
A Spectrum of Pathogenic Variants in the LAMA2 Gene in the Russian Federation

 , , , , ,
, , , , ,  , , , , ,
, , , , ,  ,
, 
Abstract
1. Introduction
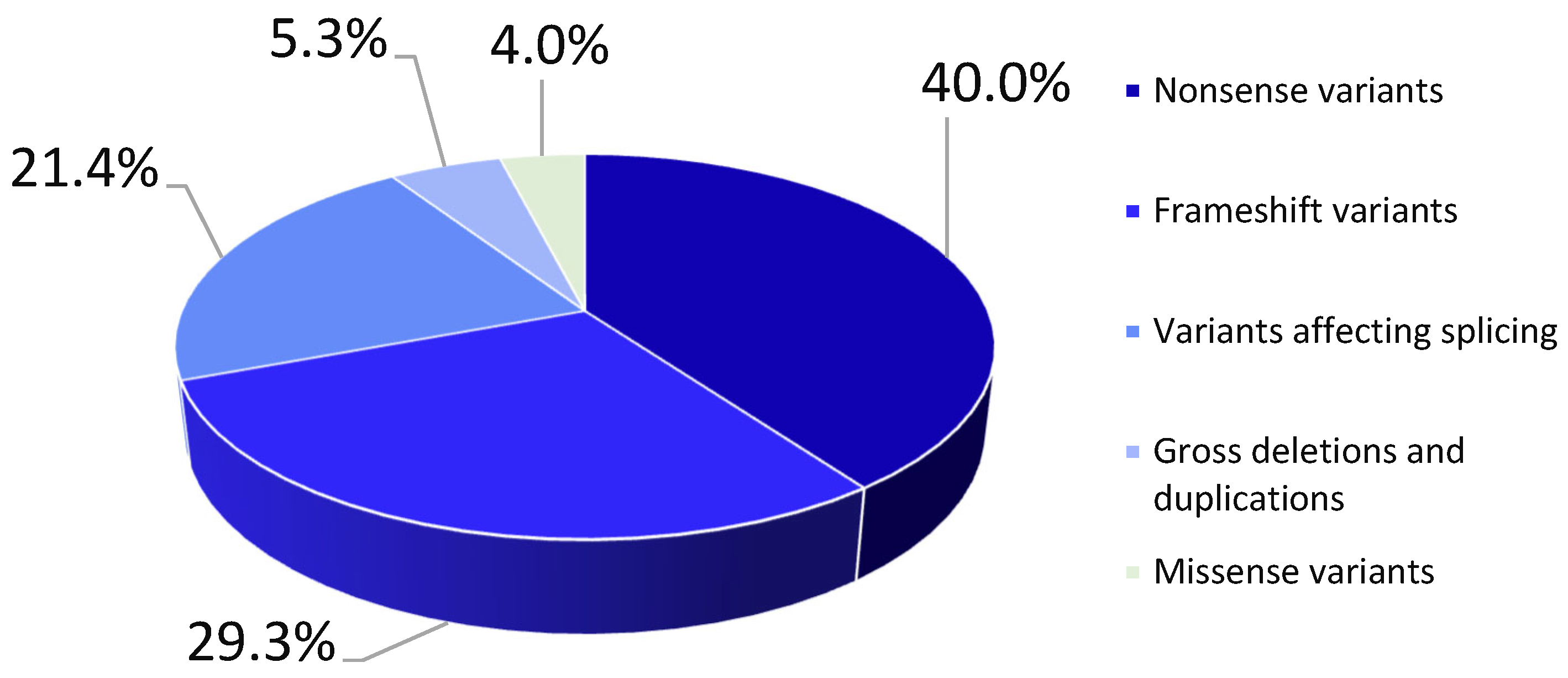
2. Results
2.1. Clinical Data
2.2. Genetic Analysis
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yurchenco, P.D.; McKee, K.K. Linker Protein Repair of LAMA2 Dystrophic Neuromuscular Basement Membranes. Front. Mol. Neurosci. 2019, 12, 305. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, A.; Foley, A.R.; Zambon, A.A.; Bönnemann, C.G.; Muntoni, F. LAMA2-Related Dystrophies: Clinical Phenotypes, Disease Biomarkers, and Clinical Trial Readiness. Front. Mol. Neurosci. 2020, 13, 123. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.; Gruber, A.; Cardoso, M.; Taipa, R.; Fineza, I.; Gonçalves, A.; Laner, A.; Winder, T.L.; Schroeder, J.; Rath, J.; et al. LAMA2 gene mutation update: Toward a more comprehensive picture of the laminin-α2 variome and its related phenotypes. Hum. Mutat. 2018, 39, 1314–1337. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Voit, T. The congenital muscular dystrophies in 2004: A century of exciting progress. Neuromuscul. Disord. 2004, 14, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Zambon, A.A.; Ridout, D.; Main, M.; Mein, R.; Phadke, R.; Muntoni, F.; Sarkozy, A. LAMA2-related muscular dystrophy: Natural history of a large pediatric cohort. Ann. Clin. Transl. Neurol. 2020, 7, 1870–1882. [Google Scholar] [CrossRef]
- Prandini, P.; Berardinelli, A.; Fanin, M.; Morello, F.; Zardini, E.; Pichiecchio, A.; Uggetti, C.; Lanzi, G.; Angelini, C.; Pegoraro, E. LAMA2 loss-of-function mutation in a girl with a mild congenital muscular dystrophy. Neurology 2004, 63, 1118–1121. [Google Scholar] [CrossRef]
- Chausova, P.A.; Ryzhkova, O.P.; Polyakov, A.V. Clinical and genetic characteristics of congenital muscular dystrophies (Part 1). Neuromuscul. Dis. 2020, 10, 10–21. [Google Scholar] [CrossRef]
- Bönnemann, C.G.; Wang, C.H.; Quijano-Roy, S.; Deconinck, N.; Bertini, E.; Ferreiro, A.; Muntoni, F.; Sewry, C.; Béroud, C.; Mathews, K.D.; et al. Members of International Standard of Care Committee for Congenital Muscular Dystrophies Diagnostic approach to the congenital muscular dystrophies. Neuromuscul. Disord. 2014, 24, 289–311. [Google Scholar] [CrossRef]
- Geranmayeh, F.; Clement, E.; Feng, L.H.; Sewry, C.; Pagan, J.; Mein, R.; Abbs, S.; Brueton, L.; Childs, A.-M.; Jungbluth, H.; et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul. Disord. 2010, 20, 241–250. [Google Scholar] [CrossRef]
