Abstract
The presence of cognitive lapses in the post-COVID-19 period, particularly among younger individuals, suggests a potential genetic predisposition. This case–control study aimed to assess the association between neurodegeneration-associated genes and cognitive declines in the post-COVID-19 Armenian population under the age of 65. In addition, we examined other contributing factors, including depressive symptoms, hypovitaminosis D, vitamin B12 and B9 deficiencies, and some viral infections, as potential confounders or effect modifiers. A total of 162 participants (ages 19–65, Med = 43), who were exposed to SARS-CoV-2 in Armenia between 2020 and 2022, participated in this study. Standardized assessments, including the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS) and the Montreal Cognitive Assessment (MoCA), were used to evaluate cognitive functions and mental status, while the Patient Health Questionnaire-9 (PHQ-9) was utilized to assess depressive symptoms. Clinical interview data, comprising yes/no self-reports regarding the presence of cognitive problems and depressive symptoms, were also included. Genetic analysis identified copy number variations (CNVs) in the APP, PSEN1, PSEN2, MAPT, and GRN genes, while viral infections (HSV-1, HSV-2, CMV, EBV, HIV, SARS-CoV-2, Hepatitis A, B, and C) and vitamin D, B12, and B9 deficiencies were measured. Lower cognitive performance was associated with CNVs in PSEN1 (exons 1, 9, 12), GRN (exons 1, 6, 12), and MAPT (exons 2, 8), along with viral infections (HSV-1, HSV-2, HAV-2). The findings indicate that post-COVID-19 cognitive problems are multifactorial and are linked to genetic mutations, viral infections, age, gender, and folic acid deficiency.
1. Introduction
Cognitive lapses, colloquially known as brain fog, impact global health, including the younger population [1]. It negatively affects quality of life, daily functionality, and has socio-economic consequences. Alzheimer’s disease (AD), the most common cause of dementia [2], typically presents in older adults, but early-onset AD (EOAD)—occurring before age 65—represents a rarer form (~2% of AD cases), primarily affecting younger individuals. This led us to investigate the main genetic mutations associated with EOAD, specifically those in the Amyloid Precursor Protein (APP), Presenilin 1 (PSEN1), and Presenilin 2 (PSEN2) genes [3]. PSEN1 and PSEN2 are integral to the γ-secretase complex, which processes APP and leads to the production of amyloid-beta (Aβ) peptides, a key factor in neuronal damage in AD [4]. Additionally, we explored two of the three main genetic mutations associated with frontotemporal dementia (FTD)—those in the Microtubule-Associated Protein tau (MAPT) and Progranulin (GRN) genes—as FTD is a neurodegenerative condition that often affects younger populations [5].
Thus, in light of the significant impact of cognitive lapses on younger populations, a primary goal of ours was to investigate the genetic predisposition to cognitive decline in individuals under the age of 65, with a particular focus on genetic copy number variations (CNVs). Given that age-related risk factors were not a concern in our selected younger cohort, we aimed to isolate the genetic components contributing to early-onset neurodegenerative diseases. To evaluate cognitive functioning, we employed both the Montreal Cognitive Assessment (MoCA) and the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS), prioritizing MoCA due to its greater cultural adaptability and routine clinical use, despite its more limited domain specificity compared to RBANS.
While SNPs have been the predominant focus in post-COVID and neurodegenerative genetic studies, CNVs represent a distinct class of structural genomic variations with potentially greater functional impact due to dosage changes. Recent large-scale analyses have underscored this: a Genome Medicine study using UK and Estonian Biobank data found that rare CNVs significantly contribute to disease risk—often showing stronger effects on clinical phenotypes compared to SNPs [6]. A comprehensive review in Human Genomics further confirmed that rare CNVs affecting dosage-sensitive genes (e.g., PSEN1, MAPT, GRN) exhibit more pronounced impacts on gene expression and cognitive traits than single-nucleotide variants [7]. Importantly, ancestry-dependent differences in CNV burden have been reported, reinforcing the need for population-specific genomic investigations [8]. However, CNVs have been minimally explored in the context of post-COVID cognitive impairment—highlighting both the novelty and necessity of our CNV-focused approach to understanding individual susceptibility to cognitive decline following SARS-CoV-2 infection.
Specifically, we sought to explore the role of CNVs as potential biomarkers in the preclinical stages of conditions like AD, as previous studies have suggested their utility in identifying at-risk individuals before the onset of symptomatic cognitive decline [9]. CNVs are DNA segments of varying lengths, ranging from 1 Kb to several Mb, that can have different numbers of copies. These CNVs, which involve deletions or duplications of genomic segments, play a significant role in the development of complex conditions, including mental disorders [10,11], cognitive decline [12], and depressive symptoms [13] in adults.
In parallel with genetic risk markers, recent research underscores the importance of neurophysiological mechanisms—particularly cortical excitability—as early indicators of cognitive vulnerability. Techniques such as transcranial magnetic stimulation (TMS) have demonstrated that altered excitatory/inhibitory dynamics in cortical circuits, especially in the prefrontal cortex, can reflect subclinical cognitive disturbances in aging and neurodegenerative disease [14,15]. These functional biomarkers may offer complementary insights into genomic data, supporting a more integrated understanding of resilience and decline in cognitive performance
In our study, we directly analyzed CNVs across 14 exons of the MAPT gene, motivated by its well-established role in tauopathies and early-onset neurodegeneration. The MAPT gene was used to assess its relationship with cognitive function, given its well-established role in tauopathies and early-onset neurodegeneration.
The MAPT gene encodes the tau protein, which has important roles in regulating axonal transport, synaptic activity, cytoskeletal dynamics, and cell signaling [16]. In humans, the tau protein undergoes alternative splicing to generate six isoforms with distinct functions [17]. MAPT gene exons 1, 4, 5, 7, 9, 11, and 13 are constitutive exons, meaning they are expressed in all six isoforms of the tau protein. Exon 1 encodes a portion of tau’s N-terminal region, which plays a critical role in axonal transport and facilitates tau’s interaction with the cytoplasmic membrane and signaling proteins [18]. Exons 4, 5, 7, and part of exon 9 encode the region between the N-terminal inserts and the first microtubule-binding repeat. This region contains proline-rich segments predominantly encoded by exons 7 and 9. The three constitutive tubulin-binding repeats are encoded by exons 9, 11, and 12, while exon 13 contributes to the C-terminal region of tau [19,20]. Conversely, exons 2, 3, 4A, 6, 8, and 10 undergo alternative splicing. As a result, six major tau isoforms are found in the human central nervous system (CNS) due to different splicing combinations of exons 2, 3, and 10 [17]. Notably, exon 8 has not been shown to be transcribed in human mature MAPT mRNA [18,21], and its exclusion is considered the default splicing pattern, as demonstrated by exon-trapping experiments [22]. Pathogenic mutations in the MAPT gene are primarily located within exons 9 to 13. In fact, even silent and intronic mutations can contribute to the development of tauopathies [23].
While pathogenic MAPT mutations are well-characterized within exons 9–13, and most GRN CNVs are known to cause haploinsufficiency in frontotemporal dementia, the specific roles of CNVs in MAPT exon 8 and GRN exon 6 remain poorly defined. For example, GRN exon-level deletions—including exon 6—have been observed in FTD families and are considered pathogenic through transcript loss and reduced Progranulin expression [24,25,26]. By contrast, exon 8 of MAPT is typically excluded from mature transcripts, and no functional consequences of its CNVs have been documented [27]. These gaps highlight the need for the functional validation and cautious interpretation of exon-level CNVs, especially under conditions of viral or neuroinflammatory stress.
The signaling pathways involved in cell growth, proliferation, metabolism, cell-to-cell interactions, regeneration, apoptosis, and stress responses play critical roles in cellular function. Under stress conditions, interactions between these pathways and tau proteins can lead to either adaptive responses or apoptosis, with outcomes that may be neuroprotective or neurodegenerative [16]. In this context, the study by Ruiz-Gabarre et al. identified the N-terminal region of tau proteins as the primary binding site for these interactions [18]. This region, which is partially encoded by exon 1 of the MAPT gene (non-alternatively spliced), suggests that mutations in exon 1 (leading to a partially deficient N-terminal region) could impair interactions with signaling systems, potentially contributing to neurodegeneration under stress. One example of such stress is exposure to SARS-CoV-2.
