Influence of Membrane Composition on the Passive Membrane Penetration of Industrially Relevant NSO-Heterocycles


Abstract
1. Introduction
2. Results and Discussion
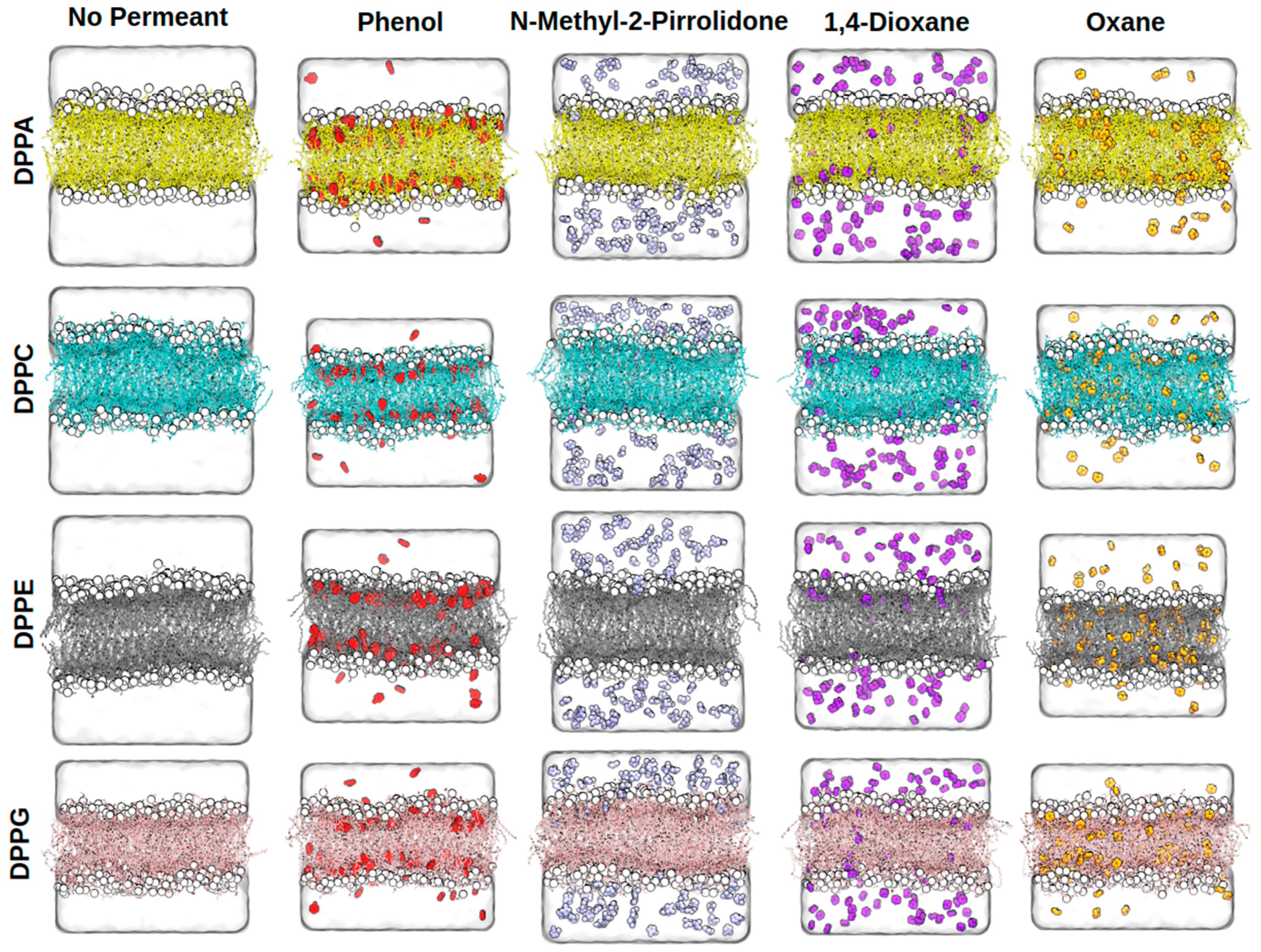
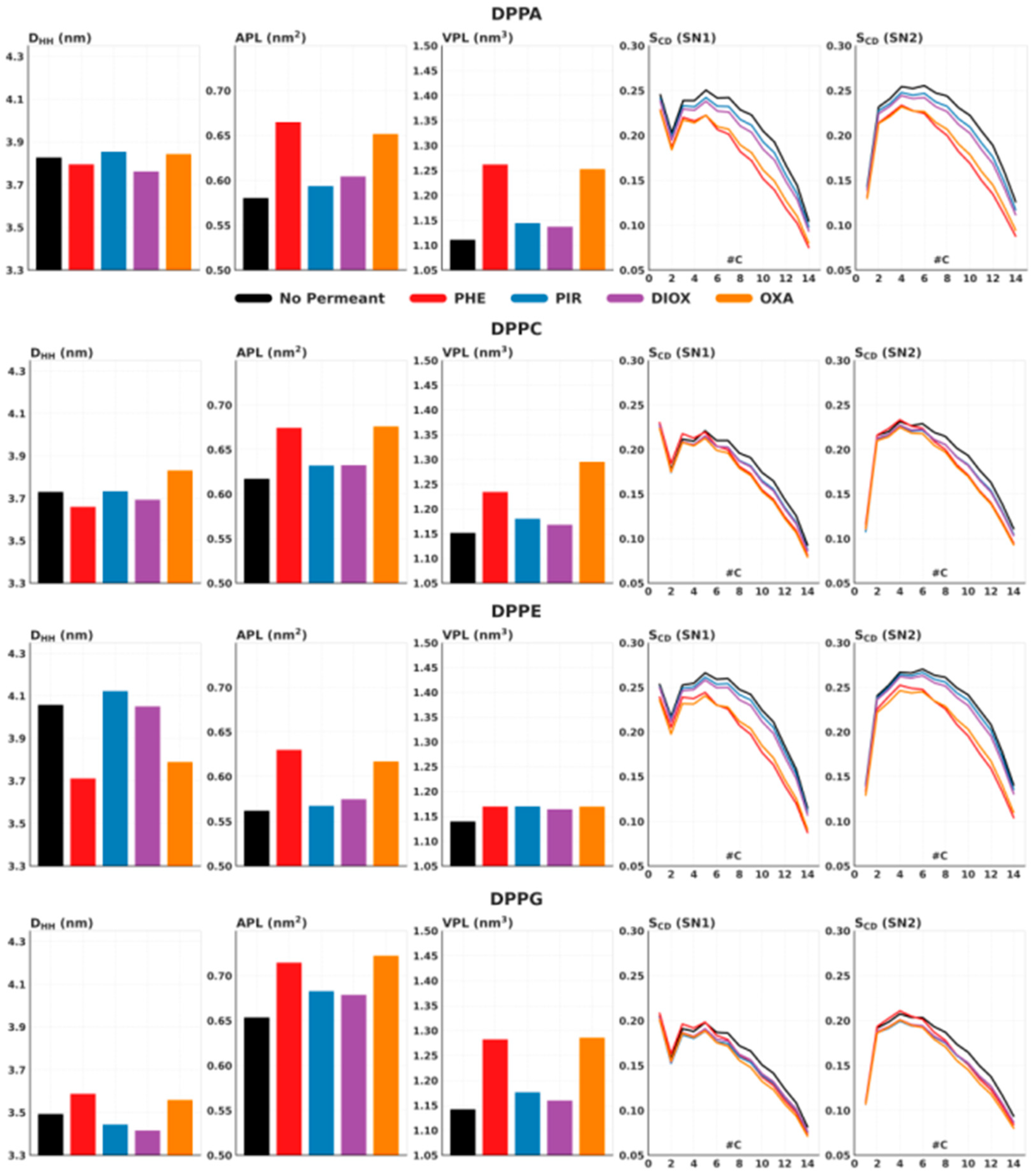
2.1. Membrane Structural Parameters
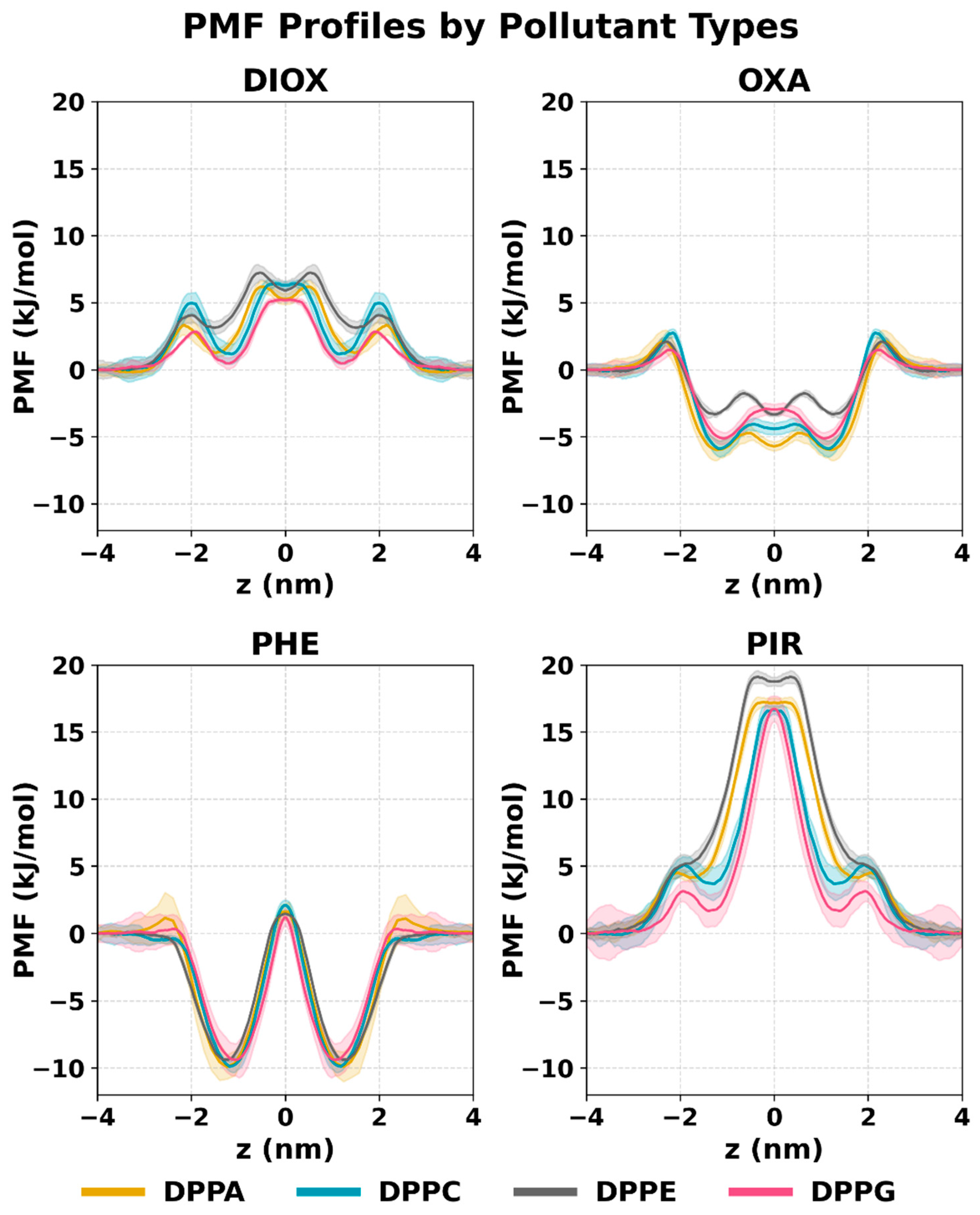
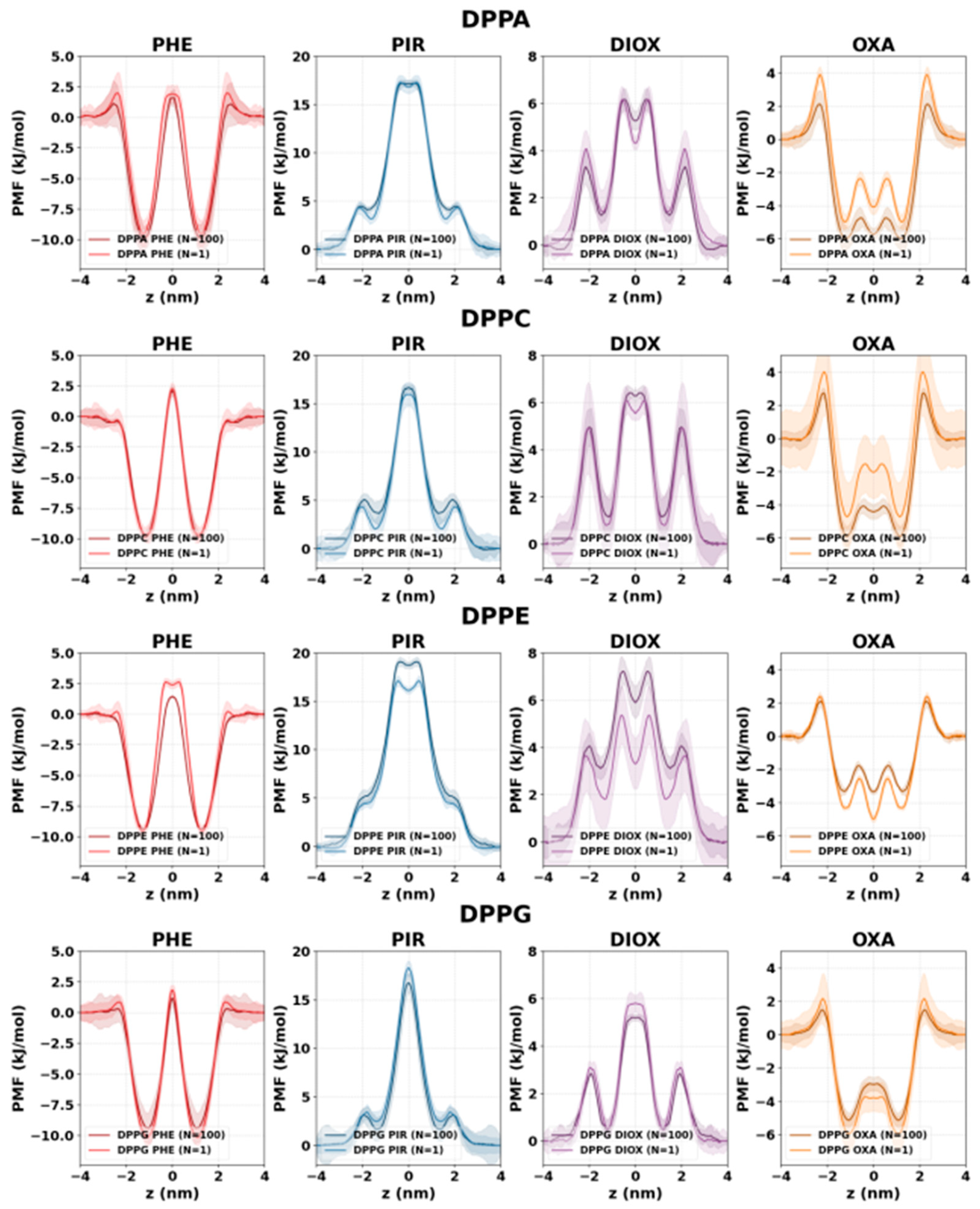
2.2. PMF Profiles of the Studied Systems
2.3. Concentration Dependence of Membrane Penetration
3. Materials and Methods
3.1. System Preparation
3.2. Parameters of MD Simulations
3.3. Analysis of Structural Membrane Parameters
3.4. Accuracy of the Simulation Protocol
3.5. Free Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palaiokostas, M.; Ding, W.; Shahane, G.; Orsi, M. Effects of Lipid Composition on Membrane Permeation. Soft Matter 2018, 14, 8496–8508. [Google Scholar] [CrossRef]
- Pogozheva, I.D.; Armstrong, G.A.; Kong, L.; Hartnagel, T.J.; Carpino, C.A.; Gee, S.E.; Picarello, D.M.; Rubin, A.S.; Lee, J.; Park, S.; et al. Comparative Molecular Dynamics Simulation Studies of Realistic Eukaryotic, Prokaryotic, and Archaeal Membranes. J. Chem. Inf. Model. 2022, 62, 1036–1051. [Google Scholar] [CrossRef]
- Santamaria, A.; Batchu, K.C.; Fragneto, G.; Laux, V.; Haertlein, M.; Darwish, T.A.; Russell, R.A.; Zaccai, N.R.; Guzmán, E.; Maestro, A. Investigation on the Relationship between Lipid Composition and Structure in Model Membranes Composed of Extracted Natural Phospholipids. J. Colloid Interface Sci. 2023, 637, 55–66. [Google Scholar] [CrossRef]
- Dowhan, W. Molecular Basis for Membrane Phospholipid Diversity: Why Are There So Many Lipids? Annu. Rev. Biochem. 1997, 66, 199–232. [Google Scholar] [CrossRef]
- Pan, J.; Heberle, F.A.; Tristram-Nagle, S.; Szymanski, M.; Koepfinger, M.; Katsaras, J.; Kučerka, N. Molecular Structures of Fluid Phase Phosphatidylglycerol Bilayers as Determined by Small Angle Neutron and X-Ray Scattering. Biochim. Biophys. Acta–Biomembr. 2012, 1818, 2135–2148. [Google Scholar] [CrossRef]
- Leekumjorn, S.; Sum, A.K. Molecular Simulation Study of Structural and Dynamic Properties of Mixed DPPC/DPPE Bilayers. Biophys. J. 2006, 90, 3951–3965. [Google Scholar] [CrossRef]
- Zhukov, A.; Popov, V. Eukaryotic Cell Membranes: Structure, Composition, Research Methods and Computational Modelling. Int. J. Mol. Sci. 2023, 24, 11226. [Google Scholar] [CrossRef]
- Uran, S.; Larsen, Å.; Jacobsen, P.B.; Skotland, T. Analysis of Phospholipid Species in Human Blood Using Normal-Phase Liquid Chromatography Coupled with Electrospray Ionization Ion-Trap Tandem Mass Spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 2001, 758, 265–275. [Google Scholar] [CrossRef]
- Leekumjorn, S.; Sum, A.K. Molecular Studies of the Gel to Liquid-Crystalline Phase Transition for Fully Hydrated DPPC and DPPE Bilayers. Biochim. Biophys. Acta–Biomembr. 2007, 1768, 354–365. [Google Scholar] [CrossRef]
- Jahn, R.; Grubmüller, H. Membrane Fusion. Curr. Opin. Cell Biol. 2002, 14, 488–495. [Google Scholar] [CrossRef]
