
Distinct Mitochondrial DNA Deletion Profiles in Pediatric B- and T-ALL During Diagnosis, Remission, and Relapse
 , , ,
, , ,
Abstract
1. Introduction
2. Results
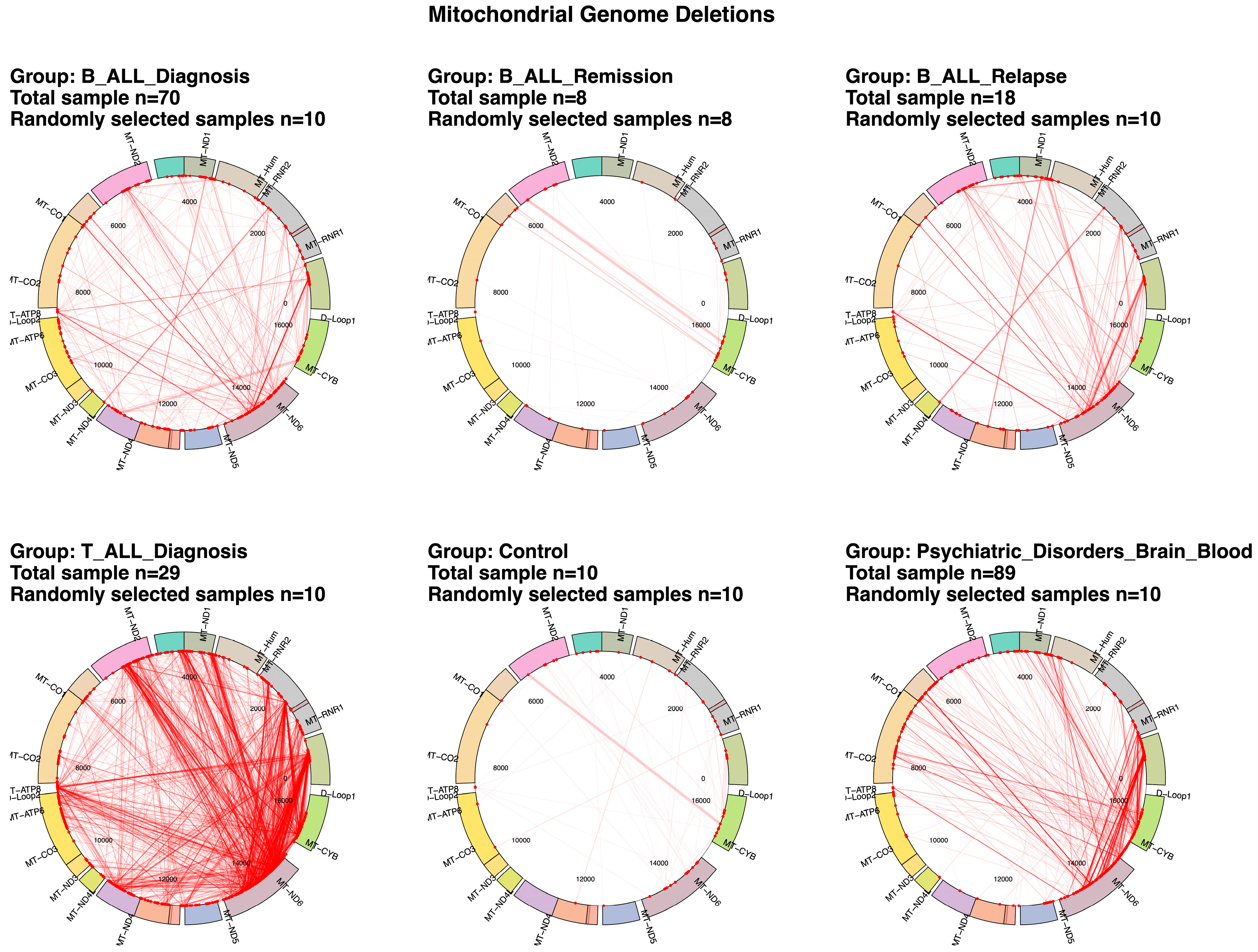
2.1. Differential mtDNA Deletion Landscapes in Acute Lymphoblastic Leukemia Subtypes and Control Groups
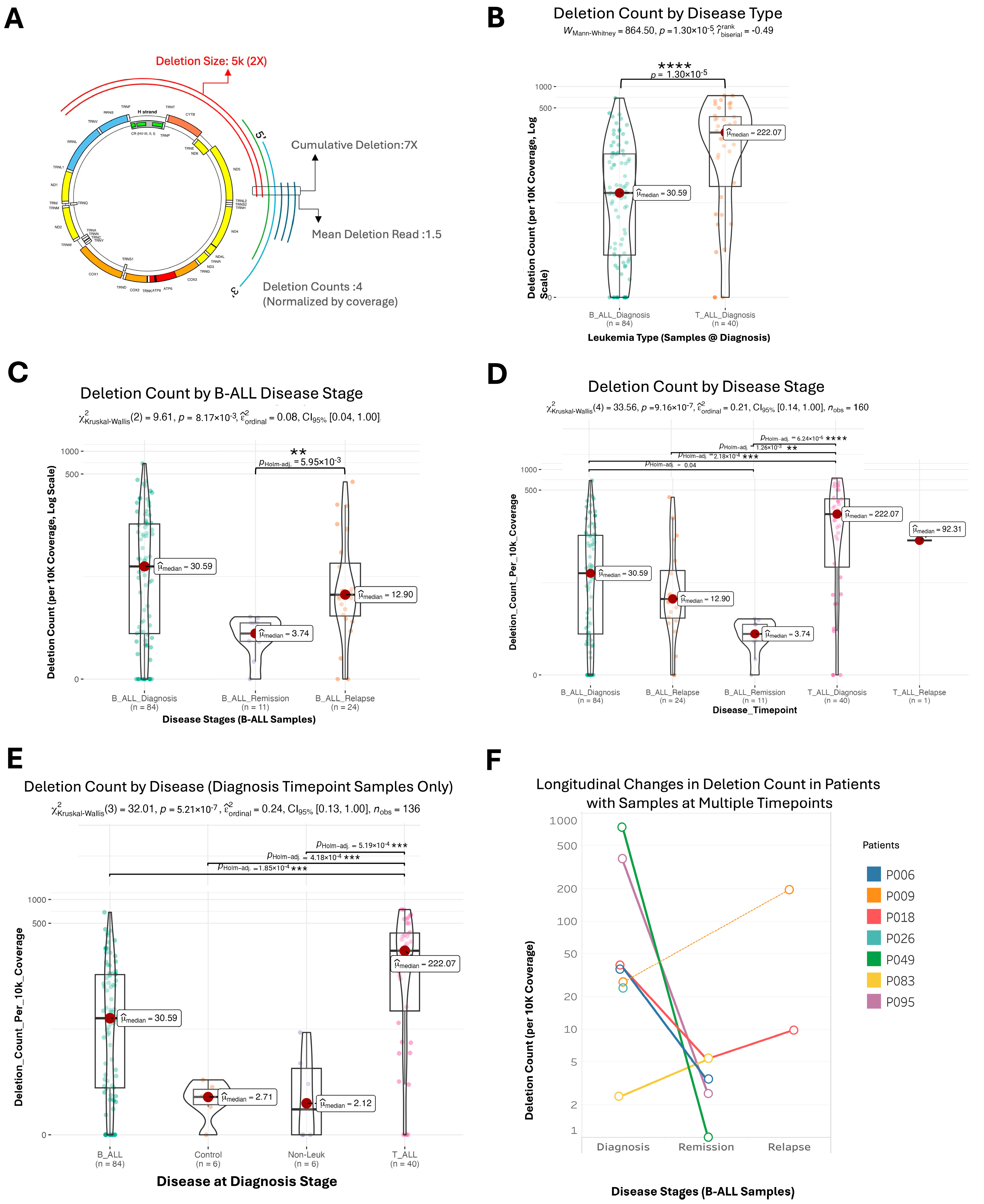
2.2. Mitochondrial Deletion Profiles Reveal Dynamic Patterns Across Disease Stages, Tumor Types, and Subtypes in Acute Lymphoblastic Leukemia
2.3. Variability and Characteristics of mtDNA Deletions in Pediatric Acute Lymphoblastic Leukemia
2.4. Clusters of Large Mitochondrial DNA Deletions in Pediatric Patients with B-ALL and T-ALL
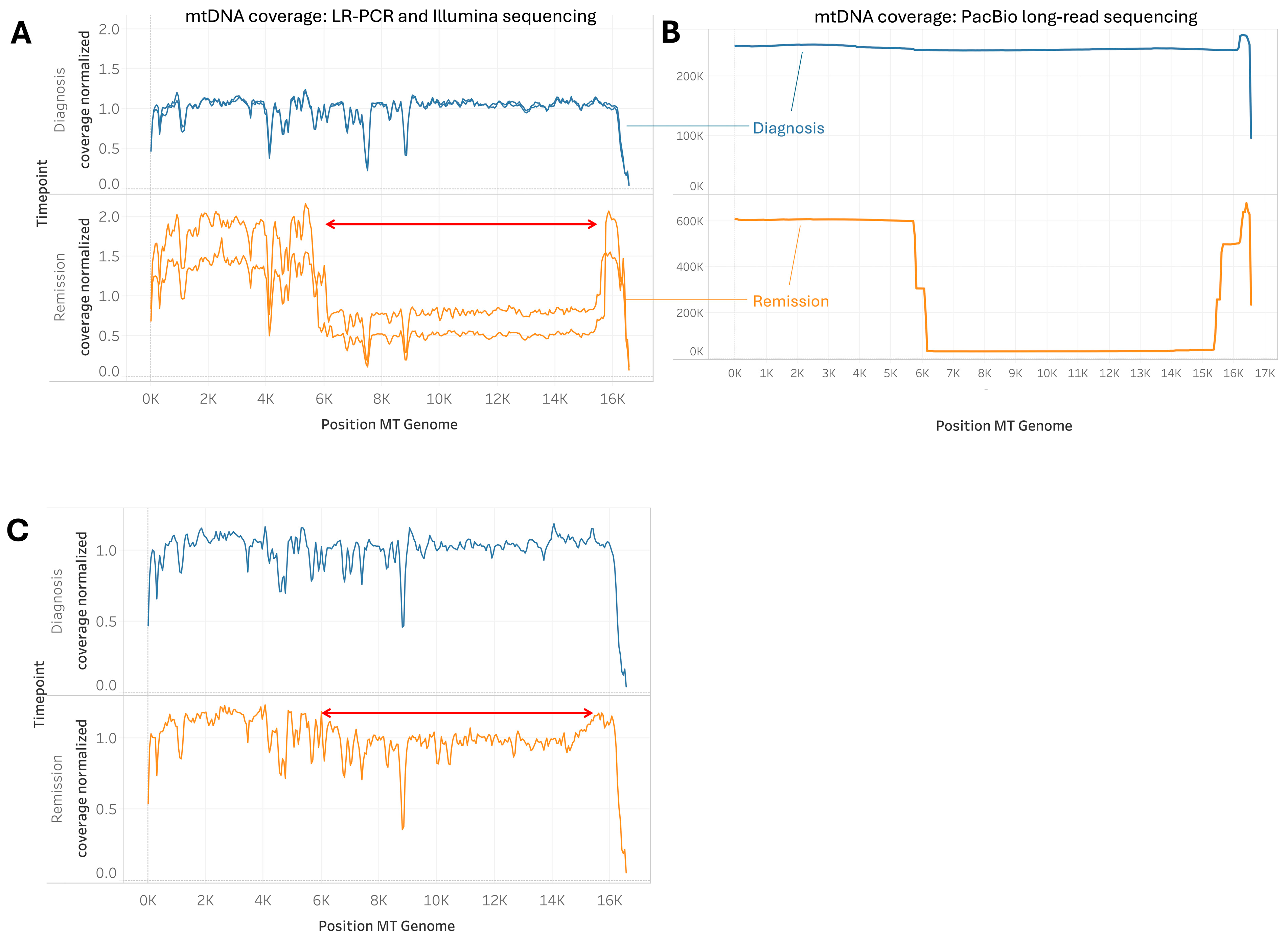
2.5. Cases of Unusually Large mtDNA Deletion During Remission
2.6. Common and Distinct Repeat Patterns at mtDNA Deletion Breakpoints in B-ALL and T-ALL
2.7. MT-ND5 as the Gene Most Frequently Implicated by mtDNA Deletions in Leukemia
2.8. Multivariate Analysis of Variance (MANOVA) Reveals Significant Effects of Disease, Timepoint, and WHO Subtype on Mitochondrial DNA Deletion Metrics
2.9. Patterns of mtDNA SNVs and Indels in B-ALL and T-ALL
3. Discussion
3.1. Mitochondrial DNA Deletion Profiles in B-ALL and T-ALL
3.2. Possible Mechanisms Underlying Mtdna Deletions
3.3. Implications of Large Mtdna Deletions During Remission
3.4. Mitochondrial DNA Deletion Patterns as Biomarkers of Disease Progression
4. Materials and Methods
4.1. Patient Samples and Study Design
4.2. DNA Extraction
4.3. Mitochondrial DNA Sequencing
4.4. Bioinformatic Analysis
4.4.1. Mitochondrial DNA Deletions
4.4.2. Mitochondrial DNA SNVs and Indels
4.4.3. Statistical Analysis
4.4.4. Repeat Pattern Analysis
4.4.5. Comparative Analysis of Deletion Profiles
4.4.6. Distance-Based Clustering and Ordination
4.5. Code Availability
4.6. Use of Large Language Model for Proof-Editing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| B-ALL | B-cell acute lymphoblastic leukemia |