According to the study by Violet et al. [28], tau plays a protective role in maintaining neuronal DNA and RNA integrity during oxidative stress. Alterations in tau can lead to neuronal damage under such stress, which may be a critical factor in the pathogenesis of SARS-CoV-2 infection, as suggested by Wieczfinska et al. [29]. Additionally, the research by Didonna et al. highlights tau’s protective role during neuroinflammation [10]. These findings may help explain why individuals with MAPT gene mutations could be more vulnerable to neuronal damage and subsequent cognitive impairment following exposure to SARS-CoV-2.
Current research on MAPT mutations primarily focuses on small nucleotide polymorphisms (SNPs) that lead to tauopathies and synucleinopathies [30,31]. However, there are a lack of comprehensive data regarding the role of exonic CNV mutations in gene regulation [21].
Our study coincided with the onset of the COVID-19 pandemic [32], during which SARS-CoV-2 emerged as a potential risk factor for cognitive impairment. In the acute phase, 52.31% of patients with COVID-19 experienced cognitive deficits [13]. These deficits manifested in a range of symptoms that significantly impacted daily functioning, including brain fog (32%), memory decline (17.5–35%), and attention impairment (22%) [33]. Post-COVID-19 cognitive impairment has been reported with prevalence rates varying from 7.2% to 59.2%. However, the underlying predisposing factors, mechanisms, and effective treatment approaches remain unclear [34]. In addition, we considered other risk factors contributing to cognitive impairment, including depression [35,36], hypovitaminosis D [37], and deficiencies in vitamin B12 [38] and B9 [39]. We also examined the potential role of human herpesvirus infections, such as herpes simplex virus type 1 and 2 (HSV-1 and HSV-2), cytomegalovirus (CMV), Epstein–Barr virus (EBV) [40], as well as Hepatitis A [41], Hepatitis B [42], Hepatitis C [43], and HIV [44]. These factors were tested as potential confounders and/or effect modifiers.
Thus, this case–control study aimed to evaluate the contribution of neurodegeneration-associated gene copy number variations to cognitive decline in the Armenian Population Following COVID-19 aged 19 to 65, while also examining the role of depressive symptoms, hypovitaminosis D, vitamin B12 and B9 deficiencies, and viral infections as potential confounders or effect modifiers.
2. Results
A total of 162 patients were included in this study, with 35% male and 65% female participants.
2.1. Virological Results and Hypovitaminosis
The following positive virological results and hypovitaminosis statuses were observed in the study population: CMV IgG (97%), HIV (0.7%), HSV-1 (67%), HSV-2 (7%), A-HCV (0.7%), EBV IgG (90%), HBSAG (3.4%), A-HAV 2 (83%), A-COV2 IgG (96%). The prevalence of hypovitaminosis was as follows: B12 (14%), D (48%), B9 (25%) (detailed information available in Supplementary Table S1).
2.2. Genetic Testing Results
Genetic testing was performed on 13 exons of MAPT, 5 exons of GRN, 18 exons of APP, 12 exons of PSEN1, and 13 exons of PSEN2 genes. The results showed the following distributions of homozygous CNV mutations: MAPT exon 1 (62%), exon 2 (2.6%), exon 6 (4.6%), exon 8 (1.3%), exon 12 (0.7%), 13 (0.7%), 14 (0.7%), GRN exon 6 (4.6%), exon 10 (7.9%), exon 12 (7.9%), APP exon 1 (64%), PSEN1 exon 1 (11%), exon 6 (1.3%), exon 12 (1.3%), PSEN2 exon 1 (84%), exon 2 (26%), exon 9 (0.7%), exon 11 (1.3%).
In addition, heterozygous CNV mutations were more common, with notable mutations in the following exons: MAPT exon 1 (26%), exon 3 (0.7%), exon 4 (0.7%), exon 5 (1.3%), exon 6 (89%), exon 7 (2.6%), exon 8 (15%), exon 9 (11%), exon 10 (42%), exon 11 (25%), exon 12 (95%), 13 (12%), 14 (20%); GRN exon 3 (11%), exon 6 (72%), exon 10 (85%), exon 12 (79%); APP exon 1 (32%), exon 3 (2.6%), exon 4 (21%), exon 5 (1.3%), exon 6 (2.0%), exon 7 (2.0%), exon 8 (4.6%), exon 12 (0.7%), exon 13 (1.3%), exon 14 (3.3%), exon 15 (12%), exon 16 (8.6%), exon 17 (43%), exon 18 (0.7%); PSEN1 exon 1 (51%), exon 2 (8.6%), exon 3 (0.7%), exon 5 (0.7%), exon 6 (6.6%), exon 7 (4.0%), exon 8 (2.0%), exon 9 (0.7%), exon 10 (0.7%), exon 12 (11%); PSEN2 exon 1 (16%), exon 2 (63%), exon 3 (11%), exon 4 (5.3%), exon 5 (13%), exon 6 (50%), exon 7 (16%), exon 8 (15%), exon 9 (56%), exon 10 (40%), exon 11 (68%), exon 12 (67%), exon 13 (60%).
No CNV mutations were detected in the following exons: APP exon 2, 9, 10, 11; PSEN1 exon 4 and 11 (Supplementary Table S1).
2.3. Cognitive Status and CNV Mutations
The distribution of CNV mutations in the APP, PSEN1, PSEN2, GRN, and MAPT genes, stratified by cognitive status as measured by the MoCA test, is shown in Figure 1. A comparison of the included risk factors (age, gender, genetic mutations, viruses, hypovitaminosis) was conducted between total neurocognitive test scores (MoCA and RBANS) and across different cognitive domains, as shown in Figure 2. The results were adjusted for multiple comparisons, revealing a significant change in the findings, as shown in Figure 3.
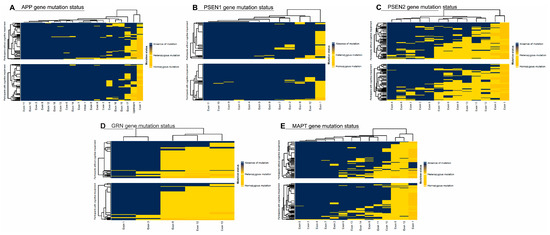
Figure 1.
(A) Amyloid Precursor Protein (APP), (B) Presenilin 1 (PSEN1), (C) Presenilin 2 (PSEN2), (D) Progranulin (GRN), and (E) Microtubule-Associated Protein tau (MAPT) gene distribution.
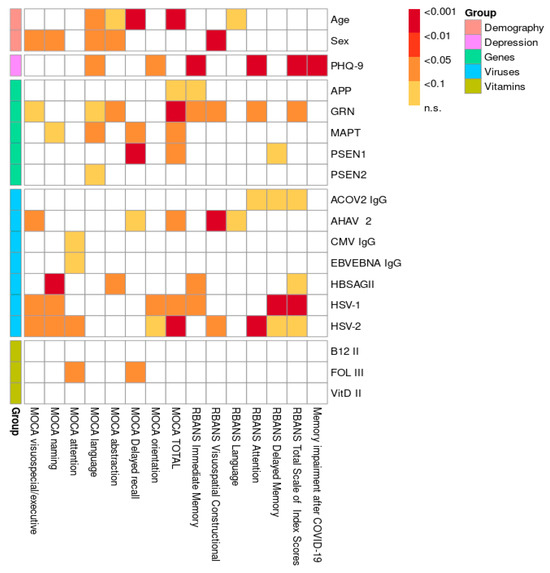
Figure 2.
Visualization of p-values without multiple comparison adjustment.
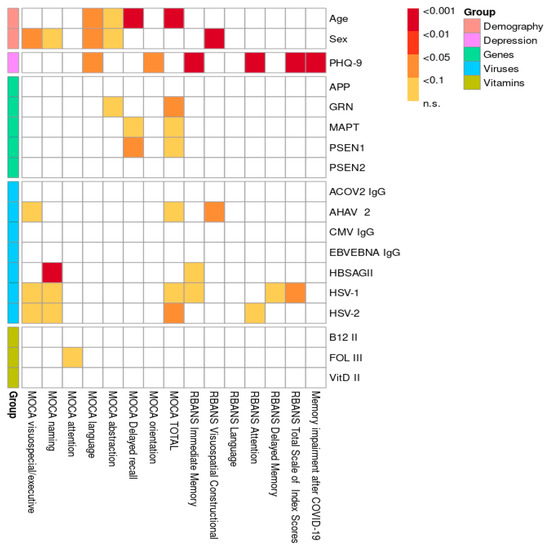
Figure 3.
