
Roles of Ion Channels in Oligodendrocyte Precursor Cells: From Physiology to Pathology

Abstract
1. Introduction
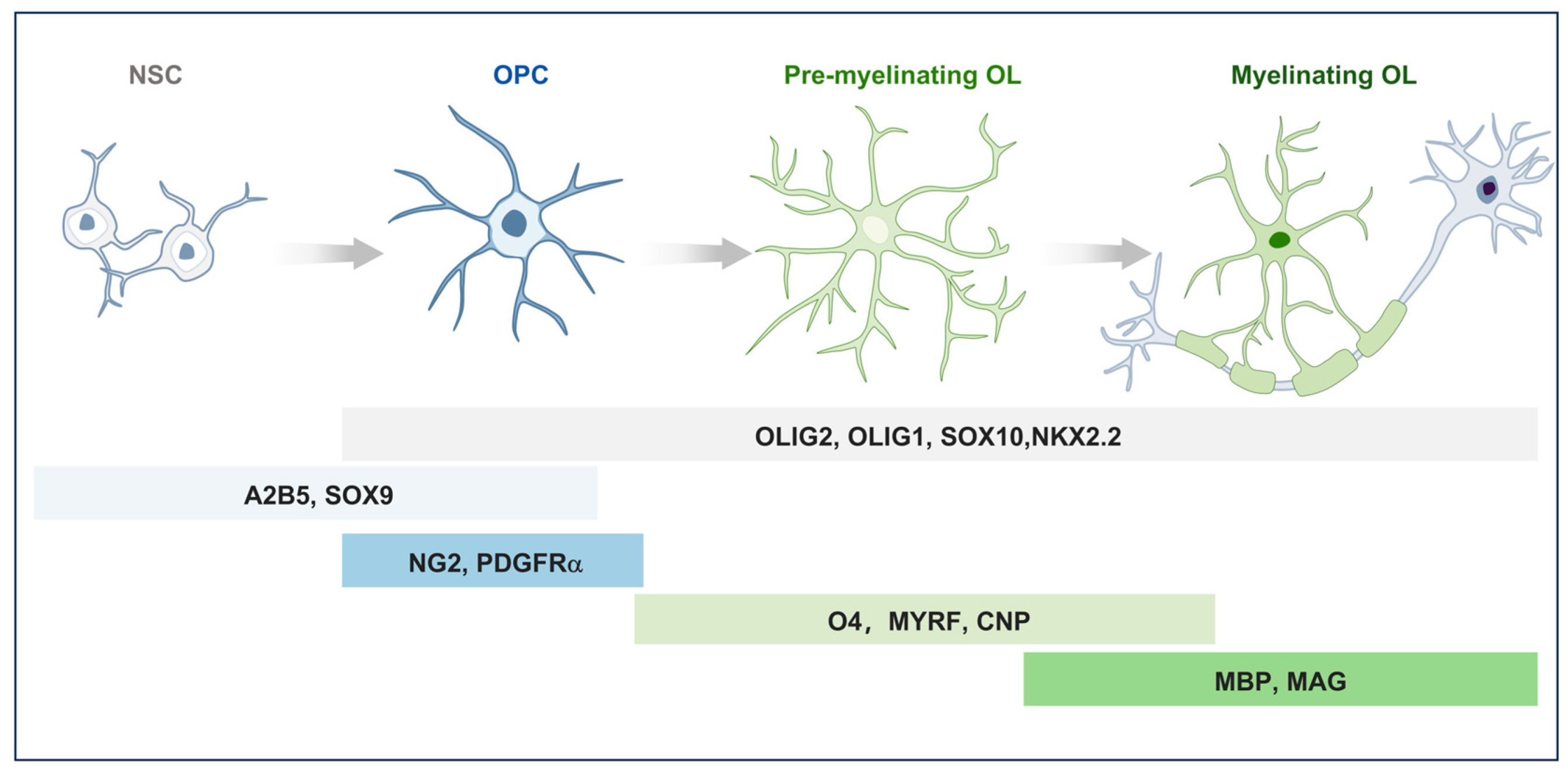
2. Distribution and Functional Role of OPCs in CNS
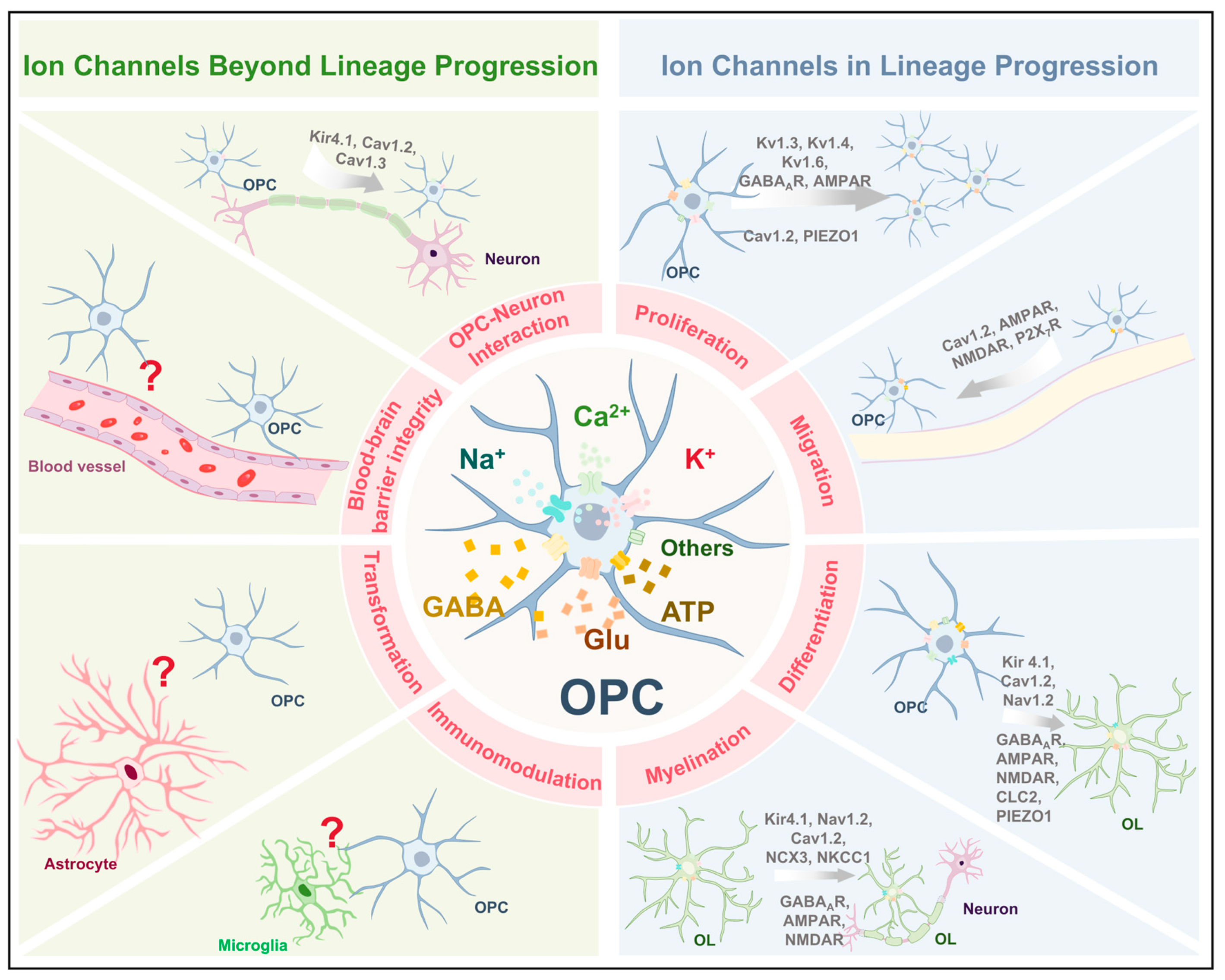
3. Ion Channels in the Physiological Functions of OPCs
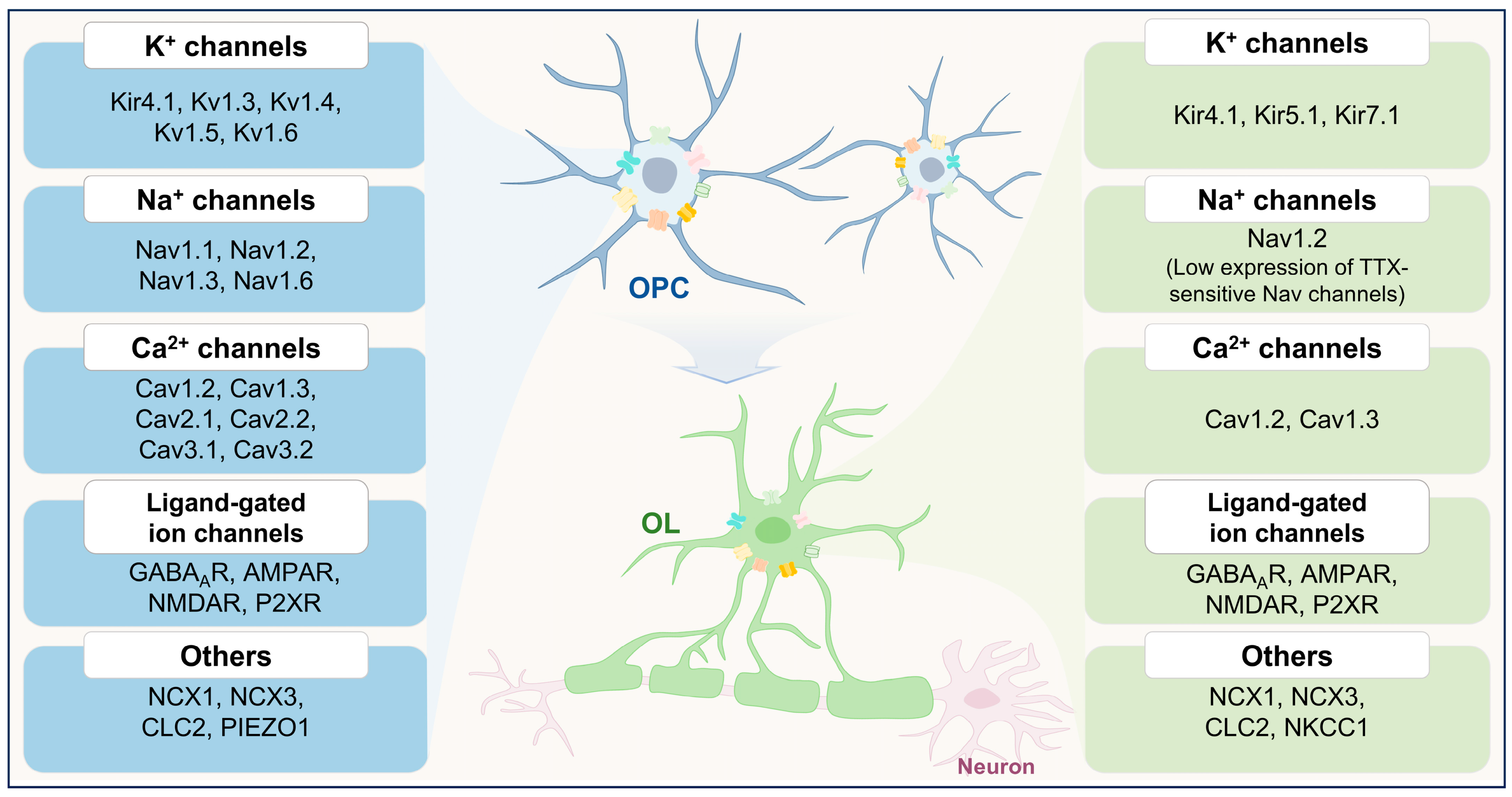

{kind=link}
{kind=link}
{kind=link}
| Ion Channel | Functions | Key Findings | |
|---|---|---|---|
| K+ Channels | Kir4.1 | Differentiation and myelination | Knockout of Kcnj10 in OPCs impairs differentiation and myelination [13]. |
| OPC-neuron interaction | Kir4.1 mediates K+ buffering between OPCs and neurons [33]. | ||
| Kv1.3, Kv1.4, and Kv1.6 | Proliferation | Overexpression of Kv1.3 or Kv1.4 increased OPC proliferation, while Kv1.6 overexpression inhibited cell cycle progression [11]. | |
| Na+ Channels | Nav1.2 | Differentiation and myelination | Specific knockout of SCN2A disrupts the maturation process of oligodendrocytes [34]. Selective knockout of the SCN2A in pre-OLs results in impaired myelin sheath formation [35]. |
| Ca2+ Channels | Cav1.2 | Proliferation, migration, differentiation, and myelination | Cav1.2 deletion in OPCs led to impaired proliferation, migration, differentiation, and myelination [36,37]. |
| Cav1.2 and Cav1.3 | OPC-neuron interaction | Conditional deletion of Cav1.2 and Cav1.3 abolished long-term depression and potentiation [38]. | |
| Ligand-gated ion channels | GABAARs | Proliferation, differentiation, and myelination | GABAARs blockade enhances OPC proliferation, differentiation, and myelination [39]. |
| AMPARs | Proliferation, migration, differentiation, and myelination | In ex vivo organotypic slice cultures, pharmacological blockade of AMPARs promoted OPC proliferation, differentiation, and myelination [40]. In the developing corpus callosum (P14—21), overexpression of GluA2 suppressed OPC differentiation and enhanced proliferation [41]. GluA2 overexpression in the adult brain (P50—P75), the same manipulation increased OPC proliferation [42]. Activation of AMPARs enhances OPC migration [43,44]. In vivo, in zebrafish, it was demonstrated that mutations in GluA4A reduced migration [45]. | |
| NMDARs | Migration, differentiation, and myelination | NMDAR-mediated calcium influx can direct OPC migration [46]. Activation of NMDARs promotes OPC differentiation [47]. | |
| P2X7Rs | Migration | High concentrations of extracellular ATP can activate P2X7 receptors to promote OPC migration [12]. | |
| Other channels | CLC-2 | Differentiation | CLC-2 inhibition suppresses OPC differentiation [48]. |
| NKCC1 | Myelination | NKCC1 overexpression enhances myelin plasticity [49]. | |
| NCX3 | Myelination | Myelination is markedly impaired in NCX3-deficient mice [32]. | |
| Piezo1 | Proliferation and differentiation | Piezo1 knockdown in aged OPCs enhances proliferation and differentiation [50]. | |
3.1. K+ Channels
3.2. Na+ Channels
3.3. Ca2+ Channels
3.4. Ligand-Gated Ion Channels
3.4.1. GABAARs
3.4.2. AMPARs
3.4.3. NMDARs
3.4.4. P2X7Rs
3.5. Other Channels
4. Pathological Roles of Ion Channels in OPCs
4.1. Multiple Sclerosis
4.2. Traumatic Injury of Central Nervous System
4.3. Neurodegenerative Disease
4.3.1. Amyotrophic Lateral Sclerosis
4.3.2. Alzheimer’s Disease
4.4. Psychiatric Disorders
4.5. Neuropathic Pain
5. Pharmacological Modulation of OPC Ion Channels: Therapeutic Potential
6. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| OPCs | Oligodendrocyte precursor cells | Cav | Voltage-gated Ca2+ channel |
| OLs | Oligodendrocytes | CPZ | Cuprizone |
| CNS | Central nervous system | NMDARs | N-methyl-D-aspartate receptors |
| MS | Multiple sclerosis | LGICs | Ligand-gated ion channels |
| ALS | Amyotrophic lateral sclerosis | AMPARs | α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors |
| AD | Alzheimer’s disease | GABA | γ-aminobutyric acid |
| NP | Neuropathic pain | GABAARs | GABAA receptors |
| BBB | Blood–brain barrier | P2XRs | P2X receptors |
| PDGFRα | Platelet-derived growth factor receptor alpha | KARs | Kainate receptors |
| NG2 | Neuron–glial antigen 2 | CLC-2 | Voltage-gated chloride channel-2 |
| Olig2 | Oligodendrocyte transcription factor 2 | NKCC1 | Na+-K+-Cl−-co-transporter 1 |
| PDGF | Platelet-derived growth factor | NCX | Sodium calcium exchanger |
| NSCs | Neural stem cells | LPS | Lipopolysaccharide |
| A2B5 | Cell surface ganglioside epitope | OGD | Oxygen-glucose deprivation |
| SOX9 | SRY-Box Transcription Factor 9 | ROS | Reactive oxygen species |
| O4 | Cell surface markers | FDA | Food and Drug Administration |
| MYRF | Myelin Regulatory Factor | ATP | Adenosine triphosphate |
| CNP | 2′, 3′-cyclic-nucleotide-phosphodiesterase | HCs | Healthy controls |
| MBP | Myelin basic protein | ADD | Acquired demyelinating disease |
| MAG | Myelin-associated glycoprotein | EAE | Experimental autoimmune encephalomyelitis |
| TTX | Tetrodotoxin | TCNSIs | Traumatic central nervous system injuries |
| Nav | Voltage-gated Na+ channels | SCI | Spinal cord injury |
| RMP | Resting membrane potential | TBI | Traumatic brain injury |
| K+ | Potassium | cFPI | Central fluid percussion injury |
| Kir | Inward rectifying K+ channels | rhEPO | Recombinant human erythropoietin |
| Kv | Voltage-gated K+ channels | CCI | Controlled cortical impact |
| K2P | Two-pore domain K+ channels | AChEI | Acetylcholinesterase inhibitor |
| tMCAO | Transient middle cerebral artery occlusion | nAChR | Nicotinic acetylcholine receptor |
| GFP | Green fluorescent protein | MDD | Major depressive disorder |
| PLP | Proteolipid protein | CSDS | Chronic social defeat stress |
| Pre-OLs | Pre-myelinating oligodendrocytes | PTSD | Posttraumatic stress disorder |
| Ca2+ | Calcium | Na+ | Sodium |
References
- Ma, Z.; Zhang, W.; Wang, C.; Su, Y.; Yi, C.; Niu, J. A New Acquaintance of Oligodendrocyte Precursor Cells in the Central Nervous System. Neurosci. Bull. 2024, 40, 1573–1589. [Google Scholar] [CrossRef]
- Fang, L.-P.; Bai, X. Oligodendrocyte Precursor Cells: The Multitaskers in the Brain. Pflügers Arch.-Eur. J. Physiol. 2023, 475, 1035–1044. [Google Scholar] [CrossRef]
- Simons, M.; Nave, K.-A. Oligodendrocytes: Myelination and Axonal Support. Cold Spring Harb. Perspect. Biol. 2015, 8, a020479. [Google Scholar] [CrossRef]
- Mishra, S.K.; Tiwari, S.P. Bioenergetics of Axon Integrity and Its Regulation by Oligodendrocytes and Schwann Cells. Mol. Neurobiol. 2024, 61, 5928–5934. [Google Scholar] [CrossRef] [PubMed]
