
Therapeutic Potential of Isoxazole–(Iso)oxazole Hybrids: Three Decades of Research


Abstract
1. Introduction
2. Treatment of Blood Disorders and Heart Diseases
2.1. WIZ Degradation Activity
2.2. GATA4 Inhibitors
2.3. P2Y1 Receptor Inhibitors
3. Treatment of Nervous System Diseases
3.1. Antidepressant and Sedative Activity
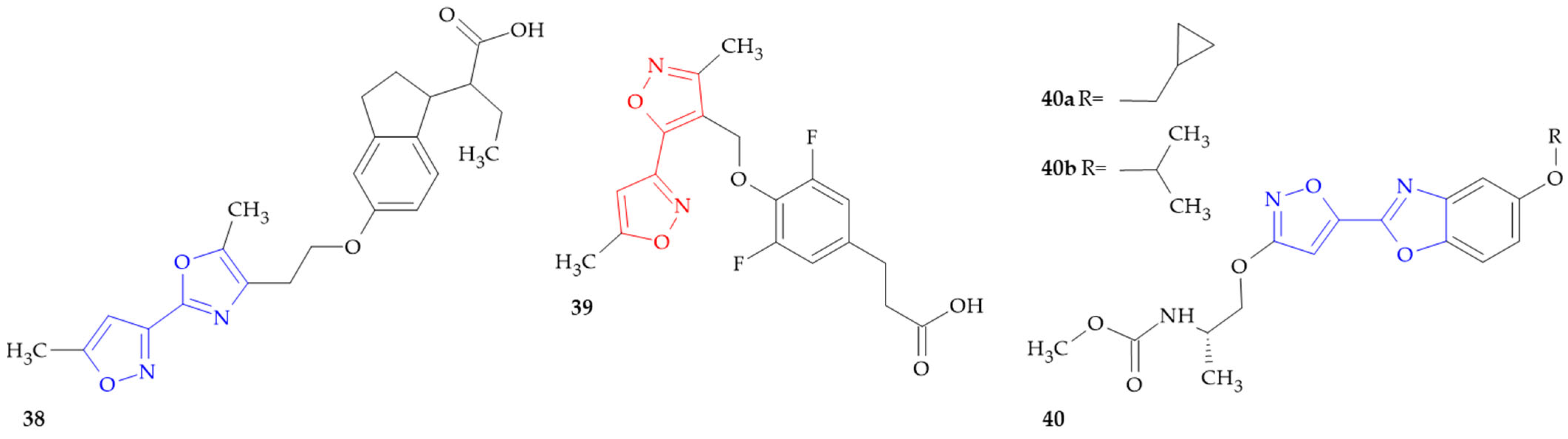
3.2. OX2R Inhibitors
3.3. GABAA α5 Receptor PAMs
3.4. nAChR Modulators
3.5. SCD Inhibitors
3.6. mGlu5 Receptor Modulator
4. Antibacterial Activity
5. Treatment of Cancer Diseases
5.1. HGF Receptor Inhibitors
5.2. ERα Receptor Modulators
5.3. TNIK Inhibitors
5.4. Antiapoptotic Protein Inhibitors
5.5. VEGFR2 Inhibitors
5.6. PARP1 Inhibitors
5.7. Hsp90 Inhibitors
5.8. PXR Antagonist
5.9. Undefined Mechanism of Action
6. Anti-Inflammatory Activity
6.1. CRTh2 Antagonists
6.2. p38 Kinase Inhibitors
6.3. IL-17 and IFN-γ Production Inhibitors
6.4. ALPK1 Inhibitors
7. Treatment of Metabolic Diseases
7.1. PPARs Agonists
7.2. GPR120 Receptor Agonists
7.3. Acetyl-CoA Carboxylase Inhibitors
8. Other Biological Activity
8.1. LPA Receptor Antagonists
8.2. Calcium-Activated K Channel Openers
8.3. FAAH Inhibitors
8.4. S1P1 Agonists
8.5. Cathepsin K Inhibitors
8.6. PRMT5 Inhibitors
8.7. RORγ Modulators
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Taylor, R.D.; MacCoss, M.; Lawson, A.D.G. Rings in drugs. J. Med. Chem. 2014, 57, 5845–5859. [Google Scholar] [CrossRef]
- Patel, S.K.; Patnayak, S. Heterocyclic compounds an potential drug and its biological activity: A review. J. Nonlinear. Anal. Optim. 2024, 15, 2705–2714. [Google Scholar]
- Sharma, P.K.; Amin, A.; Kumar, M. Synthetic Methods of Medicinally Important Heterocycles-thiazines: A Review. Open J. Med. Chem. 2020, 14, 71. [Google Scholar] [CrossRef]
- Jampilek, J. Heterocycles in Medicinal Chemistry. Molecules 2019, 24, 3839. [Google Scholar] [CrossRef]
- Luo, W.; Liu, Y.; Qin, H.; Zhao, Z.; Wang, S.; He, W.; Tang, S.; Peng, J. Nitrogen-containing heterocyclic drug products approved by the FDA in 2023: Synthesis and biological activity. Eur. J. Med. Chem. 2024, 279, 116838. [Google Scholar] [CrossRef] [PubMed]
- El-Garhy, O.H. An overview of the azoles of interest. Int. J. Curr. Pharmaceut. Res. 2015, 7, 1–6. [Google Scholar]
- Thakur, A.; Verma, M.; Sharma, R. Oxazole and isoxazole: From one-pot synthesis to medical applications. Tetrahedron 2022, 119, 132813. [Google Scholar] [CrossRef]
- Wang, Z.; Xiong, Y.; Peng, Y.; Zhang, X.; Li, S.; Peng, Y.; Peng, X.; Zhuo, L.; Jiang, W. Natural product evodiamine-inspired medicinal chemistry: Anticancer activity, structural optimization and structure-activity relationship. Eur. J. Med. Chem. 2023, 247, 115031. [Google Scholar] [CrossRef]
- Kumari, G.; Dhillon, S.; Rani, P.; Chahal, M.; Aneja, D.K.; Kinger, M. Development in the Synthesis of Bioactive Thiazole-Based Heterocyclic Hybrids Utilizing Phenacyl Bromide. ACS Omega 2024, 9, 18709–18746. [Google Scholar] [CrossRef] [PubMed]
- Bérubé, G. An overview of molecular hybrids in drug discovery. Expert Opin. Drug Discov. 2016, 11, 281–305. [Google Scholar] [CrossRef]
- Gothelf, K.V. Bis-Heterocyclic Derivatives. WIPO Patent WO9734881 A1, 14 March 1996. [Google Scholar]
- Bąchor, U.; Junka, A.; Brożyna, M.; Mączyński, M. The In Vitro Impact of Isoxazole Derivatives on Pathogenic Biofilm and Cytotoxicity of Fibroblast Cell Line. Int. J. Mol. Sci. 2023, 24, 2997. [Google Scholar] [CrossRef] [PubMed]
- Kankala, S.; Kankala, R.K.; Gundepaka, P.; Thota, N.; Nerella, S.; Gangula, M.R.; Guguloth, H.; Kagga, M.; Vadde, R.; Vasam, C.S. Regioselective synthesis of isoxazole–mercaptobenzimidazole hybrids and their in vivo analgesic and anti-inflammatory activity studies. Bioorg. Med. Chem. Lett. 2013, 23, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Mączyński, M.; Regiec, A.; Sochacka-Ćwikła, A.; Kochanowska, I.; Kocięba, M.; Zaczyńska, E.; Artym, J.; Kałas, W.; Zimecki, M. Synthesis, Physicochemical Characteristics and Plausible Mechanism of Action of an Immunosuppressive Isoxazolo[5,4-e]-1,2,4-Triazepine Derivative (RM33). Pharmaceuticals 2021, 14, 468. [Google Scholar] [CrossRef] [PubMed]
- Mazlan, M.K.N.; Mohd Tazizi, M.H.D.; Ahmad, R.; Noh, M.A.A.; Bakhtiar, A.; Wahab, H.A.; Mohd Gazzali, A. Antituberculosis Targeted Drug Delivery as a Potential Future Treatment Approach. Antibiotics 2021, 10, 908. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Park, S.M.; Kim, B.H. Synthesis of 5-isoxazol-5-yl-20-deoxyuridines exhibiting antiviral activity against HSV and several RNA viruses. Bioorg. Med. Chem. Lett. 2009, 19, 1126–1128. [Google Scholar] [CrossRef]
- Bąchor, U.; Brożyna, M.; Junka, A.; Chmielarz, M.R.; Gorczyca, D.; Mączyński, M. Novel Isoxazole-Based Antifungal Drug Candidates. Int. J. Mol. Sci. 2024, 25, 13618. [Google Scholar] [CrossRef]
- Müller-Schiffmann, A.; Sticht, H.; Korth, C. Hybrid compounds: From simple combinations to nanomachines. BioDrugs 2012, 26, 21–31. [Google Scholar] [CrossRef]