Visualization of p-values with multiple comparison adjustment.
The overall statistical comparison revealed a strong association between the total MoCA score, short-term memory, and age, which remained significant after adjustment (p < 0.001). Similarly, both the MoCA and RBANS results showed a significant association between age and speech impairment in the study population, which persisted after adjustment (p < 0.01). A weak association was observed between abstract thinking and age according to the MoCA results (p < 0.1), even after adjustment. Furthermore, both the MoCA (p < 0.05) and RBANS (p < 0.001) results indicated that men had lower scores and exhibited greater overall cognitive impairment compared to women. After adjustment for multiple comparisons, a statistically significant relationship was observed between speech dysfunction and men (p < 0.05), along with a weak association with abstract thinking and naming (p < 0.1). Our research demonstrates a significant correlation between depression and cognitive impairment in young and middle-aged individuals, as evidenced by the RBANS results both before and after adjustment. Specifically, memory (p < 0.001) and attention (p < 0.001) were particularly impaired in the context of depression. Furthermore, complaints of cognitive impairment following COVID-19 were significantly associated with a depressive state (p < 0.001). Several associations between CNV mutations in genes (APP, GRN, MAPT, PSEN1, PSEN2) and various domains of cognitive impairment were identified. A few remained significant after adjustment for multiple comparisons: GRN gene mutations were associated with abstract thinking (p < 0.1), while MAPT and PSEN1 mutations were linked to memory impairment (p < 0.1 and p < 0.05, respectively), as shown in Figure 3 and Supplementary Table S2. Additionally, all three genes were significantly associated with the MoCA total score. Interestingly, based on our research data, we suggest that the MoCA may be a more sensitive test for assessing cognitive impairment in the studied population compared to the RBANS. A detailed statistical analysis of the different exons of the PSEN1 (Table 1A,B), MAPT (Table 2A,B), and GRN (Table 3A,B) genes in relation to the significantly associated cognitive impairment domains was conducted based on MoCA data. For the PSEN1 gene, a model–submodel test showed a significant association of MoCA delayed recall with exon 1 (p = 0.0070) with an effect size compatible with 95% CI [−2.4, −0.48]. For the MoCA total score, associations were found for exon 9 and exon 12 (p = 0.026 and p = 0.027 according to model–submodel tests correspondingly). For exon 9, a 95% CI of [0.98, 15.00] was detected for a homozygous type. Unfortunately for a contrast test, exon 12 did not reach significance (CI [−9.90, 0.12]). This discrepancy may reflect limited statistical power or a true lack of effect; accordingly, definitive conclusions should await validation in larger cohorts. According to the model–submodel test, CNV mutations in GRN exon 1 (p = 0.027) and exon 6 (p = 0.0029) may be associated with a specific cognitive impairment expressed by a MoCA abstract score. However, an analysis of contrasts shows that the potential effect size is rather modest (95% CI is [0.045, 0.73] for exon 1 and [0.160, 0.810] for exon 2. For the MoCA total score, the model–submodel test highlights associations with exon 6 (p = 0.00015) and exon 12 (p = 0.00130). The test of contrasts shows that a single mutation in exon 6 is associated with a change in the MoCA total score ranging from 0.95 to 6.90, while a mutation in exon 12 is associated with a change ranging from −6.20 to −0.97. Regarding the impact of CNV mutations in the MAPT gene on cognitive impairment, a statistically significant association with the MoCA delay recall was found for exon 2 (p = 0.014) and with the MoCA total score for exon 8 (p = 0.033). The contrast analysis of effect sizes results in a 95% CI of [0.49, 4.8] and [−13, −1], correspondingly. With regard to the impact of hypovitaminosis (B9, B12, D) and viruses on objective cognitive decline (MoCA; RBANS), after adjustment, associations were found between HSV-1 (p < 0.05), HSV-2 (p < 0.05), and AHAV-2 (p < 0.01). Infections were identified as possible confounders for cognitive impairment in different domains: Hepatitis B for naming (p < 0.001) and memory (p < 0.1); Hepatitis A for executive function (p < 0.05); HSV-1 for executive function, naming, and memory (p < 0.1); and HSV-2 for executive function, naming, and attention (p < 0.1). A weak association was observed between folate (vitamin B9) deficiency and attention (p < 0.1). Hypovitaminosis B12 and D, however, did not show an effect on cognitive dysfunction in our study population (Figure 3; Supplementary Table S2).

Table 1.
Statistical associations between PSEN1 exons and cognitive impairment domains identified from MoCA scores. (A) associations between PSEN1 and MoCA delayed recall; (B) associations between PSEN1 and MoCA total score.
Table 2.
Statistical associations between MAPT exons and cognitive impairment domains identified from MoCA scores. (A) associations between MAPT and MoCA delayed recall; (B) associations between PSEN1 and MoCA total score.
Table 3.
Statistical associations between GRN exons and cognitive impairment domains identified from MoCA scores. (A) associations between MAPT and MoCA delayed recall; (B) associations between PSEN1 and MoCA total score.
3. Discussion
This study examined the association between neurodegeneration-related genetic markers and post-COVID-19 cognitive impairments. The sample consisted of a cohort of individuals of Armenian ethnicity who were exposed to SARS-CoV-2 in Armenia between 2020 and 2022. Notably, this cohort was demographically distinct, representing a younger age group (Med = 43) than typically seen in studies of neurodegeneration-related genetic markers. We investigated mutations linked to early-onset Alzheimer’s disease (EOAD), particularly in the APP, PSEN1, and PSEN2 genes, which affect amyloid-beta production and neuronal integrity. Additionally, mutations in the MAPT and GRN genes—commonly associated with frontotemporal dementia (FTD), a condition that disproportionately affects younger individuals—were analyzed. Finally, this study compared self-reported cognitive lapses with standardized assessments to evaluate how these measures correlate and whether their relationship is influenced by clinical and demographic variables.
AD is characterized by the accumulation of amyloid-β (Aβ) in plaques and abnormally phosphorylated tau in neurofibrillary tangles [12]. Aβ accumulation begins up to two decades before the onset of dementia [45]. EOAD is caused by mutations in three key genes: APP, PSEN1, and PSEN2 [46]. According to ACMG-AMP guidelines, PSEN1 mutations are the most common cause of AD in patients (89.16%), followed by APP mutations (46.97%).
The APP gene, located on chromosome 21q, contains 18 exons and encodes a protein involved in synaptic activity, transcriptional regulation, plasticity, and neuroprotection [47]. Genetic alterations in APP increase protein levels, leading to early-onset AD. Exons 7, 8, and 15 are subject to alternative splicing [48], and duplications or triplications of the APP gene can result in familial Alzheimer’s disease (fAD) [49,50]. Mutations in APP are most prevalent in exons 14, 16, and 17, and no variants are found in exons 1, 2, 3, 4, 8, 10, 15, and 18, which aligns with previous studies [51]. Mutations in exon 16 affect γ-secretase cleavage regions, influencing Aβ42 and Aβ40 ratios crucial for amyloid plaque formation [52]. As the catalytic component of the γ-secretase complex is encoded by PSEN1 and PSEN2, mutations in those genes disrupt APP processing, leading to increased Aβ accumulation [53]. It is also interesting that abnormal axodendritic proliferation has been observed in neurons with a PSEN1 mutation, which is explained by elevated levels of the cytoplasmic APP C-terminal fragments [54].
3.1. Cognitive Functioning and Depressive Symptoms
One of the broader aims of our study was to examine whether genetic factors could help explain why some COVID-19 patients experience cognitive lapses while others do not. Our findings showed that PSEN1 CNV mutations in exons 1, 9, and 12 were associated with specific cognitive deficits and helped distinguish patients based on the presence and type of cognitive impairment. The most prominent finding was mild indicators of memory problems, which are commonly reported by individuals who have had COVID-19. Other cognitive domains also showed statistically significant associations; however, our team did not assign them strong weight, given the multiple comparisons. Further specification and standardization of memory assessments is warranted, given the lack of consistency in our findings between MoCA and RBANS—partly due to different tests measure memory using varying methods and criteria.