- Orbach, R.; Park, J.; Hinkley, L.; Acquaye, N.; Alvarez, R.; Dziewczapolski, G.; Bönnemann, C.; Foley, A. FP.39 An international retrospective early natural history study of LAMA2-related dystrophies. Neuromuscul. Disord. 2022, 32, S120. [Google Scholar] [CrossRef]
- Naom, I.; D’Alessandro, M.; Sewry, C.A.; Jardine, P.; Ferlini, A.; Moss, T.; Dubowitz, V.; Muntoni, F. Mutations in the laminin alpha2-chain gene in two children with early-onset muscular dystrophy. Brain J. Neurol. 2000, 123 Pt 1, 31–41. [Google Scholar] [CrossRef]
- Tezak, Z.; Prandini, P.; Boscaro, M.; Marin, A.; Devaney, J.; Marino, M.; Fanin, M.; Trevisan, C.P.; Park, J.; Tyson, W.; et al. Clinical and molecular study in congenital muscular dystrophy with partial laminin α2 (LAMA2) deficiency. Hum. Mutat. 2003, 21, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Khodaenia, N.; Farjami, Z.; Ashnaei, A.H.; Ebrahimi, N.; Chelvarforoosh, N.; Urtizberea, A.; Razmara, E.; Houshmand, M. Novel Homozygous Pathogenic Mutations of LAMA 2 Gene in Patients with Congenital Muscular Dystrophy. Iran. J. Child Neurol. 2021, 15, 101–106. [Google Scholar] [PubMed]
- Punetha, J.; Kesari, A.; Uapinyoying, P.; Giri, M.; Clarke, N.F.; Waddell, L.B.; North, K.N.; Ghaoui, R.; O’Grady, G.L.; Oates, E.C.; et al. Targeted Re-Sequencing Emulsion PCR Panel for Myopathies: Results in 94 Cases. J. Neuromuscul. Dis. 2016, 3, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Grunseich, C.; Sarkar, N.; Lu, J.; Owen, M.; Schindler, A.; Calabresi, P.A.; Sumner, C.J.; Roda, R.H.; Chaudhry, V.; Lloyd, T.E.; et al. Improving the efficacy of exome sequencing at a quaternary care referral centre: Novel mutations, clinical presentations and diagnostic challenges in rare neurogenetic diseases. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1186–1196. [Google Scholar] [CrossRef]
- Abdel Aleem, A.; Elsaid, M.F.; Chalhoub, N.; Chakroun, A.; Mohamed, K.A.S.; AlShami, R.; Kuzu, O.; Mohamed, R.B.; Ibrahim, K.; AlMudheki, N.; et al. Clinical and genomic characteristics of LAMA2 related congenital muscular dystrophy in a patients’ cohort from Qatar. A population specific founder variant. Neuromuscul. Disord. 2020, 30, 457–471. [Google Scholar] [CrossRef]
- Liang, W.-C.; Tian, X.; Yuo, C.-Y.; Chen, W.-Z.; Kan, T.-M.; Su, Y.-N.; Nishino, I.; Wong, L.-J.C.; Jong, Y.-J. Comprehensive target capture/next-generation sequencing as a second-tier diagnostic approach for congenital muscular dystrophy in Taiwan. PLoS ONE 2017, 12, e0170517. [Google Scholar]
- Quijano-Roy, S.; Haberlova, J.; Castiglioni, C.; Vissing, J.; Munell, F.; Rivier, F.; Stojkovic, T.; Malfatti, E.; Gómez García de la Banda, M.; Tasca, G.; et al. Diagnostic interest of whole-body MRI in early-and late-onset LAMA2 muscular dystrophies: A large international cohort. J. Neurol. 2022, 269, 2414–2429. [Google Scholar] [CrossRef]