- Goetz, G.J.; Naomi, S.; Madrigal, A.M.; Chang, C.L.A.; Cornell, C.E.; Keller, S.L. Micron-Scale, Liquid-Liquid Phase Separation in Ternary Lipid Membranes Containing DPPE. bioRXiv 2025. [Google Scholar] [CrossRef]
- Molugu, T.R.; Thurmond, R.L.; Alam, T.M.; Trouard, T.P.; Brown, M.F. Phospholipid Headgroups Govern Area per Lipid and Emergent Elastic Properties of Bilayers. Biophys. J. 2022, 121, 4205–4220. [Google Scholar] [CrossRef]
- Uppulury, K.; Coppock, P.S.; Kindt, J.T. Molecular Simulation of the DPPE Lipid Bilayer Gel Phase: Coupling between Molecular Packing Order and Tail Tilt Angle. J. Phys. Chem. B 2015, 119, 8725–8733. [Google Scholar] [CrossRef]
- Maleš, P.; Butumović, M.; Erceg, I.; Brkljača, Z.; Bakarić, D. Influence of DPPE Surface Undulations on Melting Temperature Determination: UV/Vis Spectroscopic and MD Study. Biochim. Biophys. Acta–Biomembr. 2023, 1865, 184072. [Google Scholar] [CrossRef]
- Kobayashi, K.; Jimbo, H.; Nakamura, Y.; Wada, H. Biosynthesis of Phosphatidylglycerol in Photosynthetic Organisms. Prog. Lipid Res. 2024, 93, 101266. [Google Scholar] [CrossRef] [PubMed]
- Drabik, D.; Czogalla, A. Simple Does Not Mean Trivial: Behavior of Phosphatidic Acid in Lipid Mono- and Bilayers. Int. J. Mol. Sci. 2021, 22, 11523. [Google Scholar] [CrossRef]
- Shinoda, W. Permeability across Lipid Membranes. Biochim. Biophys. Acta–Biomembr. 2016, 1858, 2254–2265. [Google Scholar] [CrossRef]
- Benmameri, M.; Chantemargue, B.; Humeau, A.; Trouillas, P.; Fabre, G. MemCross: Accelerated Weight Histogram Method to Assess Membrane Permeability. Biochim. Biophys. Acta–Biomembr. 2023, 1865, 184120. [Google Scholar] [CrossRef]
- Venable, R.M.; Krämer, A.; Pastor, R.W. Molecular Dynamics Simulations of Membrane Permeability. Chem. Rev. 2019, 119, 5954–5997. [Google Scholar] [CrossRef]
- Vermaas, J.V.; Dixon, R.A.; Chen, F.; Mansfield, S.D.; Boerjan, W.; Ralph, J.; Crowley, M.F.; Beckham, G.T. Passive Membrane Transport of Lignin-Related Compounds. Proc. Natl. Acad. Sci. USA 2019, 116, 23117–23123. [Google Scholar] [CrossRef]
- Frallicciardi, J.; Melcr, J.; Siginou, P.; Marrink, S.J.; Poolman, B. Membrane Thickness, Lipid Phase and Sterol Type Are Determining Factors in the Permeability of Membranes to Small Solutes. Nat. Commun. 2022, 13, 1605. [Google Scholar] [CrossRef]
- Koyanagi, T.; Leriche, G.; Yep, A.; Onofrei, D.; Holland, G.P.; Mayer, M.; Yang, J. Effect of Headgroups on Small-Ion Permeability across Archaea-Inspired Tetraether Lipid Membranes. Chem. Eur. J. 2016, 22, 8074–8077. [Google Scholar] [CrossRef]
- DeMarco, K.R.; Bekker, S.; Clancy, C.E.; Noskov, S.Y.; Vorobyov, I. Digging into Lipid Membrane Permeation for Cardiac Ion Channel Blocker D-Sotalol with All-Atom Simulations. Front. Pharmacol. 2018, 9, 26. [Google Scholar] [CrossRef]
- Ingólfsson, H.I.; Arnarez, C.; Periole, X.; Marrink, S.J. Computational ‘Microscopy’ of Cellular Membranes. J. Cell Sci. 2016, 129, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Javanainen, M.; Martinez-Seara, H.; Vattulainen, I. Excessive Aggregation of Membrane Proteins in the Martini Model. PLoS ONE 2017, 12, e0187936. [Google Scholar] [CrossRef]