Our findings suggest that depressive symptoms contributed to post-COVID-19 cognitive decline, as reflected in both performance-based impairments in memory and individuals’ self-reported difficulties with thinking and mental clarity. This raises questions about the origin of cognitive problems in COVID-19: Is it primarily a consequence of the psychomotor slowing commonly associated with depression, which then impacts cognitive functioning, or does it reflect a direct cognitive deficit independent of mood symptoms? We hypothesize that for the general population, the former is more likely—particularly in the context of post-COVID-19 ‘cognitive fog,’ which may reflect slowed processing and attention linked to depressive symptomatology rather than isolated neurocognitive damage. However, for individuals with a genetic predisposition to neurodegeneration, COVID-19 may act as a catalyst, accelerating underlying pathological processes and resulting in more pronounced and persistent cognitive impairment.
To summarize, we identified CNV-associated cognitive impairments involving PSEN1 (exons 1, 9, and presumably 12), GRN (exons 1, 6, 12), and MAPT (exons 2 and 8), which is consistent with prior findings by Xiao et al. [29] reporting pathogenic PSEN1 variants in exons 4–8, 11, and 12.
We hypothesize that PSEN1 exon 12 CNV mutations, common in our cohort, could potentially be pathogenic for the Armenian population. However, as noted in the Results Section, the inconsistency between the model–submodel test and the contrast test for exon 12 underscores the need to investigate this phenomenon in larger cohorts. Given the limited statistical power and wide confidence intervals, the observed association should be interpreted with caution and considered hypothesis-generating until validated in independent samples. Most of the PSEN2 variants were found in exon 5, exon 7, and exon 4, and no variants were detected in exon 1, exon 2, and exon 3 [51]. However, in the current study, no significant associations were found with CNV mutations in APP, PSEN1, or PSEN2 genes and cognitive decline outside of these exons. Notably, we did not detect CNV mutations in APP exons 2, 9, 10, or 11, nor in PSEN1 exons 4 and 11, which is in line with previous reports [51].
Another important cause of early-onset dementia is frontotemporal lobar degeneration (FTLD), which is often associated with familial forms of the disease [5,55]. It is a highly heritable autosomal dominant disorder [56] and presents with different clinical forms: behavioral variant FTD (bvFTD), the semantic variant (svPPA), and the nonfluent variant (nfPPA) of primary progressive aphasia [11]. The GRN and MAPT genes are key contributors to FTLD [5,55]. In a Belgian cohort of familial FTD patients, PGRN mutations were found to be 3.5 times more prevalent than MAPT mutations, underscoring the significant role of PGRN in the etiology of FTD [57]. In contrast, the GRN and MAPT mutations are relatively uncommon in Russia [58].
In our study of the Armenian population, we identified both homozygous and heterozygous CNV mutations in the MAPT gene across several exons. Homozygous mutations were found in exon 1 (62%), exon 2 (2.6%), exon 6 (4.6%), exon 8 (1.3%), exon 12 (0.7%), exon 13 (0.7%), and exon 14 (0.7%). Heterozygous mutations were observed in exon 1 (26%), exon 3 (0.7%), exon 4 (0.7%), exon 5 (1.3%), exon 6 (89%), exon 7 (2.6%), exon 8 (15%), exon 9 (11%), exon 10 (42%), exon 11 (25%), exon 12 (95%), exon 13 (12%), and exon 14 (20%). Despite the fact that exon 8 has not been identified in cortical tau from humans [59], our study observed a statistical association between CNV mutations in exon 8 of the MAPT gene and cognitive decline. However, given the exploratory nature of this analysis and the relatively small number of individuals carrying these mutations, this association should be interpreted cautiously. According to the Ensembl database, transcripts containing exon 8 encompass all alternative exons except exon 0, which may either be included or excluded [21]. This raises a compelling question as to how CNV mutations in exon 8 could contribute to cognitive decline. While exon 8 is generally excluded in mature MAPT transcripts in the adult human cortex [48,60], its presence has been documented in certain alternative isoforms expressed under non-physiological or stress-induced conditions. It is conceivable that CNV mutations affecting exon 8 may influence splicing efficiency, RNA processing, or other regulatory mechanisms—particularly in response to viral infection or inflammatory stress, such as SARS-CoV-2. Recent studies have shown that MAPT splicing is sensitive to cellular stress and may shift toward non-canonical isoform expression in disease contexts. Thus, our findings may reflect a stress-induced reactivation of exon 8 usage or an indirect regulatory consequence of structural variation at this locus. Although further experimental validation is needed, this observation—while intriguing—remains speculative and should be viewed as a preliminary signal that warrants replication in future studies.
A weak association was observed with exon 7, which could be attributed to the small sample size or the limited sensitivity of the neurocognitive assessment tools used. While memory impairment showed low statistical significance in relation to MAPT CNV mutations in exons 8 and 9, these findings may potentially correlate with memory decline in a larger sample or with more sensitive memory assessments.
GRN is another key gene implicated in FTLD [57]. GRN-related frontotemporal dementia (GRN-FTD) is inherited in an autosomal dominant manner, with nearly 95% of affected individuals having an affected parent [61]. Heterozygous mutations are found in approximately 5–25% of familial FTLD cases and 5% of sporadic FTLD cases [55]. The GRN gene, located on chromosome 17, is primarily associated with behavioral variant frontotemporal dementia (FTD), primary progressive aphasia (PPA), atypical parkinsonism, and corticobasal syndrome when mutations occur [61]. Of the 172 identified GRN mutations, 79 are considered pathogenic [62].
In our study, we examined five exons of the GRN gene (1, 3, 6, 10, 12). CNV mutations in GRN exon 6 and exon 12 showed the strongest associations with cognitive impairment. Regarding executive dysfunction, particularly in abstract thinking, we observed significant associations with CNV mutations in GRN exon 1, exon 6, and exon 12. However, there is a gap in the data that clearly links CNV mutations to disease development, and it remains unclear how combinations of multiple CNV mutations within a single gene might influence the overall mutagenic process. In our cohort, most participants, selected using a stratified sampling technique, were carriers of genes associated with EOAD and FTD, including APP, PSEN1, PSEN2, MAPT, and GRN. At this stage, it remains unclear whether the observed results are due to natural biological processes or if specific CNV mutations are directly responsible for cognitive impairment. It is yet to be determined how many of the participants will develop EOAD or FTD with cognitive impairment in the future. Currently, there is no national registry for diseases leading to cognitive impairment, which limits our ability to provide clear data on this issue and to explore potential ethnic differences.
While the role of viruses in dementia development remains debated, a meta-analysis suggests that viral exposure could be a potential risk factor for cognitive decline [40]. Herpes viruses, for instance, may contribute to the development of late-onset AD [63,64], potentially through mechanisms such as β-amyloidosis [65]. Herpes infection is particularly linked to executive function impairment, affecting regions like the orbitofrontal and anterior cingulate cortices [66], a finding that aligns with our study.
In a similar vein, over 50% of individuals with viral Hepatitis experience cognitive impairment [67], even in the absence of cirrhosis [68] or hepatic encephalopathy [43]. Several studies have highlighted that liver pathologies can impair the liver’s ability to eliminate beta-amyloid from the bloodstream [69], promoting its accumulation in the brain and potentially contributing to Alzheimer’s disease [60]. Individuals with chronic Hepatitis B and C are at an increased risk for deficits in executive function, mental processing speed, learning capacity, memory, complex attention, perceptual–motor skills, and social cognition [70]. Our research further suggests a significant link between Hepatitis B and linguistic impairment, particularly in object-naming ability. Additionally, seropositive Hepatitis A has been associated with diminished psychomotor speed [41], and our findings indicate a similar association with executive dysfunction, particularly in visuospatial abilities.
To address the question of causality between CNV mutations and neuronal vulnerability to stressors such as SARS-CoV-2 infection or other viral exposures, future hypothesis-generating research is warranted. One possible experimental direction involves in vitro models using neuronal cultures with gene-specific knockouts to assess differential responses to viral stress. While promising, this concept remains exploratory and would require validation through a series of preclinical steps. As a more immediate translational pathway, future efforts should focus on replicating our CNV findings in larger, ethnically diverse cohorts, characterizing the functional consequences of CNVs through transcriptomic or epigenetic profiling, and developing early detection tools (e.g., biomarker panels) for individuals at risk of cognitive decline. Additionally, cost-effectiveness studies of population-level genomic screening could help assess the feasibility of incorporating such approaches into precision medicine strategies for post-COVID-19 cognitive vulnerability.
