J.M.; Zhou, X.; Zhang, Y.; Lu, H. MiR-1246: A new link of the p53 family with cancer and Down syndrome. Cell Cycle 2012, 11, 2624–2630. [Google Scholar] [CrossRef]
- Zhang, Y.; Liao, J.M.; Zeng, S.X.; Lu, H. p53 downregulates Down syndrome-associated DYRK1A through miR-1246. EMBO Rep. 2011, 12, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Hawdon, J.M.; Ward Platt, M.P.; Aynsley-Green, A. Patterns of metabolic adaptation for preterm and term infants in the first neonatal week. Arch. Dis. Child. 1992, 67, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Hosford, G.E.; Olson, D.M. Effects of hyperoxia on VEGF, its receptors, and HIF-2alpha in the newborn rat lung. Am. J. Physiology. Lung Cell. Mol. Physiol. 2003, 285, L161–L168. [Google Scholar] [CrossRef] [PubMed]
- Thébaud, B.; Abman, S.H. Bronchopulmonary dysplasia: Where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am. J. Respir. Crit. Care Med. 2007, 175, 978–985. [Google Scholar] [CrossRef]
- Alvira, C.M. Nuclear factor-kappa-B signaling in lung development and disease: One pathway, numerous functions. Birth Defects Res. Part A Clin. Mol. Teratol. 2014, 100, 202–216. [Google Scholar] [CrossRef]
- Koay, M.A.; Gao, X.; Washington, M.K.; Parman, K.S.; Sadikot, R.T.; Blackwell, T.S.; Christman, J.W. Macrophages are necessary for maximal nuclear factor-kappa B activation in response to endotoxin. Am. J. Respir. Cell Mol. Biol. 2002, 26, 572–578. [Google Scholar] [CrossRef]
- Ali, S.; Hirschfeld, A.F.; Mayer, M.L.; Fortuno, E.S., 3rd; Corbett, N.; Kaplan, M.; Wang, S.; Schneiderman, J.; Fjell, C.D.; Yan, J.; et al. Functional genetic variation in NFKBIA and susceptibility to childhood asthma, bronchiolitis, and bronchopulmonary dysplasia. J. Immunol. 2013, 190, 3949–3958. [Google Scholar] [CrossRef]
- Rajendrasozhan, S.; Chung, S.; Sundar, I.K.; Yao, H.; Rahman, I. Targeted disruption of NF-{kappa}B1 (p50) augments cigarette smoke-induced lung inflammation and emphysema in mice: A critical role of p50 in chromatin remodeling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L197–L209. [Google Scholar] [CrossRef]
- Haeusler, R.A.; Hartil, K.; Vaitheesvaran, B.; Arrieta-Cruz, I.; Knight, C.M.; Cook, J.R.; Kammoun, H.L.; Febbraio, M.A.; Gutierrez-Juarez, R.; Kurland, I.J.; et al. Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. Nat. Commun. 2014, 5, 5190. [Google Scholar] [CrossRef]
- O-Sullivan, I.; Zhang, W.; Wasserman, D.H.; Liew, C.W.; Liu, J.; Paik, J.; DePinho, R.A.; Stolz, D.B.; Kahn, C.R.; Schwartz, M.W.; et al. FoxO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nat. Commun. 2015, 6, 7079. [Google Scholar] [CrossRef]
- Xiong, X.; Tao, R.; DePinho, R.A.; Dong, X.C. Deletion of hepatic FoxO1/3/4 genes in mice significantly impacts on glucose metabolism through downregulation of gluconeogenesis and upregulation of glycolysis. PLoS ONE 2013, 8, e74340. [Google Scholar] [CrossRef] [PubMed]
- Pajvani, U.B.; Accili, D. The new biology of diabetes. Diabetologia 2015, 58, 2459–2468. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Tumaneng, K.; Guan, K.L. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 2011, 13, 877–883. [Google Scholar] [CrossRef]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef]
- Jopling, C.; Sleep, E.; Raya, M.; Martí, M.; Raya, A.; Izpisúa Belmonte, J.C. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 2010, 464, 606–609. [Google Scholar] [CrossRef]
- Kikuchi, K.; Holdway, J.E.; Werdich, A.A.; Anderson, R.M.; Fang, Y.; Egnaczyk, G.F.; Evans, T.; Macrae, C.A.; Stainier, D.Y.; Poss, K.D. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature 2010, 464, 601–605. [Google Scholar] [CrossRef]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 2011, 332, 458–461. [Google Scholar] [CrossRef]
- Sudol, M.; Harvey, K.F. Modularity in the Hippo signaling pathway. Trends Biochem. Sci. 2010, 35, 627–633. [Google Scholar] [CrossRef]
- Oka, T.; Mazack, V.; Sudol, M. Mst2 and Lats kinases regulate apoptotic function of Yes kinase-associated protein (YAP). J. Biol. Chem. 2008, 283, 27534–27546. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef] [PubMed]