- Jäkel, S.; Agirre, E.; Mendanha Falcão, A.; van Bruggen, D.; Lee, K.W.; Knuesel, I.; Malhotra, D.; ffrench-Constant, C.; Williams, A.; Castelo-Branco, G. Altered Human Oligodendrocyte Heterogeneity in Multiple Sclerosis. Nature 2019, 566, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Vanzulli, I.; Papanikolaou, M.; De-La-Rocha, I.C.; Pieropan, F.; Rivera, A.D.; Gomez-Nicola, D.; Verkhratsky, A.; Rodríguez, J.J.; Butt, A.M. Disruption of Oligodendrocyte Progenitor Cells Is an Early Sign of Pathology in the Triple Transgenic Mouse Model of Alzheimer’s Disease. Neurobiol. Aging 2020, 94, 130–139. [Google Scholar] [CrossRef]
- Bergles, D.E.; Roberts, J.D.; Somogyi, P.; Jahr, C.E. Glutamatergic Synapses on Oligodendrocyte Precursor Cells in the Hippocampus. Nature 2000, 405, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Bergles, D.E. Synaptic Signaling between GABAergic Interneurons and Oligodendrocyte Precursor Cells in the Hippocampus. Nat. Neurosci. 2004, 7, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Larson, V.A.; Zhang, Y.; Bergles, D.E. Electrophysiological Properties of NG2+ Cells: Matching Physiological Studies with Gene Expression Profiles. Brain Res. 2016, 1638, 138–160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Vautier, F.; Belachew, S.; Chittajallu, R.; Gallo, V. Shaker-Type Potassium Channel Subunits Differentially Control Oligodendrocyte Progenitor Proliferation. Glia 2004, 48, 337–345. [Google Scholar] [CrossRef]
- Feng, J.-F.; Gao, X.-F.; Pu, Y.-Y.; Burnstock, G.; Xiang, Z.; He, C. P2X7 Receptors and Fyn Kinase Mediate ATP-Induced Oligodendrocyte Progenitor Cell Migration. Purinergic Signal. 2015, 11, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-Y.; Zhou, L.; Shen, Y. Inward Rectifying Kir4.1 Channels Regulate Oligodendrocyte Precursor Cell Differentiation and CNS Myelination in Vivo. Neurosci. Lett. 2023, 807, 137278. [Google Scholar] [CrossRef] [PubMed]
- Richardson, W.D.; Kessaris, N.; Pringle, N. Oligodendrocyte Wars. Nat. Rev. Neurosci. 2006, 7, 11–18. [Google Scholar] [CrossRef]
- Bergles, D.E.; Richardson, W.D. Oligodendrocyte Development and Plasticity. Cold Spring Harb. Perspect. Biol. 2015, 8, a020453. [Google Scholar] [CrossRef]
- Tiane, A.; Schepers, M.; Rombaut, B.; Hupperts, R.; Prickaerts, J.; Hellings, N.; van den Hove, D.; Vanmierlo, T. From OPC to Oligodendrocyte: An Epigenetic Journey. Cells 2019, 8, 1236. [Google Scholar] [CrossRef] [PubMed]
- Fields, R.D. Myelin Formation and Remodeling. Cell 2014, 156, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef]
- Zou, P.; Wu, C.; Liu, T.C.-Y.; Duan, R.; Yang, L. Oligodendrocyte Progenitor Cells in Alzheimer’s Disease: From Physiology to Pathology. Transl. Neurodegener. 2023, 12, 52. [Google Scholar] [CrossRef] [PubMed]
- Fruttiger, M.; Calver, A.R.; Richardson, W.D. Platelet-Derived Growth Factor Is Constitutively Secreted from Neuronal Cell Bodies but Not from Axons. Curr. Biol. 2000, 10, 1283–1286. [Google Scholar] [CrossRef] [PubMed]
- Pringle, N.; Collarini, E.J.; Mosley, M.J.; Heldin, C.H.; Westermark, B.; Richardson, W.D. PDGF A Chain Homodimers Drive Proliferation of Bipotential (O-2A) Glial Progenitor Cells in the Developing Rat Optic Nerve. EMBO J. 1989, 8, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Đặng, T.C.; Ishii, Y.; Nguyen, V.D.; Yamamoto, S.; Hamashima, T.; Okuno, N.; Nguyen, Q.L.; Sang, Y.; Ohkawa, N.; Saitoh, Y.; et al. Powerful Homeostatic Control of Oligodendroglial Lineage by PDGFRα in Adult Brain. Cell Rep. 2019, 27, 1073–1089.e5. [Google Scholar] [CrossRef] [PubMed]
- Watzlawik, J.O.; Warrington, A.E.; Rodriguez, M. PDGF Is Required for Remyelination-Promoting IgM Stimulation of Oligodendrocyte Progenitor Cell Proliferation. PLoS ONE 2013, 8, e55149. [Google Scholar] [CrossRef]
- Emery, B. Regulation of Oligodendrocyte Differentiation and Myelination. Science 2010, 330, 779–782. [Google Scholar] [CrossRef]
- Rawji, K.S.; Gonzalez Martinez, G.A.; Sharma, A.; Franklin, R.J.M. The Role of Astrocytes in Remyelination. Trends Neurosci. 2020, 43, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Petrucco, L.; Hoodless, L.J.; Portugues, R.; Czopka, T. Oligodendrocyte Precursor Cells Sculpt the Visual System by Regulating Axonal Remodeling. Nat. Neurosci. 2022, 25, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Rivers, L.E.; Young, K.M.; Rizzi, M.; Jamen, F.; Psachoulia, K.; Wade, A.; Kessaris, N.; Richardson, W.D. PDGFRA/NG2 Glia Generate Myelinating Oligodendrocytes and Piriform Projection Neurons in Adult Mice. Nat. Neurosci. 2008, 11, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Isacoff, E.Y.; Jan, L.Y.; Minor, D.L. Conduits of Life’s Spark: A Perspective on Ion Channel Research since the Birth of Neuron. Neuron 2013, 80, 658–674. [Google Scholar] [CrossRef]
- Biase, L.M.D.; Nishiyama, A.; Bergles, D.E. Excitability and Synaptic Communication within the Oligodendrocyte Lineage. J. Neurosci. 2010, 30, 3600–3611. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-C.; Bergles, D.E. Physiological Characteristics of NG2-Expressing Glial Cells. J. Neurocytol. 2002, 31, 537–549. [Google Scholar] [CrossRef]
- Chittajallu, R.; Chen, Y.; Wang, H.; Yuan, X.; Ghiani, C.A.; Heckman, T.; McBain, C.J.; Gallo, V. Regulation of Kv1 Subunit Expression in Oligodendrocyte Progenitor Cells and Their Role in G1/S Phase Progression of the Cell Cycle. Proc. Natl. Acad. Sci. USA 2002, 99, 2350–2355. [Google Scholar] [CrossRef]
- Boscia, F.; D’Avanzo, C.; Pannaccione, A.; Secondo, A.; Casamassa, A.; Formisano, L.; Guida, N.; Sokolow, S.; Herchuelz, A.; Annunziato, L. Silencing or Knocking out the Na(+)/Ca(2+) Exchanger-3 (NCX3) Impairs Oligodendrocyte Differentiation. Cell Death Differ. 2012, 19, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, P.P.; Vélez-Fort, M.; Levavasseur, F.; Angulo, M.C. Oligodendrocyte Precursor Cells Are Accurate Sensors of Local K+ in Mature Gray Matter. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 2432–2442. [Google Scholar] [CrossRef] [PubMed]
- Gould, E.; Kim, J.H. SCN2A Contributes to Oligodendroglia Excitability and Development in the Mammalian Brain. Cell Rep. 2021, 36, 109653. [Google Scholar] [CrossRef] [PubMed]
- Berret, E.; Barron, T.; Xu, J.; Debner, E.; Kim, E.J.; Kim, J.H. Oligodendroglial Excitability Mediated by Glutamatergic Inputs and Nav1.2 Activation. Nat. Commun. 2017, 8, 557. [Google Scholar] [CrossRef]
- Cheli, V.T.; Santiago González, D.A.; Namgyal Lama, T.; Spreuer, V.; Handley, V.; Murphy, G.G.; Paez, P.M. Conditional Deletion of the L-Type Calcium Channel Cav1.2 in Oligodendrocyte Progenitor Cells Affects Postnatal Myelination in Mice. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 10853–10869. [Google Scholar] [CrossRef]