3.2. Strengths and Limitations
3.2.1. Strengths
A key strength of this study is the inclusion of the PHQ-9 to quantify depressive symptoms, which are known to influence subjective and objective measures of cognitive function. To address the potential confounding role of depressive symptoms in cognitive impairment, we included PHQ-9 scores as covariates in the adjusted regression models. The inclusion of PHQ-9 modestly attenuated some associations between CNVs and cognitive scores, particularly in RBANS subdomains, but key associations—especially involving PSEN1 exon 9 and MAPT exon 2—remained statistically significant. This suggests that while depressive mood contributes to cognitive variability, the genetic associations observed may be at least partially independent of depression.
3.2.2. Limitations
Our study cohort consists exclusively of individuals of Armenian nationality recruited from a single hospital complex, which limits the generalizability of our findings to broader populations. Genetic backgrounds, environmental factors, and healthcare access can vary significantly across different ethnic groups and geographical regions, potentially influencing the prevalence and impact of CNV mutations in the genes studied. Therefore, while our results provide valuable insights into the Armenian subpopulation, caution is warranted when extrapolating these findings to other ethnicities or populations. Future studies involving diverse cohorts from multiple geographic locations are essential to validate and expand upon our observations, ensuring broader applicability.
In addition, although two cognitive assessment tools—MoCA and RBANS—were utilized, our analysis prioritized MoCA due to its higher sensitivity to mild cognitive impairment and broader cross-cultural adaptability. The Armenian version of MoCA has been routinely applied in clinical practice, whereas RBANS, while domain-specific and comprehensive, poses challenges in non-English-speaking populations due to linguistic and normative limitations. Specifically, in the absence of Armenian national RBANS norms, we relied on cohort-specific median scores rather than standardized cutoffs, which may influence the interpretation of RBANS-derived results. This further underscores the need for culturally adapted and validated neuropsychological tools in future cognitive research in this population.
Moreover, the modest sample size (N = 162), relative to the high dimensionality of genetic and neurocognitive data, represents an additional limitation. Although our study identified statistically significant associations between specific CNV mutations and cognitive outcomes, the limited cohort size may reduce statistical power and increase the risk of both false positives and false negatives. For example, findings involving MAPT exon 8 and PSEN1 exon 12 should be interpreted with restraint, given the wide confidence intervals and the exploratory nature of these analyses. Therefore, these findings should be interpreted with caution and considered exploratory. Future studies involving larger, independent cohorts with harmonized genetic and neurocognitive assessments are essential to validate these associations and further elucidate the role of CNVs in post-COVID-19 cognitive vulnerability.
4. Materials and Methods
4.1. Study Design
A case–control design was employed for this study. The case group consisted of individuals with an objective decline in cognitive function, while the control group included individuals with a normal cognitive state. Classification into case or control groups was based solely on current objective cognitive test results, not on self-reported history. Cognitive status was assessed based on performance in key mental domains, including perception, learning, memory, understanding, awareness, reasoning, judgment, attention, and language [71]. Although 28 participants (17%) reported subjective cognitive difficulties prior to COVID-19, none had undergone formal neuropsychological evaluation before infection. Due to the lack of pre-COVID-19 cognitive data, we did not treat this subgroup separately, and allocation was based on their current post-COVID-19 cognitive status. We analyzed cognitive function in relation to various risk factors for decline, including age, gender, CNV mutations in genes associated with early-onset neurodegenerative diseases (APP, PSEN1, PSEN2, GRN, MAPT), hypovitaminosis (B12, B9, D), previous viral infections (SARS-CoV-2, HSV-1, HSV-2, CMV, EBV, HIV, Hepatitis A, B, and C), and depression status. The study flow is depicted in Figure 4.
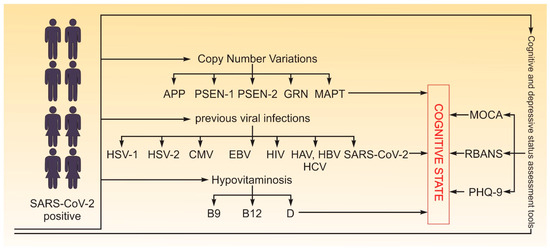
Figure 4.
Study flow. In total, 162 participants (both male and female) aged 19 to 65 years with confirmed SARS-CoV-2 were tested for cognitive state with MoCA, RBANS and PHQ-9 (for depression), CNV mutations in genes associated with early-onset neurodegenerative diseases (APP, PSEN1, PSEN2, GRN, MAPT), hypovitaminosis (B12, B9, D), and viral infections (SARS-CoV-2, HSV-1, HSV-2, CMV, EBV, HIV, Hepatitis A, B, and C).
4.2. Participants
We recruited participants from the Heratsi Hospital Complex in Yerevan, Armenia, between January 2020 and August 2022. Data collection occurred during outpatient visits and included a single physical examination, laboratory tests, and cognitive and depression assessments. A total of 162 participants (both male and female) aged 19 to 65 years confirmed SARS-CoV-2 infection via RT-PCR or rapid antigen test were included. These participants had or did not have a prior history of cognitive impairment. Patients were excluded if they exhibited an impaired level of consciousness, required mechanical ventilation, or had severe organ failure during the COVID-19 pandemic. All participants provided informed consent to participate in this study and were of Armenian nationality.
4.3. Cognitive Assessments
Cognitive function was measured using the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS) [33] and the Montreal Cognitive Assessment (MoCA) [52], with both total scores and domain-specific scores analyzed as primary outcome variables. The RBANS is a standardized neuropsychological battery designed to evaluate immediate memory, visuospatial/constructional abilities, language, attention, and delayed memory, whereas the MoCA is a brief screening tool used to detect mild cognitive impairment, assessing domains such as attention, executive function, memory, language, visuospatial skills, abstraction, calculation, and orientation. Both the RBANS and MoCA have been previously translated and culturally adapted for use in the Armenian language [72].
The results of both the MoCA and RBANS tests were converted into a nominal scale of positive or negative cognitive impairment status, based on the overall score. This transformation was made in line with the study’s aim—to determine the presence or absence of cognitive impairment rather than the level of impairment. A cutoff score of 26/30 was used for the MoCA test, with scores ≥ 26 indicating normal cognitive function and scores < 26 indicating impaired cognitive function [73]. For the RBANS test, a threshold of 90 points was selected, which varies ± 10 standard deviations (SDs) from the American norms (with a ±10–15 SD range allowed according to test guidelines) [18]. This threshold was chosen as more applicable for the Armenian population, based on our sample’s average scores and previous research findings [72].
4.4. Depression Assessment
Depression status was assessed using the PHQ-9 test, with scores of 0–4 indicating normal status and scores ≥ 5 indicating the presence of depression [74].
Clinical Interview: Participants also took part in a qualitative interview, which included a set of standardized questions about their experience with common post-COVID-19 symptoms, including fatigue, mood changes, and cognitive difficulties such as memory lapses, attention problems, and slowed thinking. Participants were asked to reflect on whether these symptoms were present before or emerged after their COVID-19 illness.
4.5. Genetic Analysis
The explanatory variables were measured using the SALSA Multiplex Ligation-Dependent Probe Amplification (MLPA) technique for the detection of CNVs. CNVs were identified as either homozygous or heterozygous deletions or duplications in the following genes: SALSA MLPA probemix P471-A1 for PSEN1 (Exons 1–12), PSEN2 (Exons 1–13), and APP (Exons 1–18) [75], and SALSA MLPA probemix P275-C3 for MAPT (Exons 1–14) and GRN (Exons 1, 3, 6, 10, 12) [76]. In the dataset, a value of 0 denotes a homozygous mutation, 1 indicates a heterozygous mutation, and 2 corresponds to individuals without CNVs in the respective exon.
4.6. Laboratory Measurements
The Electrochemiluminescence Immunoassay (ECLIA) method was used to analyze viral markers (HSV-1, HSV-2, CMV, EBV, HIV, SARS-CoV-2, Hepatitis A, B, and C) and detect deficiencies in vitamins D, B12, and B9 from blood samples. Hypovitaminosis was defined by the following thresholds: vitamin D—<30 ng/mL; vitamin B12—<191 pg/mL; and vitamin B9—<3.1 ng/mL. The following thresholds were applied for viral infections—SARS-CoV-2 IgG: >1.0 COI; HSV-1: <0.9 COI; HSV-2: <0.9 COI; CMV: <0.5 U/mL; EBV: <1.0 COI; HIV: <0.9 COI; Hepatitis A: >1.0 COI; Hepatitis B: <0.9 COI; Hepatitis C: <0.9 COI. Tests were properly calibrated and regularly controlled according to the manufacturer’s recommendations (Roche Diagnostics GmbH, Mannheim, Germany).