- Sysak, A.; Obmińska-Mrukowicz, B. Isoxazole ring as a useful scaffold in a search for new therapeutic agents. Eur. J. Med. Chem. 2017, 137, 292–309. [Google Scholar] [CrossRef]
- Kakkar, S.; Narasimhan, B. A comprehensive review on biological activities of oxazole derivatives. BMC Chem. 2019, 13, 16. [Google Scholar] [CrossRef]
- Staderini, M.; Vanni, S.; Baldeschi, A.C.; Giachin, G.; Zattoni, M.; Celauro, L.; Ferracin, C.; Bistaffa, E.; Moda, F.; Pérez, D.I.; et al. Bifunctional carbazole derivatives for simultaneous therapy and fluorescence imaging in prion disease murine cell models. Eur. J. Med. Chem. 2023, 245, 114923. [Google Scholar] [CrossRef]
- Matera, C.; Bono, F.; Pelucchi, S.; Collo, G.; Bontempi, L.; Gotti, C.; Zoli, M.; De Amici, M.; Missale, C.; Fiorentini, C.; et al. The novel hybrid agonist HyNDA-1 targets the D3R-nAChR heteromeric complex in dopaminergic neurons. Biochem. Pharmacol. 2019, 163, 154–168. [Google Scholar] [CrossRef]
- Fang, L.; Kraus, B.; Lehmann, J.; Heilmann, J.; Zhang, Y.; Decker, M. Design and synthesis of tacrine-ferulic acid hybrids as multi-potent anti-Alzheimer drug candidates. Bioorg. Med. Chem. Lett. 2008, 18, 2905–2909. [Google Scholar] [CrossRef]
- GBD 2021 Sickle Cell Disease Collaborators. Global, regional, and national prevalence and mortality burden of sickle cell disease, 2000-2021: A systematic analysis from the Global Burden of Disease Study 2021. Lancet Haematol. 2023, 10, e585–e599. [Google Scholar] [CrossRef] [PubMed]
- Ting, P.Y.; Borikar, S.; Kerrigan, J.R.; Thomsen, N.M.; Aghania, E.; Hinman, A.E.; Reyes, A.; Pizzato, N.; Fodor, B.D.; Wu, F.; et al. A molecular glue degrader of the WIZ transcription factor for fetal hemoglobin induction. Science 2024, 385, 6704. [Google Scholar] [CrossRef] [PubMed]
- Bonazzi, S.; Cernijenko, A.; Stroka Cobb, J.; Dales, N.; Kerrigan, J.R.; Lam, P.; Malik, H.A.; O’brien, G.; Patterson, A.W.; Thomsen, N.M.-F.; et al. Preparation of 3-(5-Methoxy-1-oxo-isoindolin-2-yl)piperidine-2,6-dione Derivatives for Treatment of Blood Disorders. WIPO Patent WO2021124172A1, 24 June 2021. [Google Scholar]
- Jumppanen, M.; Kinnunen, S.M.; Välimäki, M.J.; Talman, V.; Auno, S.; Bruun, T.; Boije af Gennäs, G.; Xhaard, H.; Aumüller, I.B.; Ruskoaho, H.; et al. Synthesis, Identification, and Structure−Activity Relationship Analysis of GATA4 and NKX2-5 Protein−Protein Interaction Modulators. J. Med. Chem. 2019, 62, 8284–8310. [Google Scholar] [CrossRef] [PubMed]
- Kinnunen, S.; Tölli, M.; Välimäki, M.; Jumppanen, M.; Boije af Gennäs, G.; Yli-Kauhaluoma, J.; Ruskoaho, H. Isoxazole-Amides for Treating Cardiac Diseases. WIPO Patent WO2018/055235 A1, 20 September 2018. [Google Scholar]
- Välimäki, M.J.; Tölli, M.A.; Kinnunen, S.M.; Aro, J.; Serpi, R.; Pohjolainen, L.; Talman, V.; Poso, A.; Ruskoaho, H.J. Discovery of Small Molecules Targeting the Synergy of Cardiac Transcription Factors GATA4 and NKX2-5. J. Med. Chem. 2017, 60, 7781–7798. [Google Scholar] [CrossRef]
- Zemkova, H. Purinergic P2 Receptors: Structure and Function 2.0. Int. J. Mol. Sci. 2023, 24, 5462. [Google Scholar] [CrossRef]
- Sutton, J.C.; Pi, Z.; Ruel, R.; L’heureux, A.; Thibeault, C.; Lam, P.Y.S. 2-phenoxy-n- (1, 3, 4-thiadizol-2-yl) pyridin-3-amine Derivatives and Related Compounds as p2y1 Receptor Inhibitors for the Treatment of Thromboembolic Disorders. WIPO Patent WO2006078621A2, 27 July 2006. [Google Scholar]
- Haenisch, B.; Bönisch, H. Depression and antidepressants: Insights from knockout of dopamine, serotonin or noradrenaline re-uptake transporters. Pharmacol. Ther. 2011, 129, 352–368. [Google Scholar] [CrossRef]
- Ciraulo, D.A.; Oldham, M. Sedative Hypnotics. In The Effects of Drug Abuse on the Human Nervous System; Academic Press: Cambridge, MA, USA, 2014; pp. 499–532. [Google Scholar] [CrossRef]
- Elmegeed, G.A.; Baiuomy, A.R.; Abdelhalim, M.M.; Hana, H.Y. Synthesis and antidepressant evaluation of five novel heterocyclic tryptophan-hybrid derivatives. Arch. Pharm. 2010, 343, 261–267. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Q.; Ji, B.; Pan, Y.; Xu, C.; Cheng, B.; Bai, B.; Chen, J. The Orexin/Receptor System: Molecular Mechanism and Therapeutic Potential for Neurological Diseases. Front. Mol. Neurosci. 2018, 11, 220. [Google Scholar] [CrossRef]
- Branstetter, B.J.; Letavic, M.A.; Ly, K.S.; Rudolph, D.A.; Savall, B.M.; Shah, C.R.; Shireman, B.T. Fused Heterocyclic Compounds as Orexin Receptor Modulators. WIPO Patent WO2011050200A1, 24 April 2011. [Google Scholar]
- Ghit, A.; Assal, D.; Al-Shami, A.S.; Eldin, D.; Hussein, E. GABAA receptors: Structure, function, pharmacology, and related disorders. J. Genet. Eng. Biotechnol. 2021, 19, 123. [Google Scholar] [CrossRef]
- Cecere, G.; Zbinden, K.G.; Hernandez, M.-C.; Knust, H.; Koblet, A.; Olivares Morales, M.; Patiny-Adam, A.; Pinard, E.; Runtz-Schmitt, V.; Steiner, S. New Isoxazolyl Ether Derivatives as Gaba a Alpha5 Pam. WIPO Patent WO2019238633 A1, 19 December 2019. [Google Scholar]
- Skok, M. Mitochondrial nicotinic acetylcholine receptors: Mechanisms of functioning and biological significance. Int. J. Biochem. Cell Biol. 2022, 143, 106138. [Google Scholar] [CrossRef] [PubMed]