| T-ALL | T-cell acute lymphoblastic leukemia |
| KSS | Kearns–Sayre syndrome |
| PEO | progressive external ophthalmoplegia |
| UCI/SBB | University of California Irvine/Strategic Behavioral Sciences |
| CHLA | Children’s Hospital Los Angeles |
| LR-PCR | long-range PCR |
| PERMANOVA | permutational multivariate analysis of variance |
References
- Tuppen, H.A.L.; Blakely, E.L.; Turnbull, D.M.; Taylor, R.W. Mitochondrial DNA Mutations and Human Disease. Biochim. Biophys. Acta 2010, 1797, 113–128. [Google Scholar] [CrossRef]
- Wallace, D.C.; Fan, W.; Procaccio, V. Mitochondrial Energetics and Therapeutics. Annu. Rev. Pathol. 2010, 5, 297–348. [Google Scholar] [CrossRef]
- Harrington, J.S.; Ryter, S.W.; Plataki, M.; Price, D.R.; Choi, A.M.K. Mitochondria in Health, Disease, and Aging. Physiol. Rev. 2023, 103, 2349–2422. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Wallace, D.C.; Burelle, Y. The Rise of Mitochondria in Medicine. Mitochondrion 2016, 30, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and Organization of the Human Mitochondrial Genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Wallace, D.C.; Brown, M.D.; Lott, M.T. Mitochondrial DNA Variation in Human Evolution and Disease. Gene 1999, 238, 211–230. [Google Scholar] [CrossRef]
- Vafai, S.B.; Mootha, V.K. Mitochondrial Disorders as Windows into an Ancient Organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA Mutations in Human Disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef]
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef]
- Kopinski, P.K.; Singh, L.N.; Zhang, S.; Lott, M.T.; Wallace, D.C. Mitochondrial DNA Variation and Cancer. Nat. Rev. Cancer 2021, 21, 431–445. [Google Scholar] [CrossRef]
- DiMauro, S.; Schon, E.A. Mitochondrial Respiratory-Chain Diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef] [PubMed]