- de los Angeles Beytía, M.; Dekomien, G.; Hoffjan, S.; Haug, V.; Anastasopoulos, C.; Kirschner, J. High creatine kinase levels and white matter changes: Clinical and genetic spectrum of congenital muscular dystrophies with laminin alpha-2 deficiency. Mol. Cell. Probes 2014, 28, 118–122. [Google Scholar] [CrossRef]
- Tan, D.; Ge, L.; Fan, Y.; Chang, X.; Wang, S.; Wei, C.; Ding, J.; Liu, A.; Wang, S.; Li, X.; et al. Natural history and genetic study of LAMA2-related muscular dystrophy in a large Chinese cohort. Orphanet J. Rare Dis. 2021, 16, 319. [Google Scholar] [CrossRef]
- Xiong, H.; Tan, D.; Wang, S.; Song, S.; Yang, H.; Gao, K.; Liu, A.; Jiao, H.; Mao, B.; Ding, J.; et al. Genotype/phenotype analysis in Chinese laminin-α2 deficient congenital muscular dystrophy patients. Clin. Genet. 2015, 87, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Liu, A.; Gao, K.; Du, R.; Ding, J.; Mao, B.; Hua, Y.; Zhang, X.; Tan, D.; Yang, H.; et al. Deletion of exon 4 in LAMA2 is the most frequent mutation in Chinese patients with laminin α2-related muscular dystrophy. Sci. Rep. 2018, 8, 14989. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.; Santos, R.; Soares-Silva, I.; Jorge, P.; Vieira, E.; Oliveira, M.E.; Moreira, A.; Coelho, T.; Ferreira, J.C.; Fonseca, M.J.; et al. LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clin. Genet. 2008, 74, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Milovidova, T.B.; Bulach, M.V.; Schagina, O.A.; Polyakov, A.V. Molecular genetic analysis of congenital merozin-negative muscular dystrophy in Russia. Med. Genet. 2018, 17, 38–45. [Google Scholar]
- Lake, N.J.; Phua, J.; Liu, W.; Moors, T.; Axon, S.; Lek, M. Estimating the Prevalence of LAMA2 Congenital Muscular Dystrophy using Population Genetic Databases. J. Neuromuscul. Dis. 2023, 10, 381–387. [Google Scholar] [CrossRef]
- Graziano, A.; Bianco, F.; D’Amico, A.; Moroni, I.; Messina, S.; Bruno, C.; Pegoraro, E.; Mora, M.; Astrea, G.; Magri, F.; et al. Prevalence of congenital muscular dystrophy in Italy. Neurology 2015, 84, 904–911. [Google Scholar] [CrossRef]
- Darin, N.; Tulinius, M. Neuromuscular disorders in childhood: A descriptive epidemiological study from western Sweden. Neuromuscul. Disord. 2000, 10, 1–9. [Google Scholar] [CrossRef]
- Allamand, V.; Guicheney, P. Merosin-deficient congenital muscular dystrophy, autosomal recessive (MDC1A, MIM#156225, LAMA2 gene coding for alpha2 chain of laminin). Eur. J. Hum. Genet. 2002, 10, 91–94. [Google Scholar]