- Crowley, B.M.; Campbell, B.T.; Chobanian, H.R.; Feels, J.I.; Guiadeen, D.G.; Greshock, T.J.; Leavitt, K.J.; Rada, V.L.; Bell, I.M. Spiropiperidine Allosteric Modulators of Nicotinic Acetylcholine Receptors. WIPO Patent WO2019212927 A1, 11 July 2019. [Google Scholar]
- Loix, M.; Vanherle, S.; Turri, M.; Kemp, S.; Fernandes, K.J.L.; Hendriks, J.J.A.; Bogie, J.F.J. Stearoyl-CoA desaturase-1: A potential therapeutic target for neurological disorders. Mol. Neurodegener. 2024, 19, 85. [Google Scholar] [CrossRef] [PubMed]
- Igal, R.A.; Sinner, D.I. Stearoyl-CoA desaturase 5 (SCD5), a Δ-9 fatty acyl desaturase in search of a function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2021, 1866, 158840. [Google Scholar] [CrossRef] [PubMed]
- Wrona, I.; Tivitmahaisoon, P.; Tardiff, D.; Pandya, B.; Ozboya, K.; Lucas, M.; Le Bourdonnec, B. Compounds and Uses Thereof. WIPO Patent WO2019209962 A1, 31 October 2019. [Google Scholar]
- Simonyi, A.; Schachtman, T.R.; Christoffersen, G.R.J. Metabotropic glutamate receptor subtype 5 antagonism in learning and memory. Eur. J. Pharmacol. 2010, 639, 17–25. [Google Scholar] [CrossRef]
- Burdi, D.; Spear, K.L.; Hardy, L.W. Preparation of Substituted Oxazolopyridines and Their Analogs for Treating Disorders Mediated by Metabotropic Glutamate Receptor 5. WO2010114971, 7 October 2010. [Google Scholar]
- Ishak, A.; Mazonakis, N.; Spernovasilis, N.; Akinosoglou, K.; Tsioutis, C. Bactericidal versus bacteriostatic antibacterials: Clinical significance, differences and synergistic potential in clinical practice. J. Antimicrob. Chemother. 2025, 80, 1–17. [Google Scholar] [CrossRef]
- Williams, T.L.; Yin, Y.W.; Carter, C.W., Jr. Selective Inhibition of Bacterial Tryptophanyl-tRNA Synthetases by Indolmycin Is Mechanism-based. J. Biol. Chem. 2016, 291, 255–265. [Google Scholar] [CrossRef]
- Barvian, K.; Basarab, G.S.; Gowravaram, M.R.; Hauck, S.I.; Zhou, F. Fused, Spirocyclic Heteroaromatic Compounds for the Treatment of Bacterial Infections. WIPO Patent WO2010043893 A1, 22 April 2010. [Google Scholar]
- Best, D.J.; Elder, J.S.; Osborne, N.F. Preparation of Sulfamoyl-Containing Alditols as Bactericides and t-RNA Synthetase Inhibitors. WIPO Patent WO9735859, 2 October 1997. [Google Scholar]
- Broom, N.J.P.; Elder, J.S.; Hannan, P.C.T.; Pons, J.E.; O’Hanlon, P.J.; Walker, G.; Wilson, J.; Woodall, P. The chemistry of pseudomonic acid. Part 14. Synthesis and in vivo biological activity of heterocycle substituted oxazole derivatives. J. Antibiot. 1995, 48, 1336–1344. [Google Scholar] [CrossRef]
- Wales, S.M.; Hammer, K.A.; Somphol, K.; Kemker, I.; Schroder, D.C.; Tague, A.J.; Brkic, Z.; King, A.M.; Lyras, D.; Riley, T.V.; et al. Synthesis and antimicrobial activity of binaphthyl-based, functionalized oxazole and thiazole peptidomimetics. Org. Biomol. Chem. 2015, 13, 10813–10824. [Google Scholar] [CrossRef]
- Becker, D.; Selbach, M.; Rollenhagen, C.; Ballmaier, M.; Meyer, T.F.; Mann, M.; Bumann, D. Robust Salmonella metabolism limits possibilities for new antimicrobials. Nature 2006, 440, 303–307. [Google Scholar] [CrossRef]
- Magalhães, J.; Franko, N.; Raboni, S.; Annunziato, G.; Tammela, P.; Bruno, A.; Bettati, S.; Armao, S.; Spadini, C.; Cabassi, C.S.; et al. Discovery of substituted (2-aminooxazol-4-yl)isoxazole-3-carboxylic acids as inhibitors of bacterial serine acetyltransferase in the quest for novel potential antibacterial adjuvants. Pharmaceuticals 2021, 14, 174. [Google Scholar] [CrossRef] [PubMed]
- Azzali, E.; Girardini, M.; Annunziato, G.; Pavone, M.; Vacondio, F.; Mori, G.; Rosalia, M.P.; Constantino, G.; Pieroni, M. 2-Aminooxazole as a Novel Privileged Scaffold in Antitubercular Medicinal Chemistry. ACS Med. Chem. Lett. 2020, 11, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Lilienkampf, A.; Pieroni, M.; Wan, B.; Wang, Y.; Franzblau, S.G.; Kozikowski, A.P. Rational design of 5-phenyl-3-isoxazolecarboxylic acid ethyl esters as growth inhibitors of Mycobacterium tuberculosis. a potent and selective series for further drug development. J. Med. Chem. 2010, 53, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Lilienkampf, A.; Pieroni, M.; Franzblau, S.G.; Bishai, W.R.; Kozikowski, A.P. Derivatives of 3-isoxazolecarboxylic acid esters: A potent and selective compound class against replicating and nonreplicating Mycobacterium tuberculosis. Curr. Top. Med. Chem. 2012, 12, 729–734. [Google Scholar] [CrossRef]
- Girardini, M.; Ferlenghi, F.; Annunziato, G.; Degiacomi, G.; Papotti, B.; Marchi, C.; Sammartino, J.C.; Rasheed, S.S.; Contini, A.; Pasca, M.R.; et al. Expanding the knowledge around antitubercular 5-(2-aminothiazol-4-yl)isoxazole-3-carboxamides: Hit-to-lead optimization and release of a novel antitubercular chemotype via scaffold derivatization. Eur. J. Med. Chem. 2023, 45, 114916. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer. Available online: https://www.iarc.who.int/wp-content/uploads/2024/02/pr345_E.pdf (accessed on 12 July 2025).