- Santiago González, D.A.; Cheli, V.T.; Zamora, N.N.; Lama, T.N.; Spreuer, V.; Murphy, G.G.; Paez, P.M. Conditional Deletion of the L-Type Calcium Channel Cav1.2 in NG2-Positive Cells Impairs Remyelination in Mice. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 10038–10051. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Huang, W.; Cãtãlin, B.; Scheller, A.; Kirchhoff, F. L-Type Ca2+ Channels of NG2 Glia Determine Proliferation and NMDA Receptor-Dependent Plasticity. Front. Cell Dev. Biol. 2021, 9, 759477. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.B.; Clarke, L.E.; Arancibia-Carcamo, I.L.; Kougioumtzidou, E.; Matthey, M.; Káradóttir, R.; Whiteley, L.; Bergersen, L.H.; Richardson, W.D.; Attwell, D. Endogenous GABA Controls Oligodendrocyte Lineage Cell Number, Myelination, and CNS Internode Length. Glia 2017, 65, 309–321. [Google Scholar] [CrossRef]
- Fannon, J.; Tarmier, W.; Fulton, D. Neuronal Activity and AMPA-Type Glutamate Receptor Activation Regulates the Morphological Development of Oligodendrocyte Precursor Cells. Glia 2015, 63, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-J.; Kula, B.; Nagy, B.; Barzan, R.; Gall, A.; Ehrlich, I.; Kukley, M. In Vivo Regulation of Oligodendrocyte Precursor Cell Proliferation and Differentiation by the AMPA-Receptor Subunit GluA2. Cell Rep. 2018, 25, 852–861.e7. [Google Scholar] [CrossRef]
- Khawaja, R.R.; Agarwal, A.; Fukaya, M.; Jeong, H.-K.; Gross, S.; Gonzalez-Fernandez, E.; Soboloff, J.; Bergles, D.E.; Kang, S.H. GluA2 Overexpression in Oligodendrocyte Progenitors Promotes Postinjury Oligodendrocyte Regeneration. Cell Rep. 2021, 35, 109147. [Google Scholar] [CrossRef]
- Gudz, T.I.; Komuro, H.; Macklin, W.B. Glutamate Stimulates Oligodendrocyte Progenitor Migration Mediated via an Av Integrin/Myelin Proteolipid Protein Complex. J. Neurosci. 2006, 26, 2458–2466. [Google Scholar] [CrossRef]
- Harlow, D.E.; Saul, K.E.; Komuro, H.; Macklin, W.B. Myelin Proteolipid Protein Complexes with Av Integrin and AMPA Receptors In Vivo and Regulates AMPA-Dependent Oligodendrocyte Progenitor Cell Migration through the Modulation of Cell-Surface GluR2 Expression. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 12018–12032. [Google Scholar] [CrossRef] [PubMed]
- Piller, M.; Werkman, I.L.; Brown, R.I.; Latimer, A.J.; Kucenas, S. Glutamate Signaling via the AMPAR Subunit GluR4 Regulates Oligodendrocyte Progenitor Cell Migration in the Developing Spinal Cord. J. Neurosci. Off. J. Soc. Neurosci. 2021, 41, 5353–5371. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Hu, C.; Yang, W.; Guo, D.; Li, C.; Shen, W.; Liu, X.; Aijun, H.; Dan, W.; He, C. NMDA Receptor Couples Rac1-GEF Tiam1 to Direct Oligodendrocyte Precursor Cell Migration. Glia 2013, 61, 2078–2099. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xiao, L.; Liu, X.; Yang, W.; Shen, W.; Hu, C.; Yang, G.; He, C. A Functional Role of NMDA Receptor in Regulating the Differentiation of Oligodendrocyte Precursor Cells and Remyelination. Glia 2013, 61, 732–749. [Google Scholar] [CrossRef]
- Hou, X.; Zhang, R.; Wang, J.; Li, Y.; Li, F.; Zhang, Y.; Zheng, X.; Shen, Y.; Wang, Y.; Zhou, L. CLC-2 Is a Positive Modulator of Oligodendrocyte Precursor Cell Differentiation and Myelination. Mol. Med. Rep. 2018, 17, 4515–4523. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Abe, Y.; Fujii, S.; Tanaka, K.F. Oligodendrocytic Na+-K+-Cl− Co-Transporter 1 Activity Facilitates Axonal Conduction and Restores Plasticity in the Adult Mouse Brain. Nat. Commun. 2021, 12, 5146. [Google Scholar] [CrossRef]
- Segel, M.; Neumann, B.; Hill, M.F.E.; Weber, I.P.; Viscomi, C.; Zhao, C.; Young, A.; Agley, C.C.; Thompson, A.J.; Gonzalez, G.A.; et al. Niche Stiffness Underlies the Ageing of Central Nervous System Progenitor Cells. Nature 2019, 573, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Attali, B.; Wang, N.; Kolot, A.; Sobko, A.; Cherepanov, V.; Soliven, B. Characterization of Delayed Rectifier Kv Channels in Oligodendrocytes and Progenitor Cells. J. Neurosci. 1997, 17, 8234–8245. [Google Scholar] [CrossRef] [PubMed]
- Neusch, C.; Rozengurt, N.; Jacobs, R.E.; Lester, H.A.; Kofuji, P. Kir4.1 Potassium Channel Subunit Is Crucial for Oligodendrocyte Development and in Vivo Myelination. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 5429–5438. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhou, L.; Shao, C.-Y.; Wang, X.-T.; Zhang, N.; Ma, J.; Hu, H.-L.; Wang, Y.; Qiu, M.; Shen, Y. Potassium Channel Kir4.1 Regulates Oligodendrocyte Differentiation via Intracellular pH Regulation. Glia 2022, 70, 2093–2107. [Google Scholar] [CrossRef]
- Brasko, C.; Hawkins, V.; De La Rocha, I.C.; Butt, A.M. Expression of Kir4.1 and Kir5.1 Inwardly Rectifying Potassium Channels in Oligodendrocytes, the Myelinating Cells of the CNS. Brain Struct. Funct. 2017, 222, 41–59. [Google Scholar] [CrossRef]
- Papanikolaou, M.; Butt, A.M.; Lewis, A. A Critical Role for the Inward Rectifying Potassium Channel Kir7.1 in Oligodendrocytes of the Mouse Optic Nerve. Brain Struct. Funct. 2020, 225, 925–934. [Google Scholar] [CrossRef]
- Coppi, E.; Maraula, G.; Fumagalli, M.; Failli, P.; Cellai, L.; Bonfanti, E.; Mazzoni, L.; Coppini, R.; Abbracchio, M.P.; Pedata, F.; et al. UDP-Glucose Enhances Outward K+ Currents Necessary for Cell Differentiation and Stimulates Cell Migration by Activating the GPR17 Receptor in Oligodendrocyte Precursors. Glia 2013, 61, 1155–1171. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The Blood-Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Wang, L.-P.; Pan, J.; Li, Y.; Geng, J.; Liu, C.; Zhang, L.-Y.; Zhou, P.; Tang, Y.-H.; Wang, Y.; Zhang, Z.; et al. Oligodendrocyte Precursor Cell Transplantation Promotes Angiogenesis and Remyelination via Wnt/β-Catenin Pathway in a Mouse Model of Middle Cerebral Artery Occlusion. J. Cereb. Blood Flow Metab. 2022, 42, 757–770. [Google Scholar] [CrossRef]
- Seo, J.H.; Maki, T.; Maeda, M.; Miyamoto, N.; Liang, A.C.; Hayakawa, K.; Pham, L.-D.D.; Suwa, F.; Taguchi, A.; Matsuyama, T.; et al. Oligodendrocyte Precursor Cells Support Blood-Brain Barrier Integrity via TGF-β Signaling. PLoS ONE 2014, 9, e103174. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Yang, J.; Zhu, Z.; Fang, Y.; Tian, Y.; Xie, M.; Wang, W.; Liu, Y. The Two-Pore Domain Potassium Channel TREK-1 Promotes Blood-Brain Barrier Breakdown and Exacerbates Neuronal Death After Focal Cerebral Ischemia in Mice. Mol. Neurobiol. 2022, 59, 2305–2327. [Google Scholar] [CrossRef] [PubMed]