- Hjelm, B.E.; Ramiro, C.; Rollins, B.L.; Omidsalar, A.A.; Gerke, D.S.; Das, S.C.; Sequeira, A.; Morgan, L.; Schatzberg, A.F.; Barchas, J.D.; et al. Large Common Mitochondrial DNA Deletions Are Associated with a Mitochondrial SNP T14798C near the 3′ Breakpoints. Complex Psychiatry 2023, 8, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.C.; Rosa, H.S.; Grady, J.P.; Blakely, E.L.; He, L.; Romain, N.; Haller, R.G.; Newman, J.; McFarland, R.; Ng, Y.S.; et al. Pathological Mechanisms Underlying Single Large-Scale Mitochondrial DNA Deletions. Ann. Neurol. 2018, 83, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Nissanka, N.; Minczuk, M.; Moraes, C.T. Mechanisms of Mitochondrial DNA Deletion Formation. Trends Genet. 2019, 35, 235–244. [Google Scholar] [CrossRef]
- Yamashita, S.; Nishino, I.; Nonaka, I.; Goto, Y.-I. Genotype and Phenotype Analyses in 136 Patients with Single Large-Scale Mitochondrial DNA Deletions. J. Hum. Genet. 2008, 53, 598. [Google Scholar] [CrossRef]
- Björkman, K.; Vissing, J.; Østergaard, E.; Bindoff, L.A.; de Coo, I.F.M.; Engvall, M.; Hikmat, O.; Isohanni, P.; Kollberg, G.; Lindberg, C.; et al. Phenotypic Spectrum and Clinical Course of Single Large-Scale Mitochondrial DNA Deletion Disease in the Paediatric Population: A Multicentre Study. J. Med. Genet. 2023, 60, 65–73. [Google Scholar] [CrossRef]
- Triska, P.; Kaneva, K.; Merkurjev, D.; Sohail, N.; Falk, M.J.; Triche, T.J.; Biegel, J.A.; Gai, X. Landscape of germline and somatic mitochondrial DNA mutations in pediatric malignancies. Cancer Res. 2019, 79, 1318–1330. [Google Scholar] [CrossRef]
- Smith, A.L.M.; Whitehall, J.C.; Greaves, L.C. Mitochondrial DNA Mutations in Ageing and Cancer. Mol. Oncol. 2022, 16, 3276–3294. [Google Scholar] [CrossRef]
- Vodicka, P.; Vodenkova, S.; Danesova, N.; Vodickova, L.; Zobalova, R.; Tomasova, K.; Boukalova, S.; Berridge, M.V.; Neuzil, J. Mitochondrial DNA Damage, Repair, and Replacement in Cancer. Trends Cancer 2025, 11, 62–73. [Google Scholar] [CrossRef]