- Baier, A.; Szyszka, R. Compounds from Natural Sources as Protein Kinase Inhibitors. Biomolecules 2020, 10, 1546. [Google Scholar] [CrossRef]
- Cui, J.; Bhumralkar, D.; Botrous, I.; Chu, J.Y.; Funk, L.A.; Hanau, C.E.; Harris, G.D.; Jia, L.; Johnson, J.; Kolodziej, S.A.; et al. Aminoheteroaryl Compounds as Protein Kinase Inhibitors. WIPO Patent WO2004076412A2, 26 February 2004. [Google Scholar]
- Anbalagan, M.; Rowan, B.G. Estrogen receptor alpha phosphorylation and its functional impact in human breast cancer. Mol. Cell Endocrinol. 2015, 418, 264–2672. [Google Scholar] [CrossRef]
- Dijcks, F.A.; Lusher, S.J.; Stock, H.T.; Veeneman, G.H. N-Substituted Azetidine Derivatives. WIPO Patent WO2012084711A1, 28 June 2012. [Google Scholar]
- Pohanka, M. Alpha7 Nicotinic Acetylcholine Receptor Is a Target in Pharmacology and Toxicology. Int. J. Mol. Sci. 2012, 13, 2219–2238. [Google Scholar] [CrossRef]
- Zavoronkovs, A.; Aliper, A.; Aladinskiy, V.; Kukharenko, A.; Qin, L.; Cheng, X. Preparation of Imidazole Analogs as TNIK and MAP4K4 Kinases Inhibitors for the Treatment of TNIK-Mediated Diseases. WIPO Patent WO2022179529, 1 September 2022. [Google Scholar]
- Qian, S.; Wei, Z.; Yang, W.; Huang, J.; Yang, Y.; Wang, J. The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front. Oncol. 2022, 12, 985363. [Google Scholar] [CrossRef]
- Sochacka-Ćwikła, A.; Regiec, A.; Zimecki, M.; Artym, J.; Zaczyńska, E.; Kocięba, M.; Kochanowska, I.; Bryndal, I.; Pyra, A.; Mączyński, M. Synthesis and Biological Activity of New 7-Amino-oxazolo[5,4-d]Pyrimidine Derivatives. Molecules 2020, 25, 3558. [Google Scholar] [CrossRef]
- Sochacka-Ćwikła, A.; Mączyński, M.; Czyżnikowska, Ż.; Wiatrak, B.; Jęśkowiak, I.; Czerski, A.; Regiec, A. New oxazolo[5,4-d]pyrimidines as potential anticancer agents: Their design, synthesis, and in vitro biological activity research. Int. J. Mol. Sci. 2022, 23, 11694. [Google Scholar] [CrossRef]
- Wang, X.; Bove, A.M.; Simone, G.; Ma, B. Molecular Bases of VEGFR-2-Mediated Physiological Function and Pathological Role. Front. Cell Dev. Biol. 2020, 8, 599281. [Google Scholar] [CrossRef]
- Sochacka-Ćwikła, A.; Regiec, A.; Czyżnikowska, Ż.; Śliwińska-Hill, U.; Kwiecień, A.; Wiatrak, B.; Rusak, A.; Krawczyńska, K.; Mrozowska, M.; Borska, S.; et al. Synthesis and structural proof of novel oxazolo[5,4-d]pyrimidine derivatives as potential VEGFR2 inhibitors. in vitro study of their anticancer activity. Bioorg. Chem. 2024, 153, 107958. [Google Scholar] [CrossRef]
- Kim, D.; Nam, H.J. PARP Inhibitors: Clinical Limitations and Recent Attempts to Overcome Them. Int. J. Mol. Sci. 2022, 23, 8412. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, N. Phthalazinone Compound as Parp Inhibitor. WIPO Patent WO2011007145 A1, 20 January 2011. [Google Scholar]
- Jung, J.; Kwon, J.; Hong, S.; Moon, A.-N.; Jeong, J.; Kwon, S.; Kim, J.; Lee, M.; Lee, H.; Lee, J.H.; et al. Discovery of novel heat shock protein (Hsp90) inhibitors based on luminespib with potent antitumor activity. Bioorg. Med. Chem. Lett. 2020, 30, 127165. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-H.; Lee, H.-S.; Kwon, J.-S.; Park, J.-T.; Hong, C.-S.; Shin, D.-H.; Hong, S.-J.; Moon, A.-N.; Jeong, J.-A.; Kwon, S.-W. A Novel 5-Membered Heterocycle Derivatives and Manufacturing Process Thereof. WIPO Patent WO2011102660A2, 25 August 2011. [Google Scholar]