- Sframeli, M.; Sarkozy, A.; Bertoli, M.; Astrea, G.; Hudson, J.; Scoto, M.; Mein, R.; Yau, M.; Phadke, R.; Feng, L.; et al. Congenital muscular dystrophies in the UK population: Clinical and molecular spectrum of a large cohort diagnosed over a 12-year period. Neuromuscul. Disord. 2017, 27, 793–803. [Google Scholar] [CrossRef]
- O’Grady, G.L.; Lek, M.; Lamande, S.R.; Waddell, L.; Oates, E.C.; Punetha, J.; Ghaoui, R.; Sandaradura, S.A.; Best, H.; Kaur, S.; et al. Diagnosis and etiology of congenital muscular dystrophy: We are halfway there. Ann. Neurol. 2016, 80, 101–111. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| № | Exon/Intron | cDNA Position | Protein Change (NP_000417.3) | Number of Alleles/Prevalence | HGMD® Professional (2024.3) ID/ACMG Criteria |
|---|---|---|---|---|---|
| 1 | 54 | c.7536del | p.(Asp2513IlefsTer34) | 27/15.00% | CD2117468 |
| 2 | 32 | c.4692_4695dup | p.(Arg1566CysfsTer13) | 16/8.90% | CI054467 |
| 3 | Intron 58 | c.8245-2A>G | p.? | 12/6.70% | CS1311884 |
| 4 | 36 | c.5116C>T | p.(Arg1706Ter) | 10/5.50% | CM981165 |
| 5 | 14 | c.2049_2050del | p.(Arg683SerfsTer21) | 9/5.00% | CD982727 |
| 6 | 55 | c.7732C>T | p.(Arg2578Ter) | 5/2.80% | CM032280 |
| 7 | 26 | c.3829C>T | p.(Arg1277Ter) | 4/2.20% | CM1816718 |
| 8 | 50 | c.7147C>T | p.(Arg2383Ter) | 4/2.20% | CM004723 |
| 9 | 65 | c.9235_9238dup | p.(Thr3080AsnfsTer26) | 4/2.20% | CI102066 |
| 10 | 49 | c.6955C>T | p.(Arg2319Ter) | 3/1.66% | CM981166 |
| 11 | Intron 49 | c.6992+1G>T | p.? | 3/1.66% | PM2, PVS1, PP4 |
| 12 | Intron 49 | c.6993-2A>C | p.? | 3/1.66% | CS085942 |
| 13 | 22 | c.3085C>T | p.(Arg1029Ter) | 3/1.66% | CM020725 |
| 14 | 46 | c.6466C>T | p.(Arg2156Ter) | 3/1.66% | CM142796 |
| 15 | 54 | c.7520del | p.(Asn2507IlefsTer40) | 3/1.66% | PM2, PVS1, PM3, PP4 |
| 16 | 2 | c.163A>C | p.(Asn55His) | 2/1.10% | PM2, PP3, PM3, PP4 |
| 17 | Intron 2 | c.283+1G>A | p.? | 2/1.10% | CS102090 |
| 18 | 9 | c.1303C>T | p.(Arg435Ter) | 2/1.10% | CM102052 |
| 19 | 11 | c.1522C>T | p.(Gln508Ter) | 2/1.10% | CM2136520 |
| 20 | 14 | c.1893_1897del | p.(Asp631GlufsTer8) | 2/1.10% | CD021018 |
| 21 | 15 | c.2184_2185del | p.(Gly729ValfsTer7) | 2/1.10% | PM2, PVS1 |
| 22 | 14 | del ex 14 | p.? | 2/1.10% | PM2, PVS1 |
| 23 | 32 | c.4706G>A | p.(Trp1569Ter) | 2/1.10% | PM2, PVS1 |
| 24 | Intron 35 | c.5071+1G>A | p.? | 2/1.10% | CS151383 |
| 25 | 36 | c.5212G>T | p.(Glu1738Ter) | 2/1.10% | CM2136516 |
| 26 | Intron 52 | c.7439+1G>T | p.? | 2/1.10% | PM2, PVS1 |
| 27 | 1 | c.3dup | p.(Pro2AlafsTer48) | 1/0.56% | PM2, PVS1 |
| 28 | 1 | c.79C>T | p.(Gln27Ter) | 1/0.56% | PM2, PVS1, PP4 |