4.7. Statistical Methods
All statistical analyses were performed using R version 4.3.2 (R Core Team, 2023) within the RStudio environment (version 2023.09.1+494). A significance level of p < 0.05 was applied to all tests unless otherwise specified. The following approaches were employed:
- a.
- Non-Parametric Methods
For continuous variables, Spearman’s rank correlation coefficient was used to assess correlations. Group comparisons across independent groups were evaluated using the Mann-Whitney U test or Kruskal–Wallis test depending on the number of groups. Male sex and a low level of AHAV were used as a reference level when performing a Mann–Whitney test.
- b.
- Categorical Variables
Associations between categorical variables were analyzed using the chi-square test of independence.
- c.
- Gene–Score Associations
The association between a gene and cognitive function scores, including the MoCA score, RBANS score, and self-reported cognitive impairment scores, was analyzed using model–submodel tests. In these models, cognitive scores were treated as dependent variables, while individual exons (presence/absence of mutation) were modeled as independent categorical predictors. Using the model–submodel approach in each test, we compared a full model containing all relevant exons and a trivial model, containing only an intercept.
- d.
- Multiple Comparison Adjustment
For multiple hypothesis testing within each feature group (anthropometric measurements, depression scores, genetic variables, vitamins, and viruses), Benjamini–Hochberg (BH) correction was applied to control the false discovery rate.
- e.
- Multiple Regression for Gene Validation
Significant genes identified in the initial analyses were further validated using multiple regression models. In these models, all relevant exons were included as independent categorical variables, while cognitive function scores were used as the dependent variable. A model–submodel comparison test was performed for each exon to obtain a single p-value per exon, thereby maximizing statistical power. Additionally, we assessed contrasts highlighting differences between categories 1 and 2 compared to category 0, which was treated as the reference. From this model, we derived effect sizes corresponding to different heterozygous and homozygous mutation statuses.
5. Conclusions
The significant prevalence of CNV mutations in the APP, PSEN1, PSEN2, MAPT, and GRN genes within the Armenian population may have potential pathogenic impacts on cognitive functions. We identified associations with cognitive impairment specifically in the CNV mutations of the PSEN1 (exons 1, 9, 12), GRN (exons 1, 6, 12), and MAPT (exons 2, 8) genes. Although genetic mutations do not directly influence self-reported cognitive deterioration, post-COVID-19 cognitive complaints are primarily associated with depression. Cognitive impairment in our cohort is also correlated with factors such as age, gender, depressive mood, viral infections (HSV1, HSV2, HAV, and HBV), and folic acid insufficiency. The findings of this study are primarily applicable to the subpopulation of patients exposed to SARS-CoV-2. However, further research is needed to assess the broader applicability of these results to other populations or contexts.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms26146965/s1.
Author Contributions
Y.H.—Conceptualization, data curation, formal analysis, investigation, methodology, writing—original draft, writing—review and editing; H.Y.—data curation, investigation, methodology, writing—original draft; G.H.—data curation, investigation, methodology, writing—original draft; G.P.—data curation, investigation, methodology, writing—original draft; H.H.—formal analysis, investigation, methodology, validation, writing—original draft; writing—review and editing; A.M.—funding acquisition, resources, writing— original draft; A.A.—writing—original draft, writing—review and editing; K.Y.—conceptualization, formal analysis, funding acquisition, investigation, methodology, project administration, resources, supervision, writing—original draft, writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Higher Education and Science Committee, Ministry of Education, Science, Culture and Sports of RA (25YSMU-CON-I-3A) and YSMU.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of Yerevan State Medical University (N 9; 20 May 2021).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data can be made available by the corresponding author upon reasonable request.
Acknowledgments
We express our gratitude to all patients and medical personnel for their assistance with the laboratory assessment of vitamins, viruses, and antibodies, Armine Chopikyan and Evgeny Bakin for their contributions to the statistical analyses, our students Hamlet Torosyan and Ani Grigoryan for their help with serum collection and related organizational tasks, and Hasmik Harutyunyan for the graphical illustrations.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Launer, L.J. Statistics on the burden of dementia: Need for stronger data. Lancet Neurol. 2019, 18, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Neuner, S.M.; Tcw, J.; Goate, A.M. Genetic architecture of Alzheimer’s disease. Neurobiol. Dis. 2020, 143, 104976. [Google Scholar] [CrossRef] [PubMed]
- Bagyinszky, E.; Youn, Y.C.; An, S.; Kim, S. The genetics of Alzheimer’s disease. Clin. Interv. Aging 2014, 9, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Watanabe, H.; Wu, B.; Lee, S.H.; Li, Y.; Tsvetkov, E.; Bolshakov, V.Y.; Shen, J.; Kelleher, R.J. Presenilin-1 Knockin Mice Reveal Loss-of-Function Mechanism for Familial Alzheimer’s Disease. Neuron 2015, 85, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Greaves, C.V.; Rohrer, J.D. An update on genetic frontotemporal dementia. J. Neurol. 2019, 266, 2075–2086. [Google Scholar] [CrossRef] [PubMed]
- Auwerx, C.; Jõeloo, M.; Sadler, M.C.; Tesio, N.; Ojavee, S.; Clark, C.J.; Mägi, R.; Estonian Biobank Research Team; Esko, T.; Metspalu, A.; et al. Rare copy-number variants as modulators of common disease susceptibility. Genome Med. 2024, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Cocoș, R.; Popescu, B.O. Scrutinizing neurodegenerative diseases: Decoding the complex genetic architectures through a mul-ti-omics lens. Hum. Genom. 2024, 141, 18. [Google Scholar] [CrossRef] [PubMed]
- Rahaie, Z.; Rabiee, H.R.; Alinejad-Rokny, H. CNVDeep: Deep association of copy number variants with neurocognitive disorders. BMC Bioinform. 2024, 25, 283. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-P.; Tucci, A.A.; Conery, M.; Leung, Y.Y.; Kuzma, A.B.; Valladares, O.; Chou, Y.-F.; Lu, W.; Wang, L.-S.; Schellenberg, G.D.; et al. Copy Number Variation Identification on 3800 Alzheimer’s Disease Whole Genome Sequencing Data from the Alzheimer’s Disease Sequencing Project. Front. Genet. 2021, 12, 752390. [Google Scholar] [CrossRef] [PubMed]
- Didonna, A.; Cantó, E.; Shams, H.; Isobe, N.; Zhao, C.; Caillier, S.J.; Condello, C.; Yamate-Morgan, H.; Tiwari-Woodruff, S.K.; Mofrad, M.R.; et al. Sex-specific Tau methylation patterns and synaptic transcriptional alterations are associated with neural vulnerability during chronic neuroinflammation. J. Autoimmun. 2019, 101, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Ljubenkov, P.A.; Miller, B.L. A Clinical Guide to Frontotemporal Dementias. Focus 2016, 14, 448–464. [Google Scholar] [CrossRef] [PubMed]
- Jagust, W. Imaging the evolution and pathophysiology of Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 687–700. [Google Scholar] [CrossRef] [PubMed]