- Chen, Y.K.; Nie, D.T. Pregnane X receptor and its potential role in drug resistance in cancer treatment. Recent Pat. Anti-Cancer Drug Discov. 2009, 4, 19–27. [Google Scholar] [CrossRef]
- Hodnik, Ž.; Maši, L.P.; Tomaši, T.; Smodiš, D.; D’Amore, C.; Fiorucci, S.; Kikelj, D. Bazedoxifene scaffold-based mimetics of solomonsterols A and B as novel pregnane X receptor antagonists. J. Med. Chem. 2014, 57, 4819–4833. [Google Scholar] [CrossRef]
- Premalatha, S.; Rambabu, G.; Hatti, I.; Ramachandran, D. Design, Synthesis and Biological Evaluation of 3-(3,4,5-Trimethoxyphenyl)-5-(2-(5-arylbenzo[b]thiophen-3-yl)oxazol-5-yl)isoxazole Derivatives as Anticancer Agents. Lett. Org. Chem. 2020, 17, 345–351. [Google Scholar] [CrossRef]
- Jandl, K.; Heinemann, A. The therapeutic potential of CRTH2/DP2 beyond allergy and asthma. Prostaglandins Other Lipid Mediat. 2017, 133, 42–48. [Google Scholar] [CrossRef]
- Xiao, D.; Zhu, X.; Yu, Y.; Shao, N.; Wu, J.; McCormick, K.D.; Dhondi, P.; Qin, J.; Mazzola, R.; Tang, H. Quality by design (QbD) of amide isosteres: 5,5-Disubstituted isoxazolines as potent CRTh antagonists with favorable pharmacokinetic and drug-like properties. Bioorg. Med. Chem. Lett. 2014, 24, 1615–1620. [Google Scholar] [CrossRef]
- Aslanian, R.G.; Boyce, C.W.; Mazzola, R.D., Jr.; Mckittrick, B.A.; Mccormick, K.D.; Palani, A.; Qin, J.; Tang, H.; Xiao, D.; Yu, Y.; et al. Preparation of Quinazolinone Compounds as CRTH2 Antagonists. WIPO Patent WO2012051036, 19 April 2012. [Google Scholar]
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef]
- Severance, D.L.; Gardiner, E.M.M.; Noble, S.A.; Lou, B.; Borchardt, A.J.; Kahraman, M.; Roppe, J.R.; Siegel, D.L.; Scranton, S.A. Heterocyclic Ortho-Terphenyl Analogs (Thiazoles, Oxazoles, Isoxazoles, and Pyrazoles, etc.) as Inhibitors of p38 Kinase, and Methods of Treating Inflammatory Disorders and Other Diseases Using Them. WO2006116355, 2 November 2006. [Google Scholar]
- Zenobia, C.; Hajishengallis, G. Basic biology and role of interleukin-17 in immunity and inflammation. Periodontol. 2000 2015, 69, 142–159. [Google Scholar] [CrossRef] [PubMed]
- Tau, G.; Rothman, P. Biologic functions of the IFN-gamma receptors. Allergy 1999, 54, 1233–1251. [Google Scholar] [CrossRef] [PubMed]
- Leban, J.; Baumgartner, R.; Saeb, W.; Chevrier, C. Preparation of Pyrazolylisoxazoles as IL-17 and IFN-gamma Production Inhibitors for Treating Autoimmune Inflammation. WO2012101261, 2 August 2012. [Google Scholar]
- Leban, J.; Baumgartner, R.; Saeb, W.; Chevrier, C. Preparation of Pyrazolylisoxazoles as IL-17 and IFN-gamma Production Inhibitors for Treating Autoimmune Inflammation. WO2012101263, 2 August 2012. [Google Scholar]
- Cohen, P.; Snelling, T. Diseases caused by altered specificity of a protein kinase for its allosteric activators. Trends Biochem. Sci. 2025, 50, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Du, N.; Li, Z.; Wang, H.; Tian, Z.; Ji, Z.; Chen, Y.; O’Yang, C. Preparation of (hetero)aryl carboxamides as Novel ALPK1 Inhibitors. WO2024153225, 25 July 2024. [Google Scholar]