- Looser, Z.J.; Faik, Z.; Ravotto, L.; Zanker, H.S.; Jung, R.B.; Werner, H.B.; Ruhwedel, T.; Möbius, W.; Bergles, D.E.; Barros, L.F.; et al. Oligodendrocyte-Axon Metabolic Coupling Is Mediated by Extracellular K+ and Maintains Axonal Health. Nat. Neurosci. 2024, 27, 433–448. [Google Scholar] [CrossRef]
- Schirmer, L.; Möbius, W.; Zhao, C.; Cruz-Herranz, A.; Ben Haim, L.; Cordano, C.; Shiow, L.R.; Kelley, K.W.; Sadowski, B.; Timmons, G.; et al. Oligodendrocyte-Encoded Kir4.1 Function Is Required for Axonal Integrity. eLife 2018, 7, e36428. [Google Scholar] [CrossRef] [PubMed]
- Larson, V.A.; Mironova, Y.; Vanderpool, K.G.; Waisman, A.; Rash, J.E.; Agarwal, A.; Bergles, D.E. Oligodendrocytes Control Potassium Accumulation in White Matter and Seizure Susceptibility. eLife 2018, 7, e34829. [Google Scholar] [CrossRef]
- Clarke, L.E.; Young, K.M.; Hamilton, N.B.; Li, H.; Richardson, W.D.; Attwell, D. Properties and Fate of Oligodendrocyte Progenitor Cells in the Corpus Callosum, Motor Cortex, and Piriform Cortex of the Mouse. J. Neurosci. 2012, 32, 8173–8185. [Google Scholar] [CrossRef] [PubMed]
- Tsata, V.; Kroehne, V.; Reinhardt, S.; El-Armouche, A.; Brand, M.; Wagner, M.; Reimer, M.M. Electrophysiological Properties of Adult Zebrafish Oligodendrocyte Progenitor Cells. Front. Cell. Neurosci. 2019, 13, 102. [Google Scholar] [CrossRef] [PubMed]
- Livesey, M.R.; Magnani, D.; Cleary, E.M.; Vasistha, N.A.; James, O.T.; Selvaraj, B.T.; Burr, K.; Story, D.; Shaw, C.E.; Kind, P.C.; et al. Maturation and Electrophysiological Properties of Human Pluripotent Stem Cell-Derived Oligodendrocytes. Stem Cells 2016, 34, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Káradóttir, R.; Hamilton, N.B.; Bakiri, Y.; Attwell, D. Spiking and Nonspiking Classes of Oligodendrocyte Precursor Glia in CNS White Matter. Nat. Neurosci. 2008, 11, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lynch, D.T.; Schools, G.P.; Feustel, P.J.; Kimelberg, H.K.; Zhou, M. Sodium Channel Currents in Rat Hippocampal NG2 Glia: Characterization and Contribution to Resting Membrane Potential. Neuroscience 2007, 150, 853–862. [Google Scholar] [CrossRef]
- Paez, P.M.; Fulton, D.; Colwell, C.S.; Campagnoni, A.T. Voltage-Operated Ca(2+) and Na(+) Channels in the Oligodendrocyte Lineage. J. Neurosci. Res. 2009, 87, 3259–3266. [Google Scholar] [CrossRef]
- Cheli, V.T.; Santiago González, D.A.; Spreuer, V.; Paez, P.M. Voltage-Gated Ca2+ Entry Promotes Oligodendrocyte Progenitor Cell Maturation and Myelination in Vitro. Exp. Neurol. 2015, 265, 69–83. [Google Scholar] [CrossRef]
- Simpson, P.B.; Armstrong, R.C. Intracellular Signals and Cytoskeletal Elements Involved in Oligodendrocyte Progenitor Migration. Glia 1999, 26, 22–35. [Google Scholar] [CrossRef]
- Paez, P.M.; Fulton, D.J.; Spreur, V.; Handley, V.; Campagnoni, A.T. Multiple Kinase Pathways Regulate Voltage-Dependent Ca2+ Influx and Migration in Oligodendrocyte Precursor Cells. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 6422–6433. [Google Scholar] [CrossRef]
- Habermacher, C.; Angulo, M.C.; Benamer, N. Glutamate versus GABA in Neuron–Oligodendroglia Communication. Glia 2019, 67, 2092–2106. [Google Scholar] [CrossRef] [PubMed]
- Arellano, R.O.; Sánchez-Gómez, M.V.; Alberdi, E.; Canedo-Antelo, M.; Chara, J.C.; Palomino, A.; Pérez-Samartín, A.; Matute, C. Axon-to-Glia Interaction Regulates GABAA Receptor Expression in Oligodendrocytes. Mol. Pharmacol. 2016, 89, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Swanson, G.T.; Kamboj, S.K.; Cull-Candy, S.G. Single-Channel Properties of Recombinant AMPA Receptors Depend on RNA Editing, Splice Variation, and Subunit Composition. J. Neurosci. Off. J. Soc. Neurosci. 1997, 17, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Zonouzi, M.; Scafidi, J.; Li, P.; McEllin, B.; Edwards, J.; Dupree, J.L.; Harvey, L.; Sun, D.; Hübner, C.A.; Cull-Candy, S.G.; et al. GABAergic Regulation of Cerebellar NG2 Cell Development Is Altered in Perinatal White Matter Injury. Nat. Neurosci. 2015, 18, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Nacher, J.; McEwen, B.S. The Role of N-Methyl-D-Asparate Receptors in Neurogenesis. Hippocampus 2006, 16, 267–270. [Google Scholar] [CrossRef]
- Gallo, V.; Zhou, J.; McBain, C.; Wright, P.; Knutson, P.; Armstrong, R. Oligodendrocyte Progenitor Cell Proliferation and Lineage Progression Are Regulated by Glutamate Receptor-Mediated K+ Channel Block. J. Neurosci. 1996, 16, 2659–2670. [Google Scholar] [CrossRef]
- Kougioumtzidou, E.; Shimizu, T.; Hamilton, N.B.; Tohyama, K.; Sprengel, R.; Monyer, H.; Attwell, D.; Richardson, W.D. Signalling through AMPA Receptors on Oligodendrocyte Precursors Promotes Myelination by Enhancing Oligodendrocyte Survival. eLife 2017, 6, e28080. [Google Scholar] [CrossRef]
- Erb, L.; Liao, Z.; Seye, C.I.; Weisman, G.A. P2 Receptors: Intracellular Signaling. Pflugers Arch. 2006, 452, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Agresti, C.; Meomartini, M.E.; Amadio, S.; Ambrosini, E.; Serafini, B.; Franchini, L.; Volonté, C.; Aloisi, F.; Visentin, S. Metabotropic P2 Receptor Activation Regulates Oligodendrocyte Progenitor Migration and Development. Glia 2005, 50, 132–144. [Google Scholar] [CrossRef]
- Wang, L.-Y.; Cai, W.-Q.; Chen, P.-H.; Deng, Q.-Y.; Zhao, C.-M. Downregulation of P2X7 Receptor Expression in Rat Oligodendrocyte Precursor Cells after Hypoxia Ischemia. Glia 2009, 57, 307–319. [Google Scholar] [CrossRef]
- Friess, M.; Hammann, J.; Unichenko, P.; Luhmann, H.J.; White, R.; Kirischuk, S. Intracellular Ion Signaling Influences Myelin Basic Protein Synthesis in Oligodendrocyte Precursor Cells. Cell Calcium 2016, 60, 322–330. [Google Scholar] [CrossRef]
- Zhang, S.-Z.; Wang, Q.-Q.; Yang, Q.-Q.; Gu, H.-Y.; Yin, Y.-Q.; Li, Y.-D.; Hou, J.-C.; Chen, R.; Sun, Q.-Q.; Sun, Y.-F.; et al. NG2 Glia Regulate Brain Innate Immunity via TGF-Β2/TGFBR2 Axis. BMC Med. 2019, 17, 204. [Google Scholar] [CrossRef]
- Yu, Y.; Yu, Z.; Xie, M.; Wang, W.; Luo, X. Hv1 Proton Channel Facilitates Production of ROS and Pro-Inflammatory Cytokines in Microglia and Enhances Oligodendrocyte Progenitor Cells Damage from Oxygen-Glucose Deprivation in Vitro. Biochem. Biophys. Res. Commun. 2018, 498, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Luo, Y.; Liao, P.; Zuo, Y.; Jiang, R. Role of the Voltage-Gated Proton Channel Hv1 in Nervous Systems. Neurosci. Bull. 2023, 39, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A Comprehensive Map of Molecular Drug Targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, L.; Srivastava, R.; Kalluri, S.R.; Böttinger, S.; Herwerth, M.; Carassiti, D.; Srivastava, B.; Gempt, J.; Schlegel, J.; Kuhlmann, T.; et al. Differential Loss of KIR4.1 Immunoreactivity in Multiple Sclerosis Lesions. Ann. Neurol. 2014, 75, 810–828. [Google Scholar] [CrossRef]