| 29 | 1 | c.106C>T | p.(Gln36Ter) | 1/0.56% | PM2, PVS1 |
| 30 | 1 | del ex 1 | p.? | 1/0.56% | CG1815550 CG1815547 |
| 31 | 2 | c.172T>C | p.(Cys58Arg) | 1/0.56% | CM1724080 PM2, PP3. PM3, PP4 |
| 32 | 7 | c.958C>T | p.(Gln320Ter) | 1/0.56% | PM2, PVS1, PP4 |
| 33 | 12 | c.1755del | p.(Ser585ArgfsTer12) | 1/0.56% | CD2124491 |
| 34 | 14 | c.1894_1895del | p.(Leu632GlufsTer8) | 1/0.56% | PM2, PVS1 |
| 35 | Intron 14 | c.2097-1G>A | p.? | 1/0.56% | PM2, PVS1 |
| 36 | 15 | c.2166A>T | p.(Glu722Asp) | 1/0.56% | CM2214171 PM2, PP3, PM3, PP4 |
| 37 | 16 | c.2230C>T | p.(Arg744Ter) | 1/0.56% | CM130400 CS016102 |
| 38 | Intron 20 | c.2856+2T>A | p.? | 1/0.56% | PM2, PVS1, PM3, PP4 |
| 39 | 21 | c.2962C>T, | p.(Gln988Ter) | 1/0.56% | CM981163 |
| 40 | 23 | c.3283C>T | p.(Arg1095Ter) | 1/0.56% | CM151370 |
| 41 | 25 | c.3569del | p.(Ala1190ValfsTer9) | 1/0.56% | PM2, PVS1, PM3, PP4 |
| 42 | 25 | c.3644del | p.(Pro1215GlnfsTer9) | 1/0.56% | PM2, PVS1 |
| 43 | Intron 25 | c.3736-2A>T | p.? | 1/0.56% | CS2113419 |
| 44 | 26 | c.3799_3821del | p.(Phe1267AspfsTer11) | 1/0.56% | CG077478 |
| 45 | 27 | c.4048C>T | p.(Arg1350Ter) | 1/0.56% | CM102055 |
| 46 | 27 | c.4056dup | p.(Arg1353GlnfsTer4) | 1/0.56% | PM2, PVS1, PM3, PP4 |
| 47 | 29 | C.4198C>T | p.(Arg1400Ter) | 1/0.56% | CM1311877 |
| 48 | 29 | c.4255_4258dup | p.(Cys1420SerfsTer5) | 1/0.56% | CI243376 |
| 49 | 32 | c.4645C>T | p.(Arg1549Ter) | 1/0.56% | CM001209 |
| 50 | 32 | c.4665dup | p. (Lys1556GlufsTer3) | 1/0.56% | PM2, PVS1 |
| 51 | 36 | c.5188del | p.(Arg1730GlyfsTer4) | 1/0.56% | PM2, PVS1, PM3 |
| 52 | Intron 36 | c.5234+1G>A | p.? | 1/0.56% | CS085941 |
| 53 | Intron 36 | c.5235-2A>G | p.? | 1/0.56% | PM2, PVS1, PM3, PP4 |
| 54 | 37 | c.5422C>T | p.(Gln1808Ter) | 1/0.56% | PM2, PVS1, PM3, PP4 |
| 55 | Intron 37 | c.5445+1G>A | p.? | 1/0.56% | CS206309 |
| 56 | 38 | c.5476C>T | p.(Arg1826Ter) | 1/0.56% | CM983961 |
| 57 | Intron 38 | c.5562+5G>C | p.? | 1/0.56% | CS003701 |
| 58 | Intron 38 | c.5727-2A>G | p.? | 1/0.56% | PM2, PVS1 |
| 59 | 39 | c.5706del | p.(Ser1903HisfsTer61) | 1/0.56% | PM2, PVS1 |
| 60 | 40 | del ex 40 | p.? | 1/0.56% | PM2, PVS1, PP4 |
| 61 | 46 | c.6474C>A | p.(Tyr2158Ter) | 1/0.56% | PM2, PVS1, PP4 |
| 62 | 46 | c.6560delinsTGCCA | p.(Gly2187ValfsTer8) | 1/0.56% | PM2, PVS1, PP4 |
| 63 | 48 | c.6721G>T | p.(Gly2241Ter) | 1/0.56% | PM2, PVS1, PP4 |
| 64 | Intron 49 | c.6993-1G>C | p.? | 1/0.56% | PM2, PVS1, PP4 |
| 65 | 50 | c.7074C>A | p.(Tyr2358Ter) | 1/0.56% | CM981167 |
| 66 | 51 | c.7265G>A | p.(Trp2422Ter) | 1/0.56% | PM2, PVS1 |
| 67 | 52 | c.7377dup | p.(Leu2460SerfsTer2) | 1/0.56% | CI020898 |