- Zhang, H.; Zhou, X.D.; Shapiro, M.D.; Lip, G.Y.H.; Tilg, H.; Valenti, L.; Somers, V.K.; Byrne, C.D.; Targher, G.; Yang, W.; et al. Global burden of metabolic diseases, 1990–2021. Metabolism 2024, 160, 155999. [Google Scholar] [CrossRef]
- Varga, T.; Czimmerer, Z.; Nagy, L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta 2011, 1812, 1007–1022. [Google Scholar] [CrossRef]
- Lowe, D.B.; Bifulco, N.; Bullock, W.H.; Claus, T.; Coish, P.; Dai, M.; Dela Cruz, F.E.; Dickson, D.; Fan, D.; Hoover-Litty, H.; et al. Substituted indanylacetic acids as PPAR-α-γ activators. Bioorg. Med. Chem. Lett. 2006, 16, 297–301. [Google Scholar] [CrossRef]
- Ji, G.; Guo, Q.; Xue, Q.; Kong, R.; Wang, S.; Lei, K.; Liu, R.; Wang, X. Novel GPR120 Agonists with Improved Pharmacokinetic Profiles for the Treatment of Type 2 Diabetes. Molecules 2021, 26, 6907. [Google Scholar] [CrossRef]
- Hirasawa, A.; Tsumaya, K.; Awaji, T.; Katsuma, S.; Adachi, T.; Yamada, M.; Sugimoto, Y.; Miyazaki, S.; Tsujimoto, G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 2005, 11, 90–94. [Google Scholar] [CrossRef]
- Ma, J.; Novack, A.; Nashashibi, I.; Pham, P.; Rabbat, C.J.; Song, J.; Shi, D.F.; Zhao, Z.; Choi, Y.-J.; Chen, X. Aryl gpr120 Receptor Agonists and Uses Thereof. WIPO Patent WO2010048207A2, 29 April 2010. [Google Scholar]
- Octave, M.; Pirotton, L.; Ginion, A.; Robaux, V.; Lepropre, S.; Ambroise, J.; Bouzin, C.; Guigas, B.; Giera, M.; Foretz, M.; et al. Acetyl-CoA Carboxylase Inhibitor CP640.186 Increases Tubulin Acetylation and Impairs Thrombin-Induced Platelet Aggregation. Int. J. Mol. Sci. 2021, 22, 13129. [Google Scholar] [CrossRef]
- Yasuma, T.; Kamata, M.; Yamashita, T.; Hirose, H.; Murakami, M.; Kina, A.; Yonemori, K.; Mizojiri, R.; Fujimori, I.; Fujimoto, T.; et al. Preparation of Heterocyclic Bicyclic Compounds as Acetyl-CoA Carboxylase Inhibitors. WIPO Patent WO2012074126, 7 June 2012. [Google Scholar]
- Nishikimi, M.; Choudhary, R.C.; Shoaib, M.; Yagi, T.; Becker, L.B.; Kim, J. Neurological Improvement via Lysophosphatidic Acid Administration in a Rodent Model of Cardiac Arrest-Induced Brain Injury. Int. J. Mol. Sci. 2023, 24, 17451. [Google Scholar] [CrossRef]
- Buckman, B.O.; Nicholas, J.B.; Emayan, K.; Seiwert, S.D. Preparation of N-Heterocyclylcarbamates as Lysophosphatidic Acid (LPA) Receptor Antagonists. WIPO Patent WO2013025733, 21 February 2013. [Google Scholar]
- Weaver, A.K.; Bomben, V.C.; Sontheimer, H. Expression and function of calcium-activated potassium channels in human glioma cells. Glia 2006, 54, 223–233. [Google Scholar] [CrossRef]
- Hongu, M.; Hosaka, T.; Kashiwagi, T.; Kono, R.; Kobayashi, H. Preparation of Substituted Imidazoles/Oxazoles/Thiazoles as Large Conductance Calcium-Activated K Channel Openers. WIPO Patent WO2002083111, 24 October 2002. [Google Scholar]