- Velasco-Estevez, M.; Koch, N.; Klejbor, I.; Caratis, F.; Rutkowska, A. Mechanoreceptor Piezo1 Is Downregulated in Multiple Sclerosis Brain and Is Involved in the Maturation and Migration of Oligodendrocytes in Vitro. Front. Cell. Neurosci. 2022, 16, 914985. [Google Scholar] [CrossRef] [PubMed]
- Craner, M.J.; Hains, B.C.; Lo, A.C.; Black, J.A.; Waxman, S.G. Co-Localization of Sodium Channel Nav1.6 and the Sodium-Calcium Exchanger at Sites of Axonal Injury in the Spinal Cord in EAE. Brain J. Neurol. 2004, 127, 294–303. [Google Scholar] [CrossRef]
- Casamassa, A.; La Rocca, C.; Sokolow, S.; Herchuelz, A.; Matarese, G.; Annunziato, L.; Boscia, F. Ncx3 Gene Ablation Impairs Oligodendrocyte Precursor Response and Increases Susceptibility to Experimental Autoimmune Encephalomyelitis. Glia 2016, 64, 1124–1137. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.K.; Nashmi, R.; Fehlings, M.G. Role of L- and N-Type Calcium Channels in the Pathophysiology of Traumatic Spinal Cord White Matter Injury. Neuroscience 2000, 99, 179–188. [Google Scholar] [CrossRef]
- Peric, M.; Nikolic, L.; Andjus, P.R.; Bataveljic, D. Dysfunction of Oligodendrocyte Inwardly Rectifying Potassium Channel in a Rat Model of Amyotrophic Lateral Sclerosis. Eur. J. Neurosci. 2021, 54, 6339–6354. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Wu, D.; Zhan, Y.; Li, F.; Zang, Y.-Y.; Teng, X.-Y.; Zhang, L.; Duan, G.-F.; Wang, H.; Xu, R.; et al. Cation Channel TMEM63A Autonomously Facilitates Oligodendrocyte Differentiation at an Early Stage. Neurosci. Bull. 2025, 41, 615–632. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wu, Z.; Wang, D.; Qu, Y.; Zhang, J.; Jiang, R.; Xu, X.; Xu, X.; Wang, Y.; Liu, H.; et al. Myelin-Associated Oligodendrocytic Basic Protein-Dependent Myelin Repair Confers the Long-Lasting Antidepressant Effect of Ketamine. Mol. Psychiatry 2024, 29, 1741–1753. [Google Scholar] [CrossRef]
- Yeung, M.S.Y.; Djelloul, M.; Steiner, E.; Bernard, S.; Salehpour, M.; Possnert, G.; Brundin, L.; Frisén, J. Dynamics of Oligodendrocyte Generation in Multiple Sclerosis. Nature 2019, 566, 538–542. [Google Scholar] [CrossRef]
- Tepavčević, V.; Lubetzki, C. Oligodendrocyte Progenitor Cell Recruitment and Remyelination in Multiple Sclerosis: The More, the Merrier? Brain J. Neurol. 2022, 145, 4178–4192. [Google Scholar] [CrossRef]
- Srivastava, R.; Aslam, M.; Kalluri, S.R.; Schirmer, L.; Buck, D.; Tackenberg, B.; Rothhammer, V.; Chan, A.; Gold, R.; Berthele, A.; et al. Potassium Channel KIR4.1 as an Immune Target in Multiple Sclerosis. N. Engl. J. Med. 2012, 367, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Kraus, V.; Srivastava, R.; Kalluri, S.R.; Seidel, U.; Schuelke, M.; Schimmel, M.; Rostasy, K.; Leiz, S.; Hosie, S.; Grummel, V.; et al. Potassium Channel KIR4.1-Specific Antibodies in Children with Acquired Demyelinating CNS Disease. Neurology 2014, 82, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.; Frisullo, G.; Di Sante, G.; Samengo, D.M.; Provenzano, C.; Mirabella, M.; Pani, G.; Ria, F.; Bartoccioni, E. Low Reliability of Anti-KIR4.183-120 Peptide Auto-Antibodies in Multiple Sclerosis Patients. Mult. Scler. Houndmills Basingstoke Engl. 2018, 24, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Chastre, A.; Hafler, D.A.; O’Connor, K.C. Evaluation of KIR4.1 as an Immune Target in Multiple Sclerosis. N. Engl. J. Med. 2016, 374, 1495–1496. [Google Scholar] [CrossRef] [PubMed]
- Malyavantham, K.; Weinstock-Guttman, B.; Suresh, L.; Zivadinov, R.; Shanahan, T.; Badgett, D.; Ramanathan, M. Humoral Responses to Diverse Autoimmune Disease-Associated Antigens in Multiple Sclerosis. PLoS ONE 2015, 10, e0129503. [Google Scholar] [CrossRef]
- Black, J.A.; Waxman, S.G.; Smith, K.J. Remyelination of Dorsal Column Axons by Endogenous Schwann Cells Restores the Normal Pattern of Nav1.6 and Kv1.2 at Nodes of Ranvier. Brain 2006, 129, 1319–1329. [Google Scholar] [CrossRef]
- Enders, M.; Weier, A.; Chunder, R.; An, Y.; Bremm, F.; Feigenspan, A.; Buettner, C.; Ekici, A.B.; Mingardo, E.; Odermatt, B.; et al. Impact of the Voltage-Gated Calcium Channel Antagonist Nimodipine on the Development of Oligodendrocyte Precursor Cells. Int. J. Mol. Sci. 2023, 24, 3716. [Google Scholar] [CrossRef] [PubMed]
- Ingwersen, J.; De Santi, L.; Wingerath, B.; Graf, J.; Koop, B.; Schneider, R.; Hecker, C.; Schröter, F.; Bayer, M.; Engelke, A.D.; et al. Nimodipine Confers Clinical Improvement in Two Models of Experimental Autoimmune Encephalomyelitis. J. Neurochem. 2018, 146, 86–98. [Google Scholar] [CrossRef]
- Dąbrowska-Bouta, B.; Strużyńska, L.; Chalimoniuk, M.; Frontczak-Baniewicz, M.; Sulkowski, G. The Influence of Glutamatergic Receptor Antagonists on Biochemical and Ultrastructural Changes in Myelin Membranes of Rats Subjected to Experimental Autoimmune Encephalomyelitis. Folia Neuropathol. 2015, 53, 317–326. [Google Scholar] [CrossRef]
- Wallström, E.; Diener, P.; Ljungdahl, A.; Khademi, M.; Nilsson, C.G.; Olsson, T. Memantine Abrogates Neurological Deficits, but Not CNS Inflammation, in Lewis Rat Experimental Autoimmune Encephalomyelitis. J. Neurol. Sci. 1996, 137, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Cunha, M.I.; Su, M.; Cantuti-Castelvetri, L.; Müller, S.A.; Schifferer, M.; Djannatian, M.; Alexopoulos, I.; van der Meer, F.; Winkler, A.; van Ham, T.J.; et al. Pro-Inflammatory Activation Following Demyelination Is Required for Myelin Clearance and Oligodendrogenesis. J. Exp. Med. 2020, 217, e20191390. [Google Scholar] [CrossRef] [PubMed]
- Antel, J.P.; Kennedy, T.E.; Kuhlmann, T. Seeking Neuroprotection in Multiple Sclerosis: An Ongoing Challenge. J. Clin. Investig. 2023, 133, e168595. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Brody, D.L.; Kochanek, P.M.; Levin, H.; McKee, A.; Ribbers, G.M.; Yaffe, K.; Zetterberg, H. Traumatic Brain Injuries. Nat. Rev. Dis. Primer 2016, 2, 16084. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.-Y.; Gao, G.-Y.; Feng, J.-F.; Mao, Q.; Chen, L.-G.; Yang, X.-F.; Liu, J.-F.; Wang, Y.-H.; Qiu, B.-H.; Huang, X.-J. Traumatic Brain Injury in China. Lancet Neurol. 2019, 18, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Dent, K.A.; Christie, K.J.; Bye, N.; Basrai, H.S.; Turbic, A.; Habgood, M.; Cate, H.S.; Turnley, A.M. Oligodendrocyte Birth and Death Following Traumatic Brain Injury in Adult Mice. PLoS ONE 2015, 10, e0121541. [Google Scholar] [CrossRef]