- Yusoff, A.A.M.; Abdullah, W.S.W.; Khair, S.Z.N.M.; Radzak, S.M.A. A Comprehensive Overview of Mitochondrial DNA 4977-Bp Deletion in Cancer Studies. Oncol. Rev. 2019, 13, 409. [Google Scholar] [CrossRef]
- Meissner, C.; Bruse, P.; Mohamed, S.A.; Schulz, A.; Warnk, H.; Storm, T.; Oehmichen, M. The 4977 Bp Deletion of Mitochondrial DNA in Human Skeletal Muscle, Heart and Different Areas of the Brain: A Useful Biomarker or More? Exp. Gerontol. 2008, 43, 645–652. [Google Scholar] [CrossRef]
- Vecoli, C.; Borghini, A.; Andreassi, M.G. The Molecular Biomarkers of Vascular Aging and Atherosclerosis: Telomere Length and Mitochondrial DNA4977 Common Deletion. Mutat. Res./Rev. Mutat. Res. 2020, 784, 108309. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; He, J.; Shen, L.; Fang, H.; Nie, H.; Jin, T.; Wei, X.; Xin, Y.; Jiang, Y.; Li, H.; et al. The Mitochondrial DNA 4,977-Bp Deletion and Its Implication in Copy Number Alteration in Colorectal Cancer. BMC Med. Genet. 2011, 12, 8. [Google Scholar] [CrossRef]
- Fontana, G.A.; MacArthur, M.R.; Rotankova, N.; Di Filippo, M.; Beer, H.-D.; Gahlon, H.L. The Mitochondrial DNA Common Deletion as a Potential Biomarker of Cancer-Associated Fibroblasts from Skin Basal and Squamous Cell Carcinomas. Sci. Rep. 2024, 14, 553. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Júnior, H.J.; Leeuwenburgh, C.; Bucci, C.; Marzetti, E. The Contribution of Mitochondrial DNA Alterations to Aging, Cancer, and Neurodegeneration. Exp. Gerontol. 2023, 178, 112203. [Google Scholar] [CrossRef] [PubMed]
- Kakaje, A.; Alhalabi, M.M.; Ghareeb, A.; Karam, B.; Mansour, B.; Zahra, B.; Hamdan, O. Rates and Trends of Childhood Acute Lymphoblastic Leukaemia: An Epidemiology Study. Sci. Rep. 2020, 10, 6756. [Google Scholar] [CrossRef]
- National Cancer Institute, Surveillance Research Program Acute Lymphocytic Leukemia (ALL) Recent Trends in SEER Age-Adjusted Incidence Rates, 2000–2022. Available online: https://seer.cancer.gov/statistics-network/explorer/ (accessed on 13 July 2025).