- Torrie, G.; Valleau, J. Nonphysical Sampling Distributions in Monte Carlo Free-Energy Estimation: Umbrella Sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Comer, J.; Gumbart, J.C.; Hénin, J.; Lelièvre, T.; Pohorille, A.; Chipot, C. The Adaptive Biasing Force Method: Everything You Always Wanted to Know but Were Afraid to Ask. J. Phys. Chem. B 2015, 119, 1129–1151. [Google Scholar] [CrossRef]
- Darve, E.; Rodríguez-Gómez, D.; Pohorille, A. Adaptive Biasing Force Method for Scalar and Vector Free Energy Calculations. J. Chem. Phys. 2008, 128, 144120. [Google Scholar] [CrossRef]
- Tse, C.H.; Comer, J.; Chu, S.K.S.; Wang, Y.; Chipot, C. Affordable Membrane Permeability Calculations: Permeation of Short-Chain Alcohols through Pure-Lipid Bilayers and a Mammalian Cell Membrane. J. Chem. Theory Comput. 2019, 15, 2913–2924. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Parrinello, M. Escaping Free-Energy Minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, V.; Lidmar, J.; Hess, B. Riemann Metric Approach to Optimal Sampling of Multidimensional Free-Energy Landscapes. Phys. Rev. E 2018, 98, 023312. [Google Scholar] [CrossRef]
- Lindahl, V.; Villa, A.; Hess, B. Sequence Dependency of Canonical Base Pair Opening in the DNA Double Helix. PLoS Comput. Biol. 2017, 13, e1005463. [Google Scholar] [CrossRef]
- Lindahl, V.; Gourdon, P.; Andersson, M.; Hess, B. Permeability and Ammonia Selectivity in Aquaporin TIP2;1: Linking Structure to Function. Sci. Rep. 2018, 8, 2995. [Google Scholar] [CrossRef]
- Lindahl, V.; Lidmar, J.; Hess, B. Accelerated Weight Histogram Method for Exploring Free Energy Landscapes. J. Chem. Phys. 2014, 141, 044110. [Google Scholar] [CrossRef] [PubMed]
- Lundborg, M.; Wennberg, C.; Lidmar, J.; Hess, B.; Lindahl, E.; Norlén, L. Skin Permeability Prediction with MD Simulation Sampling Spatial and Alchemical Reaction Coordinates. Biophys. J. 2022, 121, 3837–3849. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, M.; Schneider, A.L.; Bluhm, K.; Schiwy, S.; Lehmann, G.; Deutschmann, B.; Müller, A.; Tiehm, A.; Hollert, H. Ecotoxicity of Nitrogen, Sulfur, or Oxygen Heterocycles and Short-Chained Alkyl Phenols Commonly Detected in Contaminated Groundwater. Environ. Toxicol. Chem. 2019, 38, 1343–1355. [Google Scholar] [CrossRef]
- Sikkema, J.; de Bont, J.A.; Poolman, B. Interactions of Cyclic Hydrocarbons with Biological Membranes. J. Biol. Chem. 1994, 269, 8022–8028. [Google Scholar] [CrossRef]
- Sikkema, J.; de Bont, J.A.; Poolman, B. Mechanisms of Membrane Toxicity of Hydrocarbons. Microbiol. Rev. 1995, 59, 201–222. [Google Scholar] [CrossRef] [PubMed]
- De Smet, M.J.; Jaap, K.; Witholt, B. The Effect of Toluene on the Structure and Permeability of the Outer and Cytoplasmic Membranes of Escherichia coli. Biochim. Biophys. Acta–Biomembr. 1978, 506, 64–80. [Google Scholar] [CrossRef] [PubMed]