- De Alcântara, I.J.; Nuber-Champier, A.; Voruz, P.; Cionca, A.; Assal, F.; Péron, J.A. Cognitive Deficits in the Acute Phase of COVID-19: A Review and Meta-Analysis. J. Clin. Med. 2023, 12, 762. [Google Scholar] [CrossRef] [PubMed]
- Di Fazio, C.; Scaliti, E.; Stanziano, M.; Nigri, A.; Demichelis, G.; Tamietto, M.; Palermo, S. rTMS for enhancing cognitive reserve: A case report. Brain Disord. 2025, 18, 100221. [Google Scholar] [CrossRef]
- Palermo, S.; Di Fazio, C.; Scaliti, E.; Stanziano, M.; Nigri, A.; Tamietto, M. Cortical excitability and the aging brain: Toward a biomarker of cognitive resilience. Front. Psychol. 2025, 16, 1542880. [Google Scholar] [CrossRef] [PubMed]
- Sinsky, J.; Pichlerova, K.; Hanes, J. Tau Protein Interaction Partners and Their Roles in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2021, 22, 9207. [Google Scholar] [CrossRef] [PubMed]
- Andreadis, A. Tau gene alternative splicing: Expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim. Biophys. Acta. (BBA)—Mol. Basis Disease 2005, 1739, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Gabarre, D.; Carnero-Espejo, A.; Ávila, J.; García-Escudero, V. What’s in a Gene? The Outstanding Diversity of MAPT. Cells 2022, 11, 840. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, S.; Bell, M.; Klimek, J.; Zempel, H. Differential Effects of the Six Human TAU Isoforms: Somatic Retention of 2N-TAU and Increased Micro-tubule Number Induced by 4R-TAU. Front. Neurosci. 2021, 15, 643115. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Tau filaments in neurodegenerative diseases. FEBS Lett. 2018, 592, 2383–2391. [Google Scholar] [CrossRef] [PubMed]
- Caillet-Boudin, M.-L.; Buée, L.; Sergeant, N.; Lefebvre, B. Regulation of human MAPT gene expression. Mol. Neurodegener. 2015, 28, 10. [Google Scholar] [CrossRef] [PubMed]
- Andreadis, A.; Nisson, P.E.; Kosik, K.S.; Watkins, P.C. The exon trapping assay partly discriminates against alternatively spliced exons. Nucleic Acids Res. 1993, 21, 2217–2221. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Ahn, S.I.; Gallo, J.-M. Tau mis-splicing in the pathogenesis of neurodegenerative disorders. BMB Rep. 2016, 49, 405–413. [Google Scholar] [CrossRef] [PubMed]
- De Houwer, J.F.H.; Dopper, E.G.P.; Rajicic, A.; van Buuren, R.; Arcaro, M.; Galimberti, D.; Breedveld, G.J.; Wilke, M.; van Minkelen, R.; Jiskoot, L.C.; et al. Two novel variants in GRN: The relevance of CNV analysis and genetic screening in FTLD patients with a negative family history. J. Neurol. 2024, 272, 64. [Google Scholar] [CrossRef] [PubMed]
- Milan, G.; Napoletano, S.; Pappatà, S.; Gentile, M.T.; Colucci-D’Amato, L.; Della Rocca, G.; Maciag, A.; Rossetti, C.P.; Fucci, L.; Puca, A.; et al. GRN deletion in familial frontotemporal dementia showing association with clinical variability in 3 familial cases. Neurobiol. Aging 2017, 53, 193.e9–193.e16. [Google Scholar] [CrossRef] [PubMed]
- Wauters, E.; Gossye, H.; Frydas, A.; Sieben, A.; Van Broeckhoven, C. Rare exonic variant affects GRN splicing and contributes to frontotemporal lobar degeneration. Neurobiol. Aging 2023, 130, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Fischer, I. Evolutionary perspective of Big tau structure: 4a exon variants of MAPT. Front. Mol. Neurosci. 2022, 15, 1019999. [Google Scholar] [CrossRef] [PubMed]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell. Neurosci. 2014, 8, 84. [Google Scholar] [CrossRef] [PubMed]
- Wieczfinska, J.; Kleniewska, P.; Pawliczak, R.; Kouretas, D. Oxidative Stress-Related Mechanisms in SARS-CoV-2 Infections. Oxidative Med. Cell. Longev. 2022, 2022, 5589089. [Google Scholar] [CrossRef] [PubMed]
- Leveille, E.; Ross, O.A.; Gan-Or, Z. Tau and MAPT genetics in tauopathies and synucleinopathies. Park Relat. Disord. 2021, 90, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Strang, K.H.; Golde, T.E.; Giasson, B.I. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Mod. Pathol. 2019, 99, 912–928. [Google Scholar] [CrossRef] [PubMed]
- Coronavirus Disease (COVID-19) Pandemic. Available online: https://www.who.int/europe/emergencies/situations/covid-19 (accessed on 6 January 2025).
- Premraj, L.; Kannapadi, N.V.; Briggs, J.; Seal, S.M.; Battaglini, D.; Fanning, J.; Suen, J.; Robba, C.; Fraser, J.; Cho, S.-M. Mid and long-term neurological and neuropsychiatric manifestations of post-COVID-19 syndrome: A me-ta-analysis. J. Neurol. Sci. 2022, 434, 120162. [Google Scholar] [CrossRef] [PubMed]
- Quan, M.; Wang, X.; Gong, M.; Wang, Q.; Li, Y.; Jia, J. Post-COVID cognitive dysfunction: Current status and research recommendations for high risk population. Lancet Reg. Heal West Pac. 2023, 38, 100836. [Google Scholar] [CrossRef] [PubMed]
- Dobielska, M.; Bartosik, N.K.; Zyzik, K.A.; Kowalczyk, E.; Karbownik, M.S. Mechanisms of Cognitive Impairment in Depression. May Probiotics Help? Front. Psychiatry 2022, 13, 904426. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, H.; Liu, Y.; Zhao, Z.; Zang, S. Association between depression status in adolescents and cognitive performance over the subsequent six years: A longitudinal study. J. Affect. Disord. 2023, 329, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Sultan, S.; Taimuri, U.; Basnan, S.A.; Ai-Orabi, W.K.; Awadallah, A.; Almowald, F.; Hazazi, A. Low Vitamin D and Its Association with Cognitive Impairment and Dementia. J. Aging Res. 2020, 2020, 6097820. [Google Scholar] [CrossRef] [PubMed]
- Jatoi, S.; Hafeez, A.; Riaz, S.U.; Ali, A.; Ghauri, M.I.; Zehra, M. Low Vitamin B12 Levels: An Underestimated Cause Of Minimal Cognitive Impairment And Dementia. Cureus 2020, 12, e6976. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.S.; Jacques, P.F.; Rosenberg, I.H.; Selhub, J. Folate and vitamin B-12 status in relation to anemia, macrocytosis, and cognitive impairment in older Americans in the age of folic acid fortification. Am. J. Clin. Nutr. 2007, 85, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Warren-Gash, C.; Forbes, H.J.; Williamson, E.; Breuer, J.; Hayward, A.C.; Mavrodaris, A.; Ridha, B.H.; Rossor, M.N.; Thomas, S.L.; Smeeth, L. Human herpesvirus infections and dementia or mild cognitive impairment: A systematic review and meta-analysis. Sci. Rep. 2019, 9, 4743. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-F.; Liu, C.-K.; Fang, T.-J.; Yu, Y.-H.; Lai, C.-L.; Kuo, H.-K. Previous Hepatitis A Virus Infection Is Related to Slower Psychomotor Speed in Elderly Adults. J. Gerontol. Ser. A 2009, 64, 1090–1096. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tan, C.-H.; Chang, M.-C.; Tsai, W.-F.; Chuang, W.-L.; Huang, J.-F.; Lin, Z.-Y.; Dai, C.-Y.; Yeh, M.-L.; Li, C.-T.; Yu, R.-L. Different profiles of neurocognitive impairment in patients with hepatitis B and C virus infections. Sci. Rep. 2022, 12, 10625. [Google Scholar] [CrossRef] [PubMed]
- Vaghi, G.; Gori, B.; Strigaro, G.; Burlone, M.; Minisini, R.; Barbaglia, M.N.; Brigatti, E.; Varrasi, C.; Pirisi, M.; Cantello, R. Direct antivirals and cognitive impairment in hepatitis C: A clinical-neurophysiologic study. J. Neurovirol. 2020, 26, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Clifford, D.B.; Ances, B.M. HIV-associated neurocognitive disorder. Lancet Infect. Diseases 2013, 13, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, S.; Rabinovici, G.D.; Cohn-Sheehy, B.I.; Madison, C.; Ayakta, N.; Ghosh, P.M.; La Joie, R.; Arthur-Bentil, S.K.; Vogel, J.W.; Marks, S.M.; et al. Existing Pittsburgh Compound-B positron emission tomography thresholds are too high: Statistical and pathological evaluation. Brain 2015, 138, 2020–2033. [Google Scholar] [CrossRef] [PubMed]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Shariati, S.A.M.; De Strooper, B. Redundancy and divergence in the amyloid precursor protein family. FEBS Lett. 2013, 587, 2036–2045. [Google Scholar] [CrossRef] [PubMed]