- Grieco, M.; De Caris, M.G.; Maggi, E.; Armeli, F.; Coccurello, R.; Bisogno, T.; D’Erme, M.; Maccarrone, M.; Mancini, P.; Businaro, R. Fatty Acid Amide Hydrolase (FAAH) Inhibition Modulates Amyloid-Beta-Induced Microglia Polarization. Int. J. Mol. Sci. 2021, 22, 7711. [Google Scholar] [CrossRef] [PubMed]
- Genovese, T.; Duranti, A.; D’Amico, R.; Fusco, R.; Impellizzeri, D.; Peritore, A.F.; Crupi, R.; Gugliandolo, E.; Cuzzocrea, S.; Di Paola, R.; et al. Fatty Acid Amide Hydrolase (FAAH) Inhibition Plays a Key Role in Counteracting Acute Lung Injury. Int. J. Mol. Sci. 2022, 23, 2781. [Google Scholar] [CrossRef] [PubMed]
- Chobanian, H.; Lin, L.S.; Liu, P.; Chioda, M.D.; Devita, R.J.; Nargund, R.P.; Guo, Y.; Hamill, T.; Li, W.; Henze, D.A. Preparation of Pyridinylsulfanyloxazole Derivatives and Analogs for Use as FAAH Inhibitors. WIPO Patent WO2011094209, 4 August 2011. [Google Scholar]
- Chen, S.; Wu, L.; Lang, B.; Zhao, G.; Zhang, W. Sphingosine 1-phosphate receptor 1 modulators exert neuroprotective effects in central nervous system disorders. Front. Pharmacol. 2025, 16, 1516991. [Google Scholar] [CrossRef] [PubMed]
- Coyle, P.K.; Freedman, M.S.; Cohen, B.A.; Cree, B.A.C.; Markowitz, C.E. Sphingosine 1-phosphate receptor modulators in multiple sclerosis treatment: A practical review. Ann. Clin. Transl. Neurol. 2024, 11, 842–855. [Google Scholar] [CrossRef]
- Murali Dhar, T.G.; Xiao, H.-Y.; Watterson, S.H.; Ko, S.S.; Dyckman, A.J.; Langevine, C.M.; Das, J.; Cherney, R.J. Tricyclic Heterocyclic Compounds. WIPO Patent WO2011059784A1, 19 May 2011. [Google Scholar]
- Xiao, H.-Y.; Watterson, S.H.; Langevine, C.M.; Srivastava, A.S. Identification of Tricyclic Agonists of Sphingosine-1-Phosphate Receptor 1 (S1P1) Employing Ligand-Based Drug Design. J. Med. Chem. 2016, 59, 9837–9854. [Google Scholar] [CrossRef]
- Moon, D.O. Review of Cathepsin K Inhibitor Development and the Potential Role of Phytochemicals. Molecules 2025, 30, 91. [Google Scholar] [CrossRef]
- Barrett, D.G.; Catalano, J.G.; Deaton, D.N.; Miller, A.B.; Ray, J.A.; Samano, V. Derivatives of 1-(Oxoaminoacetyl) Pentylcarbamate as Cathepsin k Inhibitors for the Treatment of Bone Loss. WIPO Patent WO03086385A1, 23 October 2003. [Google Scholar]
- Kim, H.; Ronai, Z.A. PRMT5 function and targeting in cancer. Cell Stress. 2020, 4, 199–215. [Google Scholar] [CrossRef]
- Machacek, M.; Altman, M.D.; Kawamura, S.; Reutershan, M.H.; Sloman, D.L.; Siliphaivanh, P.; Schneider, S.E.; Yeung, C.S.; Witter, D.J.; Gibeau, C.R. Spiro-Isoquinoline-3,4’-Piperidine Derivatives as PRMT5 Inhibitors and Their Preparation. WIPO Patent WO2021126729A1, 24 June 2021. [Google Scholar]
- Jetten, A.M.; Takeda, Y.; Slominski, A.; Kang, H.S. Retinoic acid-related Orphan Receptor γ (RORγ): Connecting sterol metabolism to regulation of the immune system and autoimmune disease. Curr. Opin. Toxicol. 2018, 8, 66–80. [Google Scholar] [CrossRef]
- Qiu, R.; Wang, Y. Retinoic Acid Receptor-Related Orphan Receptor γt (RORγt) Agonists as Potential Small Molecule Therapeutics for Cancer Immunotherapy. J. Med. Chem. 2018, 61, 5794–5804. [Google Scholar] [CrossRef]