- Uribe, S.; Rangel, P.; Espinola, G.; Aguirre, G. Effects of Cyclohexane, an Industrial Solvent, on the Yeast Saccharomyces cerevisiae and on Isolated Yeast Mitochondria. Appl. Environ. Microbiol. 1990, 56, 2114–2119. [Google Scholar] [CrossRef]
- Uribe, S.; Ramirez, J.; Pena, A. Effects of β-Pinene on Yeast Membrane Functions. J. Bacteriol. 1985, 161, 1195–1200. [Google Scholar] [CrossRef] [PubMed]
- Raz, A.; Livne, A. Differential Effects of Lipids on the Osmotic Fragility of Erythrocytes. Biochim. Biophys. Acta–Biomembr. 1973, 311, 222–229. [Google Scholar] [CrossRef]
- Seeman, P. The Membrane Actions of Anesthetics and Tranquilizers. Pharmacol. Rev. 1972, 24, 583–655. [Google Scholar] [CrossRef]
- Shaikh, M.S.I.; Derle, N.D.; Bhamber, R. Permeability Enhancement Techniques for Poorly Permeable Drugs: A Review. J. Appl. Pharm. Sci. 2012, 2, 34–39. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, H.; Mikov, M.; Tucker, I.G. Physicochemical and Biological Characterization of Monoketocholic Acid, a Novel Permeability Enhancer. Mol. Pharm. 2009, 6, 445–456. [Google Scholar] [CrossRef]
- Gurtovenko, A.A.; Anwar, J.; Yorkshire, W. Modulating the Structure and Properties of Cell Membranes: The Molecular Mechanism of Action of Dimethyl Sulfoxide. J. Phys. Chem. B 2007, 111, 10453–10460. [Google Scholar] [CrossRef]
- Robbiano, L.; Baroni, D.; Carrozzino, R.; Mereto, E.; Brambilla, G. DNA Damage and Micronuclei Induced in Rat and Human Kidney Cells by Six Chemicals Carcinogenic to the Rat Kidney. Toxicology 2004, 204, 187–195. [Google Scholar] [CrossRef]
- Blotevogel, J.; Reineke, A.-K.; Hollender, J.; Held, T. Identifikation NSO-Heterocyclischer Prioritärsubstanzen zur Erkundung und Überwachung Teeröl-Kontaminierter Standorte. Grundwasser 2008, 13, 147–157. [Google Scholar] [CrossRef]
- Wang, J.; Chi, Q.; Zhang, R.; Wu, X.; Jiang, X.; Mu, Y.; Tu, Y.; Shen, J. Evaluation of N-Methylpyrrolidone Bio-Mineralization Mechanism and Bacterial Community Evolution under Denitrification Environment. J. Clean. Prod. 2022, 343, 130945. [Google Scholar] [CrossRef]
- Adil, N.; Jamil, Z.; Iqbal, J.; Siddiqui, A.J.; Sibt-e-Hassan, S.; Kumari, S.; Ali, S.A.; Musharraf, S.G. The Pesticide Paradox: Metabolomics Insights into N-Methyl-2-Pyrrolidone (NMP) Exposure as a Culprit of Infant Undernourishment. J. Hazard. Mater. Adv. 2025, 17, 100533. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, T.; Hou, Y.; Xue, R.; Liu, R.; Chen, F.; Zhang, Y.; Rittmann, B.E. The Roles of Methylobacterium organophilum and Sphingomonas melonis for Accelerating N-Methyl Pyrrolidone (NMP) Biodegradation. J. Water Process Eng. 2023, 56, 104327. [Google Scholar] [CrossRef]
- Ginsberg, G.; Chen, Y.; Vasiliou, V. Mechanistic Considerations in 1,4-Dioxane Cancer Risk Assessment. Curr. Opin. Environ. Sci. Health 2022, 30, 100407. [Google Scholar] [CrossRef]
- Rafat, M.; Ghazy, M.A.; Nasr, M. Phycoremediation of 1,4 Dioxane-Laden Wastewater: A Techno-Economic and Sustainable Development Approach. J. Environ. Manag. 2024, 370, 122387. [Google Scholar] [CrossRef] [PubMed]