- Hiemenz, M.C.; Oberley, M.J.; Doan, A.; Aye, L.; Ji, J.; Schmidt, R.J.; Biegel, J.A.; Bhojwani, D.; Raca, G. A Multimodal Genomics Approach to Diagnostic Evaluation of Pediatric Hematologic Malignancies. Cancer Genet. 2021, 254–255, 25–33. [Google Scholar] [CrossRef]
- Bhagat, T.; Patel, M.; Greenberg, E. Mitochondrial Dysfunction in Cancer Treatment: New Mechanisms and Therapeutic Targets. Cancer Res. 2019, 79, 4053–4062. [Google Scholar]
- Bansal, P.; Sneha, L. Chemotherapy-Induced Mitochondrial Dysfunction in Cardiac and Skeletal Muscles: Mechanisms and Therapeutic Implications. Mol. Cancer Res. 2022, 20, 339–348. [Google Scholar]
- Liu, C.-J.; Wang, L.-K.; Tsai, F.-M. The Application and Molecular Mechanisms of Mitochondria-Targeted Antioxidants in Chemotherapy-Induced Cardiac Injury. Curr. Issues Mol. Biol. 2025, 47, 176. [Google Scholar] [CrossRef]
- Hjelm, B.E.; Rollins, B.; Morgan, L.; Sequeira, A.; Mamdani, F.; Pereira, F.; Damas, J.; Webb, M.G.; Weber, M.D.; Schatzberg, A.F.; et al. Splice-Break: Exploiting an RNA-Seq Splice Junction Algorithm to Discover Mitochondrial DNA Deletion Breakpoints and Analyses of Psychiatric Disorders. Nucl. Acids Res. 2019, 47, e59. [Google Scholar] [CrossRef] [PubMed]
- Omidsalar, A.; Xu, L. Brookehjelm/Splice-Break2: Splice-Break2 v3.0.1; Zenodo: Geneva, Switzerland, 2024. [Google Scholar] [CrossRef]
- Omidsalar, A.A.; McCullough, C.G.; Xu, L.; Boedijono, S.; Gerke, D.; Webb, M.G.; Manojlovic, Z.; Sequeira, A.; Lew, M.F.; Santorelli, M.; et al. Common Mitochondrial Deletions in RNA-Seq: Evaluation of Bulk, Single-Cell, and Spatial Transcriptomic Datasets. Commun. Biol. 2024, 7, 200. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.E.; Hjelm, B.E.; Lewis-de los Angeles, W.W.; Smith, E.; Omidsalar, A.A.; Rollins, B.L.; Sherman, A.; Parade, S.; Vawter, M.P.; Tyrka, A.R. Increased Rate of Unique Mitochondrial DNA Deletion Breakpoints in Young Adults with Early Life Stress. Biol. Psychiatr. Glob. Open Sci. 2024, 5, 100422. [Google Scholar] [CrossRef] [PubMed]
- Rollins, B.L.; Morgan, L.; Hjelm, B.E.; Sequeira, A.; Schatzberg, A.F.; Barchas, J.D.; Lee, F.S.; Myers, R.M.; Watson, S.J.; Akil, H.; et al. Mitochondrial Complex I Deficiency in Schizophrenia and Bipolar Disorder and Medication Influence. Mol. Neuropsychiatry 2018, 3, 157–169. [Google Scholar] [CrossRef]
- Kaneva, K.; Merkurjev, D.; Ostrow, D.; Ryutov, A.; Triska, P.; Stachelek, K.; Cobrinik, D.; Biegel, J.A.; Gai, X. Detection of Mitochondrial DNA Variants at Low Level Heteroplasmy in Pediatric CNS and Extra-CNS Solid Tumors with Three Different Enrichment Methods. Mitochondrion 2020, 51, 97–103. [Google Scholar] [CrossRef]
- Al Maruf, A.; O’Brien, P.J.; Naserzadeh, P.; Fathian, R.; Salimi, A.; Pourahmad, J. Methotrexate Induced Mitochondrial Injury and Cytochrome c Release in Rat Liver Hepatocytes. Drug Chem. Toxicol. 2018, 41, 51–61. [Google Scholar] [CrossRef]
- Kim, M.; Mahmood, M.; Reznik, E.; Gammage, P.A. Mitochondrial DNA Is a Major Source of Driver Mutations in Cancer. Trends Cancer Res. 2022, 8, 1046–1059. [Google Scholar] [CrossRef]