| 68 | 55 | c.7701delTinsGTGTCCCTAGGTGTCCCTA | p.(Ser2567delinsArgCysProTer) | 1/0.56% | PM2, PVS1, PM3, PP4 |
| 69 | 56 | c.7814del | p.(Thr2605LysfsTer2) | 1/0.56% | PM2, PVS1, PP4 |
| 70 | 56 | c.7888C>T | p.(Arg2630Ter) | 1/0.56% | CM1618984 |
| 71 | 57,58 | del ex 57,58 | p.? | 1/0.56% | PM2, PVS1, PP4 |
| 72 | 58 | c.8244+3_8244+6del | p.? | 1/0.56% | CD151394 |
| 73 | 61 | c.8699_8700insGTAAATTCT | p.(Pro2901Ter) | 1/0.56% | PM2, PVS1, PP4 |
| 74 | 64 | c.9139G>T | p.(Glu3047Ter) | 1/0.56% | CM2320779 |
| 75 | 65 | c.9253C>T | p.(Arg3085Ter) | 1/0.56% | CM020949 |
| Total | 180/100% |
| № | cDNA Position | Protein Change (NP_000417.3) | Number of Alleles/Prevalence | Allele Frequency GnomAD (V2.1.1) | Allele Freguency GDB (V59.1) |
|---|---|---|---|---|---|
| 1 | c.7536del | p.(Asp2513IlefsTer34) | 27/15.00% | 0.000019 | 0.00043888 |
| 2 | c.4692_4695dup | p.(Arg1566CysfsTer13) | 16/8.90% | 0.000004 | 0.000086948 |
| 3 | c.8245-2A>G | Splice | 12/6.70% | n/d | 0.000074527 |
| 4 | c.5116C>T | p.(Arg1706Ter) | 10/5.50% | 0.000024 | 0.000140773 |
| 5 | c.2049_2050del | p.(Arg683SerfsTer21) | 9/5.00% | 0.000116 | 0.000062106 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chausova, P.; Cherevatova, T.; Dadali, E.; Murtazina, A.; Bulakh, M.; Kurbatov, S.; Anisimova, I.; Kanivets, I.; Udalova, V.; Rudenskaya, G.; et al. A Spectrum of Pathogenic Variants in the LAMA2 Gene in the Russian Federation. Int. J. Mol. Sci. 2025, 26, 1257. https://doi.org/10.3390/ijms26031257
Chausova P, Cherevatova T, Dadali E, Murtazina A, Bulakh M, Kurbatov S, Anisimova I, Kanivets I, Udalova V, Rudenskaya G, et al. A Spectrum of Pathogenic Variants in the LAMA2 Gene in the Russian Federation. International Journal of Molecular Sciences. 2025; 26(3):1257. https://doi.org/10.3390/ijms26031257
Chicago/Turabian StyleChausova, Polina, Tatiana Cherevatova, Elena Dadali, Aysylu Murtazina, Maria Bulakh, Sergei Kurbatov, Inga Anisimova, Ilya Kanivets, Vasilisa Udalova, Galina Rudenskaya, and et al. 2025. "A Spectrum of Pathogenic Variants in the LAMA2 Gene in the Russian Federation" International Journal of Molecular Sciences 26, no. 3: 1257. https://doi.org/10.3390/ijms26031257
APA StyleChausova, P., Cherevatova, T., Dadali, E., Murtazina, A., Bulakh, M., Kurbatov, S., Anisimova, I., Kanivets, I., Udalova, V., Rudenskaya, G., Demina, N., Sharkova, I., Monakhova, A., Tsygankova, P., Markova, T., Ryzhkova, O., Shatohina, O., Galkina, V., Borovikov, A., ... Polyakov, A. (2025). A Spectrum of Pathogenic Variants in the LAMA2 Gene in the Russian Federation. International Journal of Molecular Sciences, 26(3), 1257. https://doi.org/10.3390/ijms26031257