- Dastidar, R.G.; Kim, M.S.; Zhou, P.; Luo, Z.; Shi, C.; Barnett, K.J.; McClelland, D.J.; Chen, E.Y.-X.; Van Lehn, R.C.; Huber, G.W. Catalytic Production of Tetrahydropyran (THP): A Biomass-Derived, Economically Competitive Solvent with Demonstrated Use in Plastic Dissolution. Green Chem. 2022, 24, 9101–9113. [Google Scholar] [CrossRef]
- Hartness, S.W.; Saab, M.; Preußker, M.; Mazzotta, R.; Dewey, N.S.; Hill, A.W.; Vanhove, G.; Fenard, Y.; Heufer, K.A.; Rotavera, B. Low-Temperature Ignition and Oxidation Mechanisms of Tetrahydropyran. Proc. Combust. Inst. 2024, 40, 105528. [Google Scholar] [CrossRef]
- Mahgoub, S.A.; Qattan, S.Y.A.; Salem, S.S.; Abdelbasit, H.M.; Raafat, M.; Ashkan, M.F.; Al-Quwaie, D.A.; Motwali, E.A.; Alqahtani, F.S.; Abd El-Fattah, H.I. Characterization and Biodegradation of Phenol by Pseudomonas aeruginosa and Klebsiella variicola Strains Isolated from Sewage Sludge and Their Effect on Soybean Seeds Germination. Molecules 2023, 28, 1203. [Google Scholar] [CrossRef]
- Panigrahy, N.; Priyadarshini, A.; Sahoo, M.M.; Verma, A.K.; Daverey, A.; Sahoo, N.K. A Comprehensive Review on Eco-Toxicity and Biodegradation of Phenolics: Recent Progress and Future Outlook. Environ. Technol. Innov. 2022, 27, 102423. [Google Scholar] [CrossRef]
- Khvedelidze, M.; Mdzinarashvili, T.; Shekiladze, E.; Schneider, M.; Moersdorf, D.; Bernhardt, I. Structure of Drug Delivery DPPA and DPPC Liposomes with Ligands and Their Permeability through Cells. J. Liposome Res. 2015, 25, 20–31. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Tümmler, B.; Boggs, J.M. Use of Spin Labels to Determine the Percentage of Interdigitated Lipid in Complexes with Polymyxin B and Polymyxin B Nonapeptide. Biochim. Biophys. Acta–Biomembr. 1989, 985, 182–198. [Google Scholar] [CrossRef]
- Petrov, A.G.; Gawrisch, K.; Brezesinski, G.; Klose, G.; Möps, A. Optical Detection of Phase Transitions in Simple and Mixed Lipid–Water Phases. Biochim. Biophys. Acta–Biomembr. 1982, 690, 1–7. [Google Scholar] [CrossRef]
- Arvayo-Zatarain, J.A.; Favela-Rosales, F.; Contreras-Aburto, C.; Urrutia-Bañuelos, E.; Maldonado, A. Molecular Dynamics Simulation Study of the Effect of Halothane on Mixed DPPC/DPPE Phospholipid Membranes. J. Mol. Model. 2019, 25, 4. [Google Scholar] [CrossRef] [PubMed]
- Rózsa, Z.B.; Szőri-Dorogházi, E.; Viskolcz, B.; Szőri, M. Transmembrane Penetration Mechanism of Cyclic Pollutants Inspected by Molecular Dynamics and Metadynamics: The Case of Morpholine, Phenol, 1,4-Dioxane and Oxane. Phys. Chem. Chem. Phys. 2021, 23, 15338–15351. [Google Scholar] [CrossRef]
- Machleidt, H.; Roth, S.; Seeman, P. The Hydrophobic Expansion of Erythrocyte Membranes by the Phenol Anesthetics. Biochim. Biophys. Acta–Biomembr. 1972, 255, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Martin, Y.C. Exploring QSAR: Hydrophobic, Electronic, and Steric Constants. J. Med. Chem. 1996, 39, 1189–1190. [Google Scholar] [CrossRef]