- Kabdesh, I.M.; Arkhipova, S.S.; Mukhamedshina, Y.O.; James, V.; Rizvanov, A.A.; Chelyshev, Y.A. The Function of NG2/CSPG4-Expressing Cells in the Rat Spinal Cord Injury: An Immunoelectron Microscopy Study. Neuroscience 2021, 467, 142–149. [Google Scholar] [CrossRef]
- Kabdesh, I.M.; Mukhamedshina, Y.O.; Arkhipova, S.S.; Sabirov, D.K.; Kuznecov, M.S.; Vyshtakalyuk, A.B.; Rizvanov, A.A.; James, V.; Chelyshev, Y.A. Cellular and Molecular Gradients in the Ventral Horns With Increasing Distance From the Injury Site After Spinal Cord Contusion. Front. Cell. Neurosci. 2022, 16, 817752. [Google Scholar] [CrossRef]
- Hampton, D.W.; Rhodes, K.E.; Zhao, C.; Franklin, R.J.M.; Fawcett, J.W. The Responses of Oligodendrocyte Precursor Cells, Astrocytes and Microglia to a Cortical Stab Injury, in the Brain. Neuroscience 2004, 127, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Verkhratsky, A.; Niu, J. Pathological Potential of Oligodendrocyte Precursor Cells: Terra Incognita. Trends Neurosci. 2023, 46, 581–596. [Google Scholar] [CrossRef]
- Flygt, J.; Clausen, F.; Marklund, N. Diffuse Traumatic Brain Injury in the Mouse Induces a Transient Proliferation of Oligodendrocyte Progenitor Cells in Injured White Matter Tracts. Restor. Neurol. Neurosci. 2017, 35, 251–263. [Google Scholar] [CrossRef]
- Michalettos, G.; Clausen, F.; Özen, I.; Ruscher, K.; Marklund, N. Impaired Oligodendrogenesis in the White Matter of Aged Mice Following Diffuse Traumatic Brain Injury. Glia 2024, 72, 728–747. [Google Scholar] [CrossRef] [PubMed]
- Barbierato, M.; Facci, L.; Marinelli, C.; Zusso, M.; Argentini, C.; Skaper, S.D.; Giusti, P. Co-Ultramicronized Palmitoylethanolamide/Luteolin Promotes the Maturation of Oligodendrocyte Precursor Cells. Sci. Rep. 2015, 5, 16676. [Google Scholar] [CrossRef] [PubMed]
- Paterniti, I.; Impellizzeri, D.; Di Paola, R.; Navarra, M.; Cuzzocrea, S.; Esposito, E. A New Co-Ultramicronized Composite Including Palmitoylethanolamide and Luteolin to Prevent Neuroinflammation in Spinal Cord Injury. J. Neuroinflamm. 2013, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Shumilov, K.; Xiao, S.; Ni, A.; Celorrio, M.; Friess, S.H. Recombinant Erythropoietin Induces Oligodendrocyte Progenitor Cell Proliferation After Traumatic Brain Injury and Delayed Hypoxemia. Neurotherapeutics 2023, 20, 1859–1874. [Google Scholar] [CrossRef] [PubMed]
- Giacino, J.T.; Whyte, J.; Bagiella, E.; Kalmar, K.; Childs, N.; Khademi, A.; Eifert, B.; Long, D.; Katz, D.I.; Cho, S.; et al. Placebo-Controlled Trial of Amantadine for Severe Traumatic Brain Injury. N. Engl. J. Med. 2012, 366, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Corps, K.N.; Roth, T.L.; McGavern, D.B. Inflammation and Neuroprotection in Traumatic Brain Injury. JAMA Neurol. 2015, 72, 355–362. [Google Scholar] [CrossRef] [PubMed]
- De Biase, L.M.; Kang, S.H.; Baxi, E.G.; Fukaya, M.; Pucak, M.L.; Mishina, M.; Calabresi, P.A.; Bergles, D.E. NMDA Receptor Signaling in Oligodendrocyte Progenitors Is Not Required for Oligodendrogenesis and Myelination. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 12650–12662. [Google Scholar] [CrossRef] [PubMed]
- Nourbakhsh, B.; Revirajan, N.; Morris, B.; Cordano, C.; Creasman, J.; Manguinao, M.; Krysko, K.; Rutatangwa, A.; Auvray, C.; Aljarallah, S.; et al. Safety and Efficacy of Amantadine, Modafinil, and Methylphenidate for Fatigue in Multiple Sclerosis: A Randomised, Placebo-Controlled, Crossover, Double-Blind Trial. Lancet Neurol. 2021, 20, 38–48. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, M.; Cai, X.; Zhang, M.; Hu, M.; Hu, Y.; Yang, Y.; Zhu, J.; Du, Y.; Yang, C. Detection and Analysis of Signals of Adverse Events of Memantine Based on the US Food and Drug Administration Adverse Event Reporting System. Expert Opin. Drug Saf. 2024, 23, 617–625. [Google Scholar] [CrossRef]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic Lateral Sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, D.; Lv, C.; Gao, Z.; Qi, Y.; Wu, H.; Tian, Y.; Guo, Y. Activation of the Notch Signaling Pathway and Cellular Localization of Notch Signaling Molecules in the Spinal Cord of SOD1-G93A ALS Model Mice. Neuroscience 2020, 432, 84–93. [Google Scholar] [CrossRef]
- Lorente Pons, A.; Higginbottom, A.; Cooper-Knock, J.; Alrafiah, A.; Alofi, E.; Kirby, J.; Shaw, P.J.; Wood, J.D.; Highley, J.R. Oligodendrocyte Pathology Exceeds Axonal Pathology in White Matter in Human Amyotrophic Lateral Sclerosis. J. Pathol. 2020, 251, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chung, A.-Y.; Na, J.E.; Lee, S.J.; Jeong, S.H.; Kim, E.; Sun, W.; Rhyu, I.J.; Park, H.-C. Myelin Degeneration Induced by Mutant Superoxide Dismutase 1 Accumulation Promotes Amyotrophic Lateral Sclerosis. Glia 2019, 67, 1910–1921. [Google Scholar] [CrossRef]
- Hayes, K.C. The Use of 4-Aminopyridine (Fampridine) in Demyelinating Disorders. CNS Drug Rev. 2004, 10, 295–316. [Google Scholar] [CrossRef]
- Zhu, Y.; Burg, T.; Neyrinck, K.; Vervliet, T.; Nami, F.; Vervoort, E.; Ahuja, K.; Sassano, M.L.; Chai, Y.C.; Tharkeshwar, A.K.; et al. Disruption of MAM Integrity in Mutant FUS Oligodendroglial Progenitors from hiPSCs. Acta Neuropathol. 2024, 147, 6. [Google Scholar] [CrossRef] [PubMed]
- Better, M.A. Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2023, 19, 1598–1695. [Google Scholar] [CrossRef]
- van der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-Independent Regulators of Tau Pathology in Alzheimer Disease. Nat. Rev. Neurosci. 2020, 21, 21–35. [Google Scholar] [CrossRef]
- Hanseeuw, B.J.; Betensky, R.A.; Jacobs, H.I.L.; Schultz, A.P.; Sepulcre, J.; Becker, J.A.; Cosio, D.M.O.; Farrell, M.; Quiroz, Y.T.; Mormino, E.C.; et al. Association of Amyloid and Tau With Cognition in Preclinical Alzheimer Disease. JAMA Neurol. 2019, 76, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Quintela-López, T.; Ortiz-Sanz, C.; Serrano-Regal, M.P.; Gaminde-Blasco, A.; Valero, J.; Baleriola, J.; Sánchez-Gómez, M.V.; Matute, C.; Alberdi, E. Aβ Oligomers Promote Oligodendrocyte Differentiation and Maturation via Integrin Β1 and Fyn Kinase Signaling. Cell Death Dis. 2019, 10, 445. [Google Scholar] [CrossRef]
- Qiu, S.; Palavicini, J.P.; Wang, J.; Gonzalez, N.S.; He, S.; Dustin, E.; Zou, C.; Ding, L.; Bhattacharjee, A.; Van Skike, C.E.; et al. Adult-Onset CNS Myelin Sulfatide Deficiency Is Sufficient to Cause Alzheimer’s Disease-like Neuroinflammation and Cognitive Impairment. Mol. Neurodegener. 2021, 16, 64. [Google Scholar] [CrossRef] [PubMed]