- Kotoku, M.; Takaki, M.; Noriyosh, S.; Shintaro, H.; Shingo, F.; Shingo, O.; Hiroshi, Y.; Masahiro, Y.; Takayuki, S.; Kazuyuki, H.; et al. Preparation of Isoxazoles and Their Use as ROR-γ Antagonists and Pharmaceutical. WIPO Patent WO2014065413A1, 5 January 2014. [Google Scholar]
- Kotoku, M.; Maeba, T.; Fujioka, S.; Yokota, M.; Seki, N.; Ito, K.; Suwa, Y.; Ikenogami, T.; Hirata, K.; Hase, Y.; et al. Discovery of Second Generation RORγ Inhibitors Composed of an Azole Scaffold. J. Med. Chem. 2019, 62, 2837–2842. [Google Scholar] [CrossRef]
- Baloglu, E.; Bohnert, G.J.; Ghosh, S.; Lobera, M.; Schmidt, D.R.; Sung, L. Preparation of Phenylphenylmethylisoxazolylbenzofuranylactamide Derivatives and Analogs for Use as Retinoid-Related Orphan Receptor Gamma Modulators. WO2013019682, 7 February 2013. [Google Scholar]
- Morais, T.S. Recent Advances in the Development of Hybrid Drugs. Pharmaceutics 2024, 16, 889. [Google Scholar] [CrossRef] [PubMed]
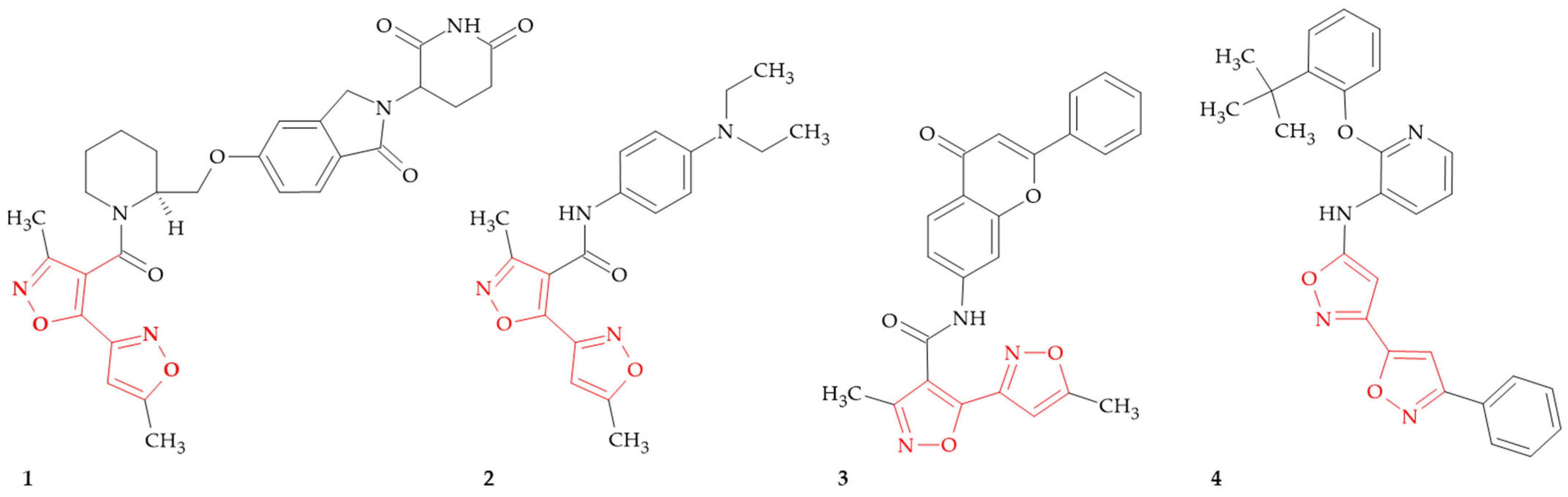

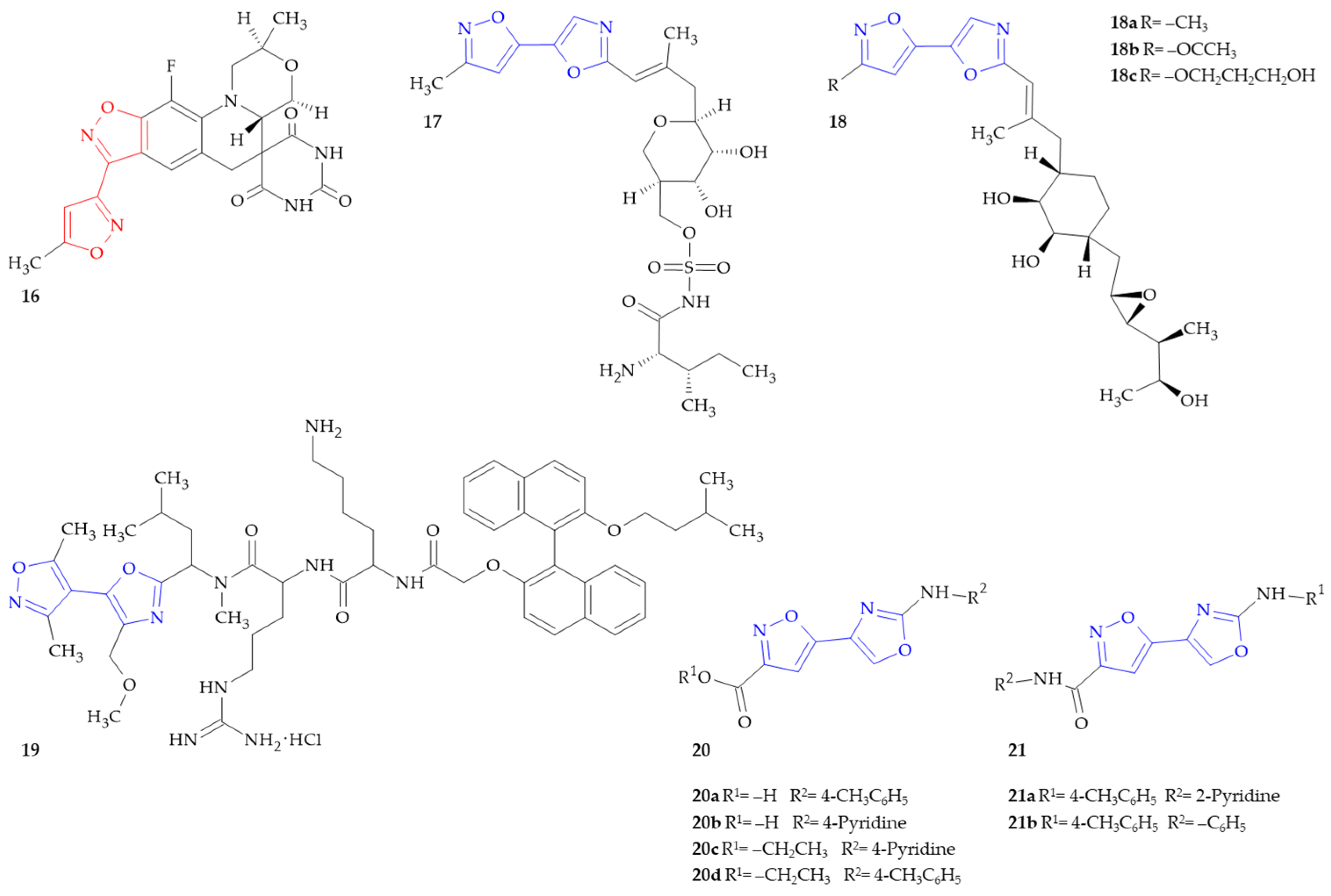

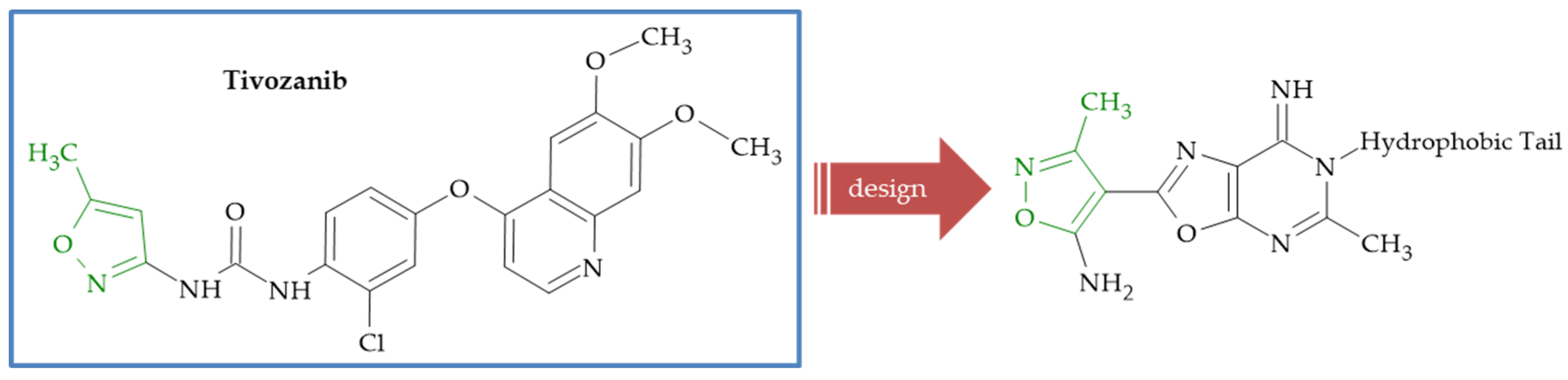
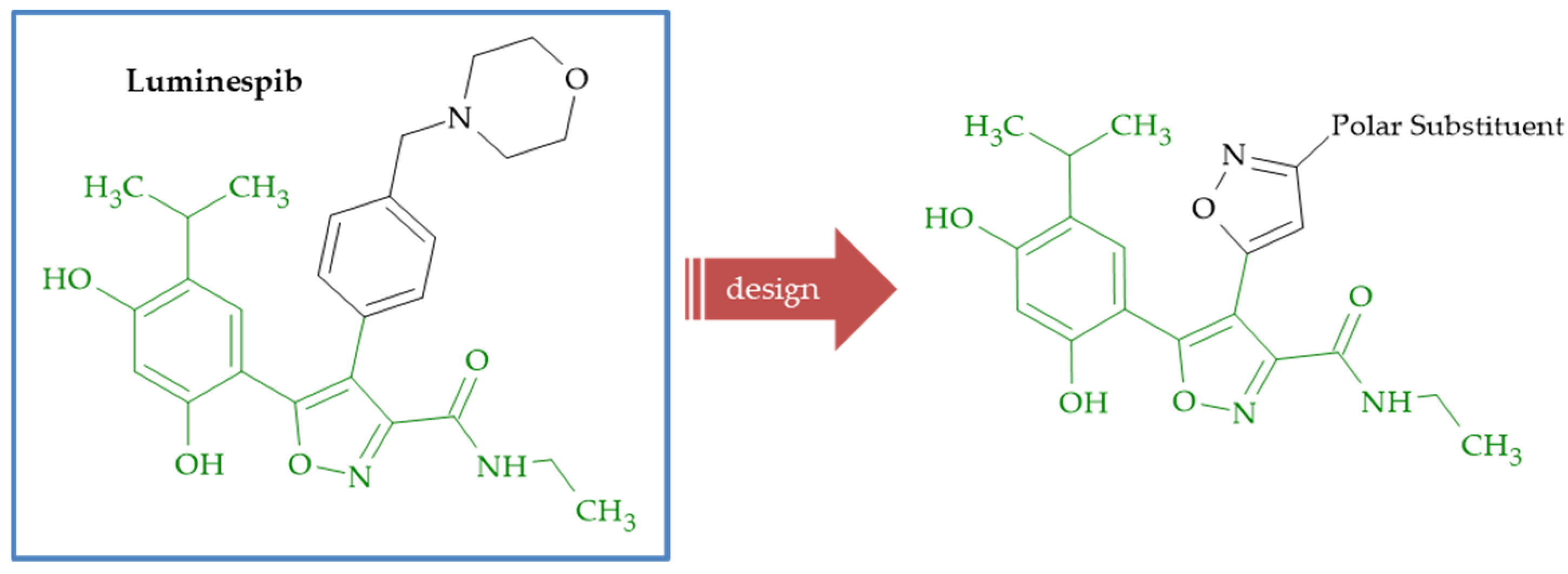




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 ± SD * [µM] | ||||||
|---|---|---|---|---|---|---|
| A549 | HT-29 | A375 | MCF7 | NHDF | VEGFR2 | |
| Tivozanib | 10.03 ± 4.25 | 8.38 ± 4.04 | 7.28 ± 3.35 | 7.44 ± 3.51 | 7.10 ± 3.74 | 15.7 ± 5.9 |
| 27a | 20.91 ± 7.60 | 19.70 ± 3.55 | 9.63 ± 2.41 | 17.43 ± 4.63 | 20.71 ± 6.92 | 3.73 ± 2.8 |
| 27b | 23.58 ± 8.67 | 38.36 ± 5.98 | 40.16 ± 9.21 | 5.33 ± 2.45 | 27.85 ± 10.92 | 51.07 ± 10.1 |
| 27c | 61.02 ± 17.04 | 149.06 ± 60.33 | 111.75 ± 21.21 | 6.12 ± 2.87 | >100 | 18.65 ± 6.8 |
| IC50 [µM] | ||||
|---|---|---|---|---|
| ATPase | Her2 | A2780 | HCT116 | |
| Luminespib | 0.500 | 0.012 | 0.006 | 0.02 |
| 29a | 0.279 | 0.042 | 0.005 | 0.013 |
| 29b | 0.284 | 0.028 | 0.043 | 0.014 |
| 29c | 0.657 | 0.052 | 0.015 | 0.079 |
| IC50 [µM] | ||||
|---|---|---|---|---|
| MCF-7 | A549 | DU-145 | MDA MB-231 | |
| Etoposide | 2.11 ± 0.024 | 3.08 ± 0.135 | 1.97 ± 0.45 | 1.91 ± 0.84 |
| 32a | 0.33 ± 0.08 | 0.48 ± 0.016 | 0.20 ± 0.06 | 0.95 ± 0.022 |
| 32b | 0.11 ± 0.033 | 0.17 ± 0.089 | 0.09 ± 0.005 | 0.16 ± 0.07 |
| 32c | 0.50 ± 0.07 | 1.54 ± 0.49 | 0.37 ± 0.09 | 0.51 ± 0.041 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bąchor, U.; Mączyński, M.; Sochacka-Ćwikła, A. Therapeutic Potential of Isoxazole–(Iso)oxazole Hybrids: Three Decades of Research. Int. J. Mol. Sci. 2025, 26, 7082. https://doi.org/10.3390/ijms26157082
Bąchor U, Mączyński M, Sochacka-Ćwikła A. Therapeutic Potential of Isoxazole–(Iso)oxazole Hybrids: Three Decades of Research. International Journal of Molecular Sciences. 2025; 26(15):7082. https://doi.org/10.3390/ijms26157082
Chicago/Turabian StyleBąchor, Urszula, Marcin Mączyński, and Aleksandra Sochacka-Ćwikła. 2025. "Therapeutic Potential of Isoxazole–(Iso)oxazole Hybrids: Three Decades of Research" International Journal of Molecular Sciences 26, no. 15: 7082. https://doi.org/10.3390/ijms26157082
APA StyleBąchor, U., Mączyński, M., & Sochacka-Ćwikła, A. (2025). Therapeutic Potential of Isoxazole–(Iso)oxazole Hybrids: Three Decades of Research. International Journal of Molecular Sciences, 26(15), 7082. https://doi.org/10.3390/ijms26157082