- Cumming, H.; Rücker, C. Octanol–Water Partition Coefficient Measurement by a Simple 1H NMR Method. ACS Omega 2017, 2, 6244–6249. [Google Scholar] [CrossRef]
- Sasaki, H.; Kojima, M.; Mori, Y.; Nakamura, J.; Shibasaki, J. Enhancing Effect of Pyrrolidone Derivatives on Transdermal Drug Delivery II. Effect of Application Concentration and Pre-Treatment of Enhancer. Int. J. Pharm. 1990, 60, 177–183. [Google Scholar] [CrossRef]
- Shafer, W.E.; Schönherr, J. Accumulation and transport of phenol, 2-nitrophenol, and 4-nitrophenol in plant cuticles. Ecotoxicol. Environ. Saf. 1985, 10, 239–252. [Google Scholar] [CrossRef]
- Ingram, T.; Storm, S.; Kloss, L.; Mehling, T.; Jakobtorweihen, S.; Smirnova, I. Prediction of micelle/water and liposome/water partition coefficients based on molecular dynamics simulations, COSMO-RS, and COSMOmic. Langmuir 2013, 29, 3527–3537. [Google Scholar] [CrossRef]
- Marrink, S.J.; Berendsen, H.J.C. Permeation process of small molecules across lipid membranes studied by molecular dynamics simulations. J. Phys. Chem. 1996, 100, 16729–16738. [Google Scholar] [CrossRef]
- Wang, Y.; Gallagher, E.; Jorgensen, C.; Troendle, E.P.; Hu, D.; Searson, P.C.; Ulmschneider, M.B. An experimentally validated approach to calculate the blood-brain barrier permeability of small molecules. Sci. Rep. 2019, 9, 6117. [Google Scholar] [CrossRef] [PubMed]
- MacCallum, J.L.; Tieleman, D.P. Computer simulation of the distribution of hexane in a lipid bilayer: Spatially resolved free energy, entropy, and enthalpy profiles. J. Am. Chem. Soc. 2006, 128, 125–130. [Google Scholar] [CrossRef]
- Su, C.-F.; Merlitz, H.; Rabbel, H.; Sommer, J.-U. Nanoparticles of various degrees of hydrophobicity interacting with lipid membranes. J. Phys. Chem. Lett. 2017, 8, 4069–4076. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Lee, J.; Patel, D.S.; Ståhle, J.; Park, S.-J.; Kern, N.R.; Kim, S.; Lee, J.; Cheng, X.; Valvano, M.A.; Holst, O.; et al. CHARMM-GUI membrane builder for complex biological membrane simulations with glycolipids and lipoglycans. J. Chem. Theory Comput. 2019, 15, 775–786. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI membrane builder—Toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef]
- Kumar, N.; Sastry, G.N. Study of lipid heterogeneity on bilayer membranes using molecular dynamics simulations. J. Mol. Graph. Model. 2021, 108, 108000. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, J.; Qiu, Z.; Cui, Z.; Li, N.; Li, X.; Wang, Y.; Zhang, H.; Zhao, C. Effects of polyethylene microplastics on cell membranes: A combined study of experiments and molecular dynamics simulations. J. Hazard. Mater. 2022, 429, 128323. [Google Scholar] [CrossRef]