- Ihara, M.; Polvikoski, T.M.; Hall, R.; Slade, J.Y.; Perry, R.H.; Oakley, A.E.; Englund, E.; O’Brien, J.T.; Ince, P.G.; Kalaria, R.N. Quantification of Myelin Loss in Frontal Lobe White Matter in Vascular Dementia, Alzheimer’s Disease, and Dementia with Lewy Bodies. Acta Neuropathol. 2010, 119, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, G.; Baer, K.; Buffo, A.; Curtis, M.A.; Faull, R.L.; Rees, M.I.; Götz, M.; Dimou, L. Dynamic Changes in Myelin Aberrations and Oligodendrocyte Generation in Chronic Amyloidosis in Mice and Men. Glia 2013, 61, 273–286. [Google Scholar] [CrossRef]
- Nielsen, H.M.; Ek, D.; Avdic, U.; Orbjörn, C.; Hansson, O.; Netherlands Brain Bank; Veerhuis, R.; Rozemuller, A.J.; Brun, A.; Minthon, L.; et al. NG2 Cells, a New Trail for Alzheimer’s Disease Mechanisms? Acta Neuropathol. Commun. 2013, 1, 7. [Google Scholar] [CrossRef]
- Chacon-De-La-Rocha, I.; Fryatt, G.; Rivera, A.D.; Verkhratsky, A.; Raineteau, O.; Gomez-Nicola, D.; Butt, A.M. Accelerated Dystrophy and Decay of Oligodendrocyte Precursor Cells in the APP/PS1 Model of Alzheimer’s-Like Pathology. Front. Cell. Neurosci. 2020, 14, 575082. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, Q.; Zhu, H.; Hou, L.; Liu, W.; Yang, Q.; Shen, H.; Chai, G.; Zhang, B.; Chen, S.; et al. Microglial Piezo1 Senses Aβ Fibril Stiffness to Restrict Alzheimer’s Disease. Neuron 2023, 111, 15–29.e8. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Guo, Y.; Fang, J.; Shi, C.; Suo, N.; Zhang, R.; Xie, X. Donepezil, a Drug for Alzheimer’s Disease, Promotes Oligodendrocyte Generation and Remyelination. Acta Pharmacol. Sin. 2019, 40, 1386–1393. [Google Scholar] [CrossRef]
- Imamura, O.; Arai, M.; Dateki, M.; Ogata, T.; Uchida, R.; Tomoda, H.; Takishima, K. Nicotinic Acetylcholine Receptors Mediate Donepezil-Induced Oligodendrocyte Differentiation. J. Neurochem. 2015, 135, 1086–1098. [Google Scholar] [CrossRef] [PubMed]
- Fern, R.F.; Matute, C.; Stys, P.K. White Matter Injury: Ischemic and Nonischemic. Glia 2014, 62, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.G.; Fern, R. NMDA Receptors Are Expressed in Developing Oligodendrocyte Processes and Mediate Injury. Nature 2005, 438, 1167–1171. [Google Scholar] [CrossRef]
- Bakiri, Y.; Hamilton, N.B.; Káradóttir, R.; Attwell, D. Testing NMDA Receptor Block as a Therapeutic Strategy for Reducing Ischaemic Damage to CNS White Matter. Glia 2008, 56, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.M.; De La Rocha, I.C.; Rivera, A. Oligodendroglial Cells in Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1175, 325–333. [Google Scholar] [CrossRef]
- Mind Matters. Nature 2016, 532, 6. [CrossRef]
- Kolomeets, N.S.; Uranova, N.A. Numerical Density of Oligodendrocytes and Oligodendrocyte Clusters in the Anterior Putamen in Major Psychiatric Disorders. Eur. Arch. Psychiatry Clin. Neurosci. 2020, 270, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Uranova, N.A.; Vikhreva, O.V.; Rachmanova, V.I.; Orlovskaya, D.D. Ultrastructural Alterations of Myelinated Fibers and Oligodendrocytes in the Prefrontal Cortex in Schizophrenia: A Postmortem Morphometric Study. Schizophr. Res. Treat. 2011, 2011, e325789. [Google Scholar] [CrossRef]
- Uranova, N.A.; Vostrikov, V.M.; Orlovskaya, D.D.; Rachmanova, V.I. Oligodendroglial Density in the Prefrontal Cortex in Schizophrenia and Mood Disorders: A Study from the Stanley Neuropathology Consortium. Schizophr. Res. 2004, 67, 269–275. [Google Scholar] [CrossRef]
- Liu, S.-H.; Du, Y.; Chen, L.; Cheng, Y. Glial Cell Abnormalities in Major Psychiatric Diseases: A Systematic Review of Postmortem Brain Studies. Mol. Neurobiol. 2022, 59, 1665–1692. [Google Scholar] [CrossRef] [PubMed]
- Nagy, C.; Maitra, M.; Tanti, A.; Suderman, M.; Théroux, J.-F.; Davoli, M.A.; Perlman, K.; Yerko, V.; Wang, Y.C.; Tripathy, S.J.; et al. Single-Nucleus Transcriptomics of the Prefrontal Cortex in Major Depressive Disorder Implicates Oligodendrocyte Precursor Cells and Excitatory Neurons. Nat. Neurosci. 2020, 23, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yang, H.-J.; Li, X.-M. Differential Effects of Antipsychotics on the Development of Rat Oligodendrocyte Precursor Cells Exposed to Cuprizone. Eur. Arch. Psychiatry Clin. Neurosci. 2014, 264, 121–129. [Google Scholar] [CrossRef]
- Steiner, J.; Martins-de-Souza, D.; Schiltz, K.; Sarnyai, Z.; Westphal, S.; Isermann, B.; Dobrowolny, H.; Turck, C.W.; Bogerts, B.; Bernstein, H.-G.; et al. Clozapine Promotes Glycolysis and Myelin Lipid Synthesis in Cultured Oligodendrocytes. Front. Cell. Neurosci. 2014, 8, 384. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, H.; Niu, J.; Mei, F.; Li, X.; Kong, J.; Cai, W.; Xiao, L. Haloperidol Activates Quiescent Oligodendroglia Precursor Cells in the Adult Mouse Brain. Schizophr. Res. 2010, 119, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Luo, K.; Zhu, X.; Zheng, R.; Wang, Y.; Yu, G.; Wang, X.; She, F.; Chen, X.; Li, T.; et al. Fluoxetine Rescues Excessive Myelin Formation and Psychological Behaviors in a Murine PTSD Model. Neurosci. Bull. 2024, 40, 1037–1052. [Google Scholar] [CrossRef] [PubMed]
- Hilker, R.; Helenius, D.; Fagerlund, B.; Skytthe, A.; Christensen, K.; Werge, T.M.; Nordentoft, M.; Glenthøj, B. Heritability of Schizophrenia and Schizophrenia Spectrum Based on the Nationwide Danish Twin Register. Biol. Psychiatry 2018, 83, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Finnerup, N.B.; Kuner, R.; Jensen, T.S. Neuropathic Pain: From Mechanisms to Treatment. Physiol. Rev. 2021, 101, 259–301. [Google Scholar] [CrossRef]
- Tian, H.; Cheng, J.; Zhao, X.; Xia, Z. Exploring Causal Correlations between Blood Inflammatory Cytokines and Low Back Pain: A Mendelian Randomization. Anesthesiol. Perioper. Sci. 2024, 2, 25. [Google Scholar] [CrossRef]
- Gritsch, S.; Lu, J.; Thilemann, S.; Wörtge, S.; Möbius, W.; Bruttger, J.; Karram, K.; Ruhwedel, T.; Blanfeld, M.; Vardeh, D.; et al. Oligodendrocyte Ablation Triggers Central Pain Independently of Innate or Adaptive Immune Responses in Mice. Nat. Commun. 2014, 5, 5472. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.; Chan, L.; Flynn, S. A Systematic Review of the Incidence, Prevalence, Costs, and Activity and Work Limitations of Amputation, Osteoarthritis, Rheumatoid Arthritis, Back Pain, Multiple Sclerosis, Spinal Cord Injury, Stroke, and Traumatic Brain Injury in the United States: A 2019 Update. Arch. Phys. Med. Rehabil. 2021, 102, 115–131. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.W.; Sadowsky, C. Spinal-Cord Injury. Lancet 2002, 359, 417–425. [Google Scholar] [CrossRef]