- Kaneva, K.; O’Halloran, K.; Triska, P.; Liu, X.; Merkurjev, D.; Bootwalla, M.; Ryutov, A.; Cotter, J.A.; Ostrow, D.; Biegel, J.A.; et al. The Spectrum of Mitochondrial DNA (MtDNA) Mutations in Pediatric CNS Tumors. Neurooncol. Adv. 2021, 3, vdab074. [Google Scholar] [CrossRef]
- O’Halloran, K.; Hakimjavadi, H.; Bootwalla, M.; Ostrow, D.; Kerawala, R.; Cotter, J.A.; Yellapantula, V.; Kaneva, K.; Wadhwani, N.R.; Treece, A.; et al. Pediatric Chordoma: A Tale of Two Genomes. Mol. Cancer Res. 2024, 22, 721–729. [Google Scholar] [CrossRef]
- Wang, J.; Balciuniene, J.; Diaz-Miranda, M.A.; McCormick, E.M.; Aref-Eshghi, E.; Muir, A.M.; Cao, K.; Troiani, J.; Moseley, A.; Fan, Z.; et al. Advanced Approach for Comprehensive MtDNA Genome Testing in Mitochondrial Disease. Mol. Genet. Metab. 2022, 135, 93–101. [Google Scholar] [CrossRef]
- Palculict, M.E.; Zhang, V.W.; Wong, L.-J.; Wang, J. Comprehensive Mitochondrial Genome Analysis by Massively Parallel Sequencing. Methods Mol. Biol. 2016, 1351, 3–17. [Google Scholar]
- Chinnery, P.F.; Hudson, G. Mitochondrial Genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef]
- Dahal, S.; Siddiqua, H.; Katapadi, V.K.; Iyer, D.; Raghavan, S.C. Characterization of G4 DNA Formation in Mitochondrial DNA and Their Potential Role in Mitochondrial Genome Instability. FEBS J. 2022, 289, 163–182. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Nambiar, M. G-Quadruplexes in the Mitochondrial Genome—A Cause for Instability. FEBS J. 2022, 289, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Laricchia, K.M.; Lake, N.J.; Watts, N.A.; Shand, M.; Haessly, A.; Gauthier, L.; Benjamin, D.; Banks, E.; Soto, J.; Garimella, K.; et al. Mitochondrial DNA variation across 56,434 individuals in gnomAD. Genome. Res. 2021, 32, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Pölönen, P.; Di Giacomo, D.; Seffernick, A.E.; Elsayed, A.; Kimura, S.; Benini, F.; Montefiori, L.E.; Wood, B.L.; Xu, J.; Chen, C.; et al. The Genomic Basis of Childhood T-Lineage Acute Lymphoblastic Leukaemia. Nature 2024, 632, 1082–1091. [Google Scholar] [CrossRef]
- Carew, J.S.; Zhou, Y.; Albitar, M.; Carew, J.D.; Keating, M.J.; Huang, P. Mitochondrial DNA Mutations in Primary Leukemia Cells after Chemotherapy: Clinical Significance and Therapeutic Implications. Leukemia 2003, 17, 1437–1447. [Google Scholar] [CrossRef]
- Kwok, C.S.-N.; Quah, T.C.; Ariffin, H.; Tay, S.K.-H.; Yeoh, A.E.-J. Mitochondrial D-Loop Polymorphisms and Mitochondrial DNA Content in Childhood Acute Lymphoblastic Leukemia. J. Pediatr. Hematol. Oncol. 2011, 33, e239–e244. [Google Scholar] [CrossRef]
- Jain, A.; Katiyar, A.; Singh, R.; Bakhshi, S.; Singh, H.; Palanichamy, J.K.; Singh, A. Implications of Mitochondrial DNA Variants in Pediatric B-Cell Acute Lymphoblastic Leukemia. Egypt. J. Med. Hum. Genet. 2022, 23, 133. [Google Scholar] [CrossRef]
- Sprason, C.; Tucker, T.; Clancy, D. MtDNA Deletions and Aging. Front. Aging 2024, 5, 1359638. [Google Scholar] [CrossRef]
- Kang, E.; Wang, X.; Tippner-Hedges, R.; Ma, H.; Folmes, C.D.L.; Gutierrez, N.M.; Lee, Y.; Van Dyken, C.; Ahmed, R.; Li, Y.; et al. Age-Related Accumulation of Somatic Mitochondrial DNA Mutations in Adult-Derived Human IPSCs. Cell Stem Cell 2016, 18, 625–636. [Google Scholar] [CrossRef]