- Talandashti, R.; Mehrnejad, F.; Rostamipour, K.; Doustdar, F.; Lavasanifar, A. Molecular insights into pore formation mechanism, membrane perturbation, and water permeation by the antimicrobial peptide pleurocidin: A combined all-atom and coarse-grained molecular dynamics simulation study. J. Phys. Chem. B 2021, 125, 7163–7176. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Patel, S. Thermodynamics of cell-penetrating HIV1 TAT peptide insertion into PC/PS/CHOL model bilayers through transmembrane pores: The roles of cholesterol and anionic lipids. Soft Matter 2016, 12, 6716–6727. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.J.; Klein, M.L. Effects of halothane on dimyristoylphosphatidylcholine lipid bilayer structure: A molecular dynamics simulation study. Bull. Korean Chem. Soc. 2009, 30, 2087–2092. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [PubMed]
- Pastor, R.W.; MacKerell, A.D. Development of the CHARMM force field for lipids. J. Phys. Chem. Lett. 2011, 2, 1526–1532. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.; Jo, S.; Brooks, C.L.; Lee, H.S.; Im, W. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Dickson, C.J.; Rosso, L.; Betz, R.M.; Walker, R.C.; Gould, I.R. GAFFlipid: A general amber force field for the accurate molecular dynamics simulation of phospholipid. Soft Matter 2012, 8, 9617. [Google Scholar] [CrossRef]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The amber lipid force field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Nagle, J.F. Area/lipid of bilayers from NMR. Biophys. J. 1993, 64, 1476–1481. [Google Scholar] [CrossRef]
- Petrache, H.I.; Dodd, S.W.; Brown, M.F. Area per lipid and acyl length distributions in fluid phosphatidylcholines determined by 2H NMR spectroscopy. Biophys. J. 2000, 79, 3172–3192. [Google Scholar] [CrossRef] [PubMed]
- Miñones, J.; Rodríguez Patino, J.M.; Dynarowicz-Latka, P.; Carrera, C. Structural and topographical characteristics of dipalmitoyl phosphatidic acid in Langmuir monolayers. J. Colloid Interface Sci. 2002, 249, 388–397. [Google Scholar] [CrossRef]
- Stidder, B.; Fragneto, G.; Roser, S.J. Structure and stability of DPPE planar bilayers. Soft Matter 2007, 3, 214–222. [Google Scholar] [CrossRef]
- Tiwary, P.; Parrinello, M. A time-independent free energy estimator for metadynamics. J. Phys. Chem. B 2015, 119, 736–742. [Google Scholar] [CrossRef]
- Dama, J.F.; Parrinello, M.; Voth, G.A. Well-tempered metadynamics converges asymptotically. Phys. Rev. Lett. 2014, 112, 240602. [Google Scholar] [CrossRef] [PubMed]
- Nencini, R.; Ollila, O.H.S. Charged small molecule binding to membranes in MD simulations evaluated against NMR experiments. J. Phys. Chem. B 2022, 126, 6955–6963. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Ghysels, A.; Wang, E.; Venable, R.M.; Klauda, J.B.; Brooks, B.R.; Pastor, R.W. Membrane permeability of small molecules from unbiased molecular dynamics simulations. J. Chem. Phys. 2020, 153, 124107. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LogPmemb/bulk | DPPA | DPPC | DPPE | DPPG |
|---|---|---|---|---|
| PHE | 0.89 | 0.93 | 0.72 | 0.99 |
| PIR | −0.87 | −0.71 | −1.08 | −0.54 |
| DIOX | −0.37 | −0.31 | −0.55 | −0.20 |
| OXA | 0.67 | 0.75 | 0.54 | 0.79 |
| Name | Phospholipid | NSO-HET | T (K) |
|---|---|---|---|
| DPPC | DPPC | - | 330 |
| DPPC-DIOX | DPPC | 1,4-Dioxane | 330 |
| DPPC-OXA | DPPC | Oxane | 330 |