- Rockenstein, E.M.; McConlogue, L.; Tan, H.; Power, M.; Masliah, E.; Mucke, L. Levels and Alternative Splicing of Amyloid β Protein Precursor (APP) Transcripts in Brains of APP Transgenic Mice and Humans with Alzheimer’s Disease. J. Biol. Chem. 1995, 270, 28257–28267. [Google Scholar] [CrossRef] [PubMed]
- Grangeon, L.; Cassinari, K.; Rousseau, S.; Croisile, B.; Formaglio, M.; Moreaud, O.; Boutonnat, J.; Le Meur, N.; Miné, M.; Coste, T.; et al. Early-Onset Cerebral Amyloid Angiopathy and Alzheimer Disease Related to an APP Locus Triplication. Neurol. Genet. 2021, 7, e609. [Google Scholar] [CrossRef] [PubMed]
- Rovelet-Lecrux, A.; Hannequin, D.; Raux, G.; Le Meur, N.; Laquerrière, A.; Vital, A.; Dumanchin, C.; Feuillette, S.; Brice, A.; Vercelletto, M.; et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 2006, 38, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Liu, H.; Liu, X.; Zhang, W.; Zhang, S.; Jiao, B. APP, PSEN1, and PSEN2 Variants in Alzheimer’s Disease: Systematic Re-evaluation According to ACMG Guidelines. Front. Aging Neurosci. 2021, 13, 695808. [Google Scholar] [CrossRef] [PubMed]
- Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N–terminus of β–amyloid. Nat. Genet. 1992, 1, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Szaruga, M.; Munteanu, B.; Lismont, S.; Veugelen, S.; Horré, K.; Mercken, M.; Saido, T.C.; Ryan, N.S.; De Vos, T.; Savvides, S.N.; et al. Alzheimer’s-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell 2017, 170, 443–456.e14. [Google Scholar] [CrossRef] [PubMed]
- Deyts, C.; Clutter, M.; Herrera, S.; Jovanovic, N.; Goddi, A.; Parent, A.T.; States, U. Loss of presenilin function is associated with a selective gain of APP function. eLife 2016, 5, e15645. [Google Scholar] [CrossRef] [PubMed]
- Grossman, M.; Seeley, W.W.; Boxer, A.L.; Hillis, A.E.; Knopman, D.S.; Ljubenov, P.A.; Miller, B.; Piguet, O.; Rademakers, R.; Whitwell, J.L.; et al. Frontotemporal lobar degeneration. Nat. Rev. Dis. Prim. 2023, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Sellami, L.; Saracino, D.; Le Ber, I. Genetic forms of frontotemporal lobar degeneration: Current diagnostic approach and new directions in therapeutic strategies. Rev. Neurol. 2020, 176, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Cruts, M.; Gijselinck, I.; van der Zee, J.; Engelborghs, S.; Wils, H.; Pirici, D.; Rademakers, R.; Vandenberghe, R.; Dermaut, B.; Martin, J.-J.; et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006, 442, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Shpilyukova, Y.; Abramycheva, N.; Grishina, D.; Fedotova, E.; Illarioshkin, S. Deletions/duplications in GRN and MAPT genes is not common in frontotemporal dementia. Alzheimer’s Dement. 2023, 19, e065342. [Google Scholar] [CrossRef]
- Nelson, P.T.; Stefansson, K.; Gulcher, J.; Saper, C.B. Molecular evolution of τ protein: Implications for Alzheimer’s disease. J. Neurochem. 1996, 67, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
- Bassendine, M.F.; Taylor-Robinson, S.D.; Fertleman, M.; Khan, M.; Neely, D. Is Alzheimer’s Disease a Liver Disease of the Brain? Journal of Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 75, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hsiung, G.-Y.R.; Feldman, H.H. GRN Frontotemporal Dementia. In GeneReviews; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Cruts, M.; Van Broeckhoven, C. Data Mining: Applying the AD&FTD Mutation Database to Progranulin. In Progranulin: Methods and Protocols; Bateman, A., Bennett, H.P.J., Cheung, S.T., Eds.; Springer: New York, NY, USA, 2018; pp. 81–92. [Google Scholar] [CrossRef]
- Itzhaki, R.F. Corroboration of a Major Role for Herpes Simplex Virus Type 1 in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 324. [Google Scholar] [CrossRef] [PubMed]
- Readhead, B.; Haure-Mirande, J.-V.; Funk, C.C.; Richards, M.A.; Shannon, P.; Haroutunian, V.; Sano, M.; Liang, W.S.; Beckmann, N.D.; Price, N.D.; et al. Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron. Neuron 2018, 99, 64–82.e7. [Google Scholar] [CrossRef] [PubMed]
- Cairns, D.M.; Rouleau, N.; Parker, R.N.; Walsh, K.G.; Gehrke, L.; Kaplan, D.L. A 3D human brain–like tissue model of herpes-induced Alzheimer’s disease. Sci. Adv. 2020, 6, eaay8828. [Google Scholar] [CrossRef] [PubMed]
- Yong, S.J.; Yong, M.H.; Teoh, S.L.; Soga, T.; Parhar, I.; Chew, J.; Lim, W.L. The Hippocampal Vulnerability to Herpes Simplex Virus Type I Infection: Relevance to Alzheimer’s Disease and Memory Impairment. Front. Cell. Neurosci. 2021, 15, 695738. [Google Scholar] [CrossRef] [PubMed]
- Mazzaro, C.; Quartuccio, L.; Adinolfi, L.E.; Roccatello, D.; Pozzato, G.; Nevola, R.; Tonizzo, M.; Gitto, S.; Andreone, P.; Gattei, V. A Review on Extrahepatic Manifestations of Chronic Hepatitis C Virus Infection and the Impact of Di-rect-Acting Antiviral Therapy. Viruses 2021, 13, 2249. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez-Samaniego, L.; Rapado-Castro, M.; Cabrero, L.; Navarrete, C.; García-Mulas, S.; Ahumada, A.; Marquez, L.; Pérez, M.D.; Rincon, D.; Bañares, R.; et al. Hepatitis C eradication improves cognitive function in patients with or without cirrhosis: A pro-spective real-life study. Eur. J. Neurol. 2022, 29, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-H.; Wang, Y.-R.; Xiang, Y.; Zhou, H.-D.; Giunta, B.; Mañucat-Tan, N.B.; Tan, J.; Zhou, X.-F.; Wang, Y.-J. Clearance of Amyloid-Beta in Alzheimer’s Disease: Shifting the Action Site from Center to Periphery. Mol. Neurobiol. 2015, 51, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Solinas, A. Cognitive dysfunction and hepatitis C virus infection. World J. Hepatol. 2015, 7, 922–925. [Google Scholar] [CrossRef] [PubMed]
- Kiely, K.M. Cognitive Function. In Encyclopedia of Quality of Life and Well-Being Research; Maggino, F., Michalos, A.C., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 974–978. [Google Scholar] [CrossRef]
- Azizian, A.; Yeghiyan, M.; Ishkhanyan, B.; Manukyan, Y.; Khandanyan, L. Clinical Validity of the Repeatable Battery for the Assessment of Neuropsychological Status among Patients with Schizophrenia in the Republic of Armenia. Arch. Clin. Neuropsychol. 2011, 26, 89–97. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nasreddine, Z.S.; Phillips, N.A.; Bédirian, V.; Charbonneau, S.; Whitehead, V.; Collin, I.; Cummings, J.L.; Chertkow, H. The Montreal Cognitive Assessment, MoCA: A Brief Screening Tool For Mild Cognitive Impairment. J. Am. Geriatr. Soc. 2005, 53, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Kroenke, K.; Spitzer, R.L.; Williams, J.B. The PHQ-9: Validity of a brief depression severity measure. J. Gen. Intern. Med. 2001, 16, 606–613. [Google Scholar] [CrossRef] [PubMed]
- The SALSA MLPA Probemix P471-A1 EOFAD; Product Description Version A1-01; Issued 27 September 2019. Available online: https://www.mrcholland.com/products/33513/Product%20description%20P471-A1%20EOFAD-v01.pdf (accessed on 17 July 2025).
- The SALSA MLPA Probemix P275 MAPT-GRN; Product Description Version C4-01; Issued 5 July 2021. Available online: https://www.mrcholland.com/products/36500/Product%20description%20P275-C4%20MAPT-GRN-v01.pdf (accessed on 17 July 2025).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).