- Yang, J.; Xiong, L.-L.; Wang, Y.-C.; He, X.; Jiang, L.; Fu, S.-J.; Han, X.-F.; Liu, J.; Wang, T.-H. Oligodendrocyte Precursor Cell Transplantation Promotes Functional Recovery Following Contusive Spinal Cord Injury in Rats and Is Associated with Altered microRNA Expression. Mol. Med. Rep. 2018, 17, 771–782. [Google Scholar] [CrossRef]
- Tao, F.; Li, Q.; Liu, S.; Wu, H.; Skinner, J.; Hurtado, A.; Belegu, V.; Furmanski, O.; Yang, Y.; McDonald, J.W.; et al. Role of Neuregulin-1/ErbB Signaling in Stem Cell Therapy for Spinal Cord Injury-Induced Chronic Neuropathic Pain. Stem Cells 2013, 31, 83–91. [Google Scholar] [CrossRef]
- Plemel, J.R.; Keough, M.B.; Duncan, G.J.; Sparling, J.S.; Yong, V.W.; Stys, P.K.; Tetzlaff, W. Remyelination after Spinal Cord Injury: Is It a Target for Repair? Prog. Neurobiol. 2014, 117, 54–72. [Google Scholar] [CrossRef]
- Yuan, X.; Han, S.; Manyande, A.; Gao, F.; Wang, J.; Zhang, W.; Tian, X. Spinal Voltage-Gated Potassium Channel Kv1.3 Contributes to Neuropathic Pain via the Promotion of Microglial M1 Polarization and Activation of the NLRP3 Inflammasome. Eur. J. Pain 2023, 27, 289–302. [Google Scholar] [CrossRef]
- Baggio, D.F.; Gambeta, E.; Souza, I.A.; Huang, S.; Zamponi, G.W.; Chichorro, J.G. CaV3.2 T-Type Calcium Channels Contribute to CGRP- Induced Allodynia in a Rodent Model of Experimental Migraine. J. Headache Pain 2024, 25, 219. [Google Scholar] [CrossRef] [PubMed]
- Ou, M.; Chen, Y.; Liu, J.; Zhang, D.; Yang, Y.; Shen, J.; Miao, C.; Tang, S.-J.; Liu, X.; Mulkey, D.K.; et al. Spinal Astrocytic MeCP2 Regulates Kir4.1 for the Maintenance of Chronic Hyperalgesia in Neuropathic Pain. Prog. Neurobiol. 2023, 224, 102436. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Lyu, D.; Gao, J. Suzetrigine: The First Nav1.8 Inhibitor Approved for the Treatment of Moderate to Severe Acute Pain. Drug Discov. Ther. 2025, 19, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Bannerman, C.A.; Douchant, K.; Segal, J.P.; Knezic, M.; Mack, A.E.; Lundell-Creagh, C.; Silva, J.R.; Duggan, S.; Sheth, P.; Ghasemlou, N. Spinal Cord Injury in Mice Affects Central and Peripheral Pathology in a Severity-Dependent Manner. Pain 2022, 163, 1172–1185. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yang, K.; Li, J.; Xu, X.; Gong, L.; Yue, S.; Wei, H.; Yue, Z.; Wu, Y.; Yin, S. Single-Cell Sequencing Reveals Glial Cell Involvement in Development of Neuropathic Pain via Myelin Sheath Lesion Formation in the Spinal Cord. J. Neuroinflamm. 2024, 21, 213. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Tsai, H.-H.; Hoi, K.K.; Huang, N.; Yu, G.; Kim, K.; Baranzini, S.E.; Xiao, L.; Chan, J.R.; Fancy, S.P.J. Aberrant Oligodendroglial-Vascular Interactions Disrupt the Blood-Brain Barrier, Triggering CNS Inflammation. Nat. Neurosci. 2019, 22, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Huang, L.; Fu, J.; Han, C.; Fang, J.; Liao, P.; Chen, Z.; Mo, Y.; Sun, P.; Liao, D.; et al. TREK Channel Family Activator with a Well-Defined Structure-Activation Relationship for Pain and Neurogenic Inflammation. J. Med. Chem. 2020, 63, 3665–3677. [Google Scholar] [CrossRef]
- Djillani, A.; Pietri, M.; Mazella, J.; Heurteaux, C.; Borsotto, M. Fighting against Depression with TREK-1 Blockers: Past and Future. A Focus on Spadin. Pharmacol. Ther. 2019, 194, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Castelo-Branco, G.; Kukanja, P.; Guerreiro-Cacais, A.O.; Rubio Rodríguez-Kirby, L.A. Disease-Associated Oligodendroglia: A Putative Nexus in Neurodegeneration. Trends Immunol. 2024, 45, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Thornton, M.A.; Futia, G.L.; Stockton, M.E.; Budoff, S.A.; Ramirez, A.N.; Ozbay, B.; Tzang, O.; Kilborn, K.; Poleg-Polsky, A.; Restrepo, D.; et al. Long-Term in Vivo Three-Photon Imaging Reveals Region-Specific Differences in Healthy and Regenerative Oligodendrogenesis. Nat. Neurosci. 2024, 27, 846–861. [Google Scholar] [CrossRef]
| Diseases | Associated Ion Channel | Supporting Experimental/ Clinical Evidence | Potentially Associated Ion Channel | Supporting Evidence for Potential Associations |
|---|---|---|---|---|
| Multiple Sclerosis | Kir4.1 | In MS lesions, a selective loss of Kir4.1 immunoreactivity specifically on oligodendrocytes is evident [88]. OL-expressed Kir4.1 plays a crucial role in maintaining white matter integrity during demyelinating injury [62]. | Piezo1 | The expression of Piezo1 in the white matter is markedly reduced in the brain affected by multiple sclerosis [89]. |
| Nav1.2, Nav1.6 | Upregulation of Nav1.2 and Nav1.6 has been consistently observed in demyelinated axonal segments within the experimental autoimmune encephalomyelitis (EAE) model [90]. | |||
| NCX3 | Deletion of NCX3 resulted in impaired oligodendrocyte myelination and exacerbated the clinical symptoms of EAE [91]. | |||
| Traumatic Injury of Central Nervous System | None | None | Cav | Traumatic white matter injury is associated with the influx of calcium ions through Cav expressed on astrocytes and oligodendrocytes surrounding the axons [92]. |
| Amyotrophic lateral sclerosis | Kir4.1 | The SOD1G93A rat model has revealed a downregulation and impaired functionality of Kir4.1 in oligodendrocytes [93]. | None | None |
| Alzheimer’s disease | None | None | Piezo1 | The specific inhibition of Piezo1 in aging animals significantly promotes the proliferation and differentiation of OPCs [50]. |
| Psychiatric disorders | TMEM63A | TMEM63A deficiency disrupts OPC differentiation and is associated with ASD and ADHD [94]. | ||
| AMPARs | Ketamine attenuates CSDS-induced myelin loss and depression-like behavior by acting on AMPARs expressed in OPCs [95]. | |||
| Neuropathic pain | None |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Shen, Y.; Liao, P.; Yang, B.; Jiang, R. Roles of Ion Channels in Oligodendrocyte Precursor Cells: From Physiology to Pathology. Int. J. Mol. Sci. 2025, 26, 7336. https://doi.org/10.3390/ijms26157336
Wang J, Shen Y, Liao P, Yang B, Jiang R. Roles of Ion Channels in Oligodendrocyte Precursor Cells: From Physiology to Pathology. International Journal of Molecular Sciences. 2025; 26(15):7336. https://doi.org/10.3390/ijms26157336
Chicago/Turabian StyleWang, Jianing, Yu Shen, Ping Liao, Bowen Yang, and Ruotian Jiang. 2025. "Roles of Ion Channels in Oligodendrocyte Precursor Cells: From Physiology to Pathology" International Journal of Molecular Sciences 26, no. 15: 7336. https://doi.org/10.3390/ijms26157336
APA StyleWang, J., Shen, Y., Liao, P., Yang, B., & Jiang, R. (2025). Roles of Ion Channels in Oligodendrocyte Precursor Cells: From Physiology to Pathology. International Journal of Molecular Sciences, 26(15), 7336. https://doi.org/10.3390/ijms26157336