| DPPC-PHE | DPPC | Phenol | 330 |
| DPPC-PIR | DPPC | N-Methyl-2-Pyrrolidone | 330 |
| DPPG | DPPG | - | 330 |
| DPPG-DIOX | DPPG | 1,4-Dioxane | 330 |
| DPPG-OXA | DPPG | Oxane | 330 |
| DPPG-PHE | DPPG | Phenol | 330 |
| DPPG-PIR | DPPG | N-Methyl-2-Pyrrolidone | 330 |
| DPPE | DPPE | - | 345 |
| DPPE-DIOX | DPPE | 1,4-Dioxane | 345 |
| DPPE-OXA | DPPE | Oxane | 345 |
| DPPE-PHE | DPPE | Phenol | 345 |
| DPPE-PIR | DPPE | N-Methyl-2-Pyrrolidone | 345 |
| DPPA | DPPA | - | 345 |
| DPPA-DIOX | DPPA | 1,4-Dioxane | 345 |
| DPPA-OXA | DPPA | Oxane | 345 |
| DPPA-PHE | DPPA | Phenol | 345 |
| DPPA-PIR | DPPA | N-Methyl-2-Pyrrolidone | 345 |
| Membrane Parameter | Definition | Formula | Abbreviations |
|---|---|---|---|
| APL | Area per Lipid [94] | L: average x and y dimensions of the simulation box | |
| DHH | Membrane Thickness [95] | Distance between peaks in the membrane electron density profile | - |
| VPL | Volume per Lipid [94] | - | |
| SCD | Deuterium Order Parameter, measures the orientation of C-H bonds with respect to the bilayer normal [95] | Θ: the angle between bilayer normal (Z) and the vector between Ci-Hi |
| Membrane Parameter | DPPC (exp) | DPPC (sim) T = 330 K | DPPE (exp) | DPPE (sim) T = 345 K | DPPA (exp) | DPPA (sim) T = 345 K | DPPG (exp) | DPPG (sim) T = 330 K |
|---|---|---|---|---|---|---|---|---|
| APL (nm2) | 0.621 ± 0.02 (323.15 K) [96] | 0.618 ± 0.014 | 0.605 (342.15 K) [97] 0.610 ± 0.32 (342.15 K) [12] | 0.562 ± 0.009 | 0.393 ± 0.13 (298.15 K) [16] ~0.42 (298.15 K) [98] | 0.581 ± 0.010 | 0.670 (323.15 K) [5] | 0.654 ± 0.013 |
| DHH (nm) | 3.850 (323.15 K) [96] | 3.732 ± 0.25 | 4.100 ± 0.10 (343.15 K) [99] | 4.059 ± 0.31 | 4.640 ± 0.02 (303.15 K) [16] | 3.828 ± 0.29 | 3.550 (323.15 K) [5] | 3.495 ± 0.27 |
| VPL (nm3) | 1.232
(323.15 K) [96] | 1.152 ± 0.02 | - | 1.141 ± 0.05 | - | 1.111 ± 0.06 | 1.189 (323.15 K) [5] | 1.143 ± 0.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rózsa, Z.B.; Horváth, T.; Viskolcz, B.; Szőri, M. Influence of Membrane Composition on the Passive Membrane Penetration of Industrially Relevant NSO-Heterocycles. Int. J. Mol. Sci. 2025, 26, 7427. https://doi.org/10.3390/ijms26157427
Rózsa ZB, Horváth T, Viskolcz B, Szőri M. Influence of Membrane Composition on the Passive Membrane Penetration of Industrially Relevant NSO-Heterocycles. International Journal of Molecular Sciences. 2025; 26(15):7427. https://doi.org/10.3390/ijms26157427
Chicago/Turabian StyleRózsa, Zsófia Borbála, Tamás Horváth, Béla Viskolcz, and Milán Szőri. 2025. "Influence of Membrane Composition on the Passive Membrane Penetration of Industrially Relevant NSO-Heterocycles" International Journal of Molecular Sciences 26, no. 15: 7427. https://doi.org/10.3390/ijms26157427
APA StyleRózsa, Z. B., Horváth, T., Viskolcz, B., & Szőri, M. (2025). Influence of Membrane Composition on the Passive Membrane Penetration of Industrially Relevant NSO-Heterocycles. International Journal of Molecular Sciences, 26(15), 7427. https://doi.org/10.3390/ijms26157427