- Minami, K.; Uehara, T.; Morikawa, Y.; Omura, K.; Kanki, M.; Horinouchi, A.; Ono, A.; Yamada, H.; Ohno, Y.; Urushidani, T. miRNA expression atlas in male rat. Sci. Data 2014, 1, 140005. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Catalucci, D.; Bauersachs, J. MicroRNAs: Novel regulators in cardiac development and disease. Cardiovasc. Res. 2008, 79, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Callis, T.E.; Chen, J.F.; Wang, D.Z. MicroRNAs in skeletal and cardiac muscle development. DNA Cell Biol. 2007, 26, 219–225. [Google Scholar] [CrossRef]
- Nordlie, R.C.; Foster, J.D.; Lange, A.J. Regulation of glucose production by the liver. Annu. Rev. Nutr. 1999, 19, 379–406. [Google Scholar] [CrossRef]
- Pilkis, S.J.; el-Maghrabi, M.R.; Claus, T.H. Hormonal regulation of hepatic gluconeogenesis and glycolysis. Annu. Rev. Biochem. 1988, 57, 755–783. [Google Scholar] [CrossRef]
- Oh, K.J.; Han, H.S.; Kim, M.J.; Koo, S.H. CREB and FoxO1: Two transcription factors for the regulation of hepatic gluconeogenesis. BMB Rep. 2013, 46, 567–574. [Google Scholar] [CrossRef]
- Ramírez, C.M.; Goedeke, L.; Rotllan, N.; Yoon, J.H.; Cirera-Salinas, D.; Mattison, J.A.; Suárez, Y.; de Cabo, R.; Gorospe, M.; Fernández-Hernando, C. MicroRNA 33 regulates glucose metabolism. Mol. Cell. Biol. 2013, 33, 2891–2902. [Google Scholar] [CrossRef]
- Fang, Z.; Li, P.; Jia, W.; Jiang, T.; Wang, Z.; Xiang, Y. miR-696 plays a role in hepatic gluconeogenesis in ob/ob mice by targeting PGC-1α. Int. J. Mol. Med. 2016, 38, 845–852. [Google Scholar] [CrossRef]
- Wei, S.; Zhang, M.; Yu, Y.; Xue, H.; Lan, X.; Liu, S.; Hatch, G.; Chen, L. HNF-4α regulated miR-122 contributes to development of gluconeogenesis and lipid metabolism disorders in Type 2 diabetic mice and in palmitate-treated HepG2 cells. Eur. J. Pharmacol. 2016, 791, 254–263. [Google Scholar] [CrossRef]
- Zhuo, S.; Yang, M.; Zhao, Y.; Chen, X.; Zhang, F.; Li, N.; Yao, P.; Zhu, T.; Mei, H.; Wang, S.; et al. MicroRNA-451 Negatively Regulates Hepatic Glucose Production and Glucose Homeostasis by Targeting Glycerol Kinase-Mediated Gluconeogenesis. Diabetes 2016, 65, 3276–3288. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.F.; Li, Y.; Wang, L. Vaccinium bracteatum Thunb. Leaves’ polysaccharide alleviates hepatic gluconeogenesis via the downregulation of miR-137. Biomed. Pharmacother. 2017, 95, 1397–1403. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Wang, J.; Chen, Z.; Chen, J.; Meng, Y.; Chen, L.; Chang, Y.; Geng, B.; Sun, L.; Dou, L.; et al. NFE2 Induces miR-423-5p to Promote Gluconeogenesis and Hyperglycemia by Repressing the Hepatic FAM3A-ATP-Akt Pathway. Diabetes 2017, 66, 1819–1832. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Pre-miRNAs | miRNA-5p | miRNA-3p | Both | Mature miRNAs |
|---|---|---|---|---|---|
| Known | 239 | 21 | 13 | 205 | 444 |
| Conserved | 482 | 129 | 138 | 215 | 697 |
| Novel | 250 | 53 | 77 | 120 | 370 |
| Total | 971 | 203 | 228 | 540 | 1511 |
| Tissues | Heart | Lung | Spleen | Muscle | Liver | Duodenum | Total |
|---|---|---|---|---|---|---|---|
| Number of DE miRNAs | 34 | 38 | 33 | 21 | 37 | 60 | 223 |
| Number of Tissues | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Number of overlapping DE miRNAs | 119 | 33 | 10 | 2 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, M.; Li, J.; Qiu, W.; Zhang, J.; Long, K.; Lu, L.; Jin, L.; Sun, J.; Ge, L.; Li, X.; et al. Identification and Functional Analysis of Key microRNAs in the Early Extrauterine Environmental Adaptation of Piglets. Int. J. Mol. Sci. 2025, 26, 1316. https://doi.org/10.3390/ijms26031316
Wen M, Li J, Qiu W, Zhang J, Long K, Lu L, Jin L, Sun J, Ge L, Li X, et al. Identification and Functional Analysis of Key microRNAs in the Early Extrauterine Environmental Adaptation of Piglets. International Journal of Molecular Sciences. 2025; 26(3):1316. https://doi.org/10.3390/ijms26031316
Chicago/Turabian StyleWen, Mingxing, Jing Li, Wanling Qiu, Jinwei Zhang, Keren Long, Lu Lu, Long Jin, Jing Sun, Liangpeng Ge, Xuewei Li, and et al. 2025. "Identification and Functional Analysis of Key microRNAs in the Early Extrauterine Environmental Adaptation of Piglets" International Journal of Molecular Sciences 26, no. 3: 1316. https://doi.org/10.3390/ijms26031316
APA StyleWen, M., Li, J., Qiu, W., Zhang, J., Long, K., Lu, L., Jin, L., Sun, J., Ge, L., Li, X., Li, M., & Ma, J. (2025). Identification and Functional Analysis of Key microRNAs in the Early Extrauterine Environmental Adaptation of Piglets. International Journal of Molecular Sciences, 26(3), 1316. https://doi.org/10.3390/ijms26031316