- Yao, Y.G.; Kajigaya, S.; Young, N.S. Mitochondrial DNA Mutations in Single Human Blood Cells. Mutat. Res. 2015, 779, 68–77. [Google Scholar] [CrossRef]
- Sturm, G.; Karan, K.R.; Monzel, A.S.; Santhanam, B.; Taivassalo, T.; Bris, C.; Ware, S.A.; Cross, M.; Towheed, A.; Higgins-Chen, A.; et al. OxPhos Defects Cause Hypermetabolism and Reduce Lifespan in Cells and in Patients with Mitochondrial Diseases. Commun. Biol. 2023, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Fujiura, Y.; Mayumi, F.; Nozuhara, A.; Sugiu, Y.; Sakanashi, T.; Hidaka, T.; Toshima, H. A Deletion of Mitochondrial DNA in Murine Doxorubicin-Induced Cardiotoxicity. Biochem. Biophys. Res. Commun. 1993, 195, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Ikeda, M.; Ide, T.; Tadokoro, T.; Miyamoto, H.D.; Furusawa, S.; Tsutsui, Y.; Miyake, R.; Ishimaru, K.; Watanabe, M.; et al. Doxorubicin Causes Ferroptosis and Cardiotoxicity by Intercalating into Mitochondrial DNA and Disrupting Alas1-Dependent Heme Synthesis. Sci. Signal. 2022, 15, eabn8017. [Google Scholar] [CrossRef] [PubMed]
- Osataphan, N.; Phrommintikul, A.; Chattipakorn, S.C.; Chattipakorn, N. Effects of Doxorubicin-Induced Cardiotoxicity on Cardiac Mitochondrial Dynamics and Mitochondrial Function: Insights for Future Interventions. J. Cell. Mol. Med. 2020, 24, 6534–6557. [Google Scholar] [CrossRef]
- Dong, L.-F.; Rohlena, J.; Zobalova, R.; Nahacka, Z.; Rodriguez, A.-M.; Berridge, M.V.; Neuzil, J. Mitochondria on the Move: Horizontal Mitochondrial Transfer in Disease and Health. J. Cell Biol. 2023, 222, e202211044. [Google Scholar] [CrossRef]
- Cui, H.; Li, F.; Chen, D.; Wang, G.; Truong, C.K.; Enns, G.M.; Graham, B.; Milone, M.; Landsverk, M.L.; Wang, J.; et al. Comprehensive Next-Generation Sequence Analyses of the Entire Mitochondrial Genome Reveal New Insights into the Molecular Diagnosis of Mitochondrial DNA Disorders. Genet. Med. 2013, 15, 388–394. [Google Scholar] [CrossRef]
- Zhang, W.; Cui, H.; Wong, L.-J.C. Comprehensive One-Step Molecular Analyses of Mitochondrial Genome by Massively Parallel Sequencing. Clin. Chem. 2012, 58, 1322–1331. [Google Scholar] [CrossRef]
- Wang, K.; Singh, D.; Zeng, Z.; Coleman, S.J.; Huang, Y.; Savich, G.L.; He, X.; Mieczkowski, P.; Grimm, S.A.; Perou, C.M.; et al. MapSplice: Accurate Mapping of RNA-Seq Reads for Splice Junction Discovery. Nucleic Acids Res. 2010, 38, e178. [Google Scholar] [CrossRef]
- Illumina DRAGEN Bio-IT Platform Support. Available online: https://support.illumina.com/sequencing/sequencing_software/dragen-bio-it-platform.html (accessed on 30 December 2024).
- Bandelt, H.J.; Kloss-Brandstätter, A.; Richards, M.B.; Yao, Y.G.; Logan, I. The Case for the Continuing Use of the Revised Cambridge Reference Sequence (RCRS) and the Standardization of Notation in Human Mitochondrial DNA Studies. J. Hum. Genet. 2014, 59, 66–77. [Google Scholar] [CrossRef]
- Larsson, J. Area-Proportional Euler and Venn Diagrams with Ellipses [R Package Eulerr Version 7.0.2]. 2024. Available online: https://cran.r-project.org/web/packages/eulerr/eulerr.pdf (accessed on 28 March 2024).
- Dixon, P. VEGAN, a Package of R Functions for Community Ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Anderson, M.J. A New Method for Non-parametric Multivariate Analysis of Variance: Non-Parametric Manova For Ecology. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Gu, Z. Complex Heatmap Visualization. Imeta 2022, 1, e43. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MapSplice Breakpoints | Gene Location | Perfect Repeat | MapSplice Break | Imperfect_5p | Imperfect_3p | Frequency | Mean Deletion Percent | Perfect Length | GC Content |
|---|---|---|---|---|---|---|---|---|---|
| 14054-14377 | (MT-ND5-MT-ND6) | CACAGCACCAA | GC | AAAACAATTTCACAGCACCAAATCTCCACCT | CTCTTTCACCCACAGCACCAATCCTACCTCC | 35 | 0.16 | 11 | 54.5 |
| 8471-13449 | (MT-ATP8-MT-ND5) | ACCTCCCTCACCA | CC | ACTACCACCTACCTCCCTCACCAAAGCCCATAA | TCTCACTTCAACCTCCCTCACCATTGGCAGCCT | 34 | 0.15 | 13 | 61.5 |
| 6545-13846 | (MT-CO1-MT-ND5) | ACCTCAAC | TC | ACAGACCGCAACCTCAACACCACCTTCT | ACAGCCCTAGACCTCAACTACCTAACCA | 30 | 0.15 | 8 | 50.0 |
| 6335-13999 | (MT-CO1-MT-ND5) | TAGACCTAACC | CC | GGAGCCTCCGTAGACCTAACCATCTTCTCCT | CTACTCCTCCTAGACCTAACCTGACTAGAAA | 29 | 0.11 | 11 | 45.5 |
| 14066-14413 | (MT-ND5-MT-ND6) | ACCTCAACCC | CA | CTCCATCATCACCTCAACCCAAAAAGGCAT | ACTCACCAAGACCTCAACCCCTGACCCCCA | 27 | 0.10 | 10 | 60.0 |
| 5368-15335 | (MT-ND2-MT-CYB) | ACTCCACCTC | CA | CGCCTAATCTACTCCACCTCAATCACACTA | CCCTAGCAACACTCCACCTCCTATTCTTGC | 22 | 0.10 | 10 | 60.0 |
| 511-13926 | (D-Loop2-MT-ND5) | CACACACCGC | CA | ACCCAGCACACACACACCGCTGCTAACCCC | ACCCTAGCATCACACACCGCACAATCCCCT | 21 | 0.26 | 10 | 70.0 |
| 2224-13792 | (MT-RNR2-MT-ND5) | CTACCTAAAA | CT | TCAACACCCACTACCTAAAAAATCCCAAAC | CAATCCCCCTCTACCTAAAACTCACAGCCC | 21 | 0.10 | 10 | 30.0 |
| 1105-13846 | (MT-RNR1-MT-ND5) | ACCTCAAC | TC | TTAGCCCTAAACCTCAACAGTTAAATCA | ACAGCCCTAGACCTCAACTACCTAACCA | 21 | 0.10 | 8 | 50.0 |
| Category | Subcategory | Patient Count (Sample Count) |
|---|---|---|
| Age Group | unknown | 6 |
| 0–4 Years | 30 | |
| 5–10 Years | 42 | |
| 11–15 Years | 23 | |
| 16–20 Years | 16 | |
| Gender * | unknown | 6 |
| Female | 45 | |
| Male | 66 | |
| Disease | B_ALL | 93 (105) |
| Control (Non-Leuk) | 12 (12) | |
| T_ALL | 28 (31) | |
| Timepoint | Control | 12 (12) |
| Diagnosis | 111 (111) | |
| Relapse | 20 (23) | |
| Remission | 6 (8) | |
| Totals | 129 (148) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hakimjavadi, H.; Eom, E.; Christodoulou, E.; Hjelm, B.E.; Omidsalar, A.A.; Ostrow, D.; Biegel, J.A.; Gai, X. Distinct Mitochondrial DNA Deletion Profiles in Pediatric B- and T-ALL During Diagnosis, Remission, and Relapse. Int. J. Mol. Sci. 2025, 26, 7117. https://doi.org/10.3390/ijms26157117
Hakimjavadi H, Eom E, Christodoulou E, Hjelm BE, Omidsalar AA, Ostrow D, Biegel JA, Gai X. Distinct Mitochondrial DNA Deletion Profiles in Pediatric B- and T-ALL During Diagnosis, Remission, and Relapse. International Journal of Molecular Sciences. 2025; 26(15):7117. https://doi.org/10.3390/ijms26157117
Chicago/Turabian StyleHakimjavadi, Hesamedin, Elizabeth Eom, Eirini Christodoulou, Brooke E. Hjelm, Audrey A. Omidsalar, Dejerianne Ostrow, Jaclyn A. Biegel, and Xiaowu Gai. 2025. "Distinct Mitochondrial DNA Deletion Profiles in Pediatric B- and T-ALL During Diagnosis, Remission, and Relapse" International Journal of Molecular Sciences 26, no. 15: 7117. https://doi.org/10.3390/ijms26157117
APA StyleHakimjavadi, H., Eom, E., Christodoulou, E., Hjelm, B. E., Omidsalar, A. A., Ostrow, D., Biegel, J. A., & Gai, X. (2025). Distinct Mitochondrial DNA Deletion Profiles in Pediatric B- and T-ALL During Diagnosis, Remission, and Relapse. International Journal of Molecular Sciences, 26(15), 7117. https://doi.org/10.3390/ijms26157117