
Comparative Genomics and Draft Genome Assembly of the Elite Tunisian Date Palm Cultivar Deglet Nour: Insights into the Genetic Variations Linked to Fruit Ripening and Quality Traits

 , , ,
, , ,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results
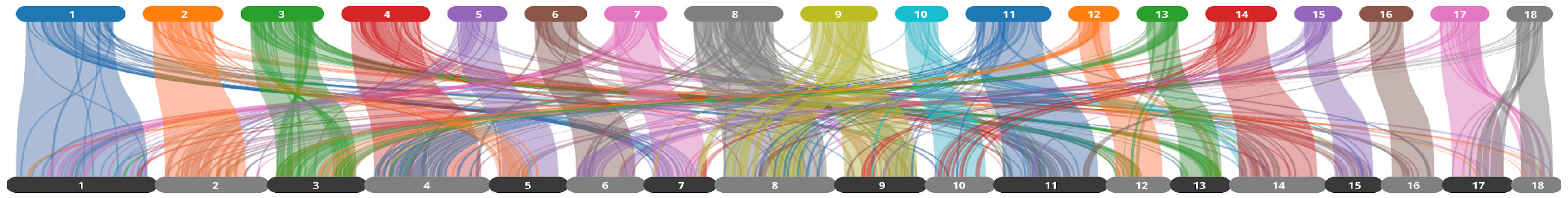
2.1. Draft Genome Assembly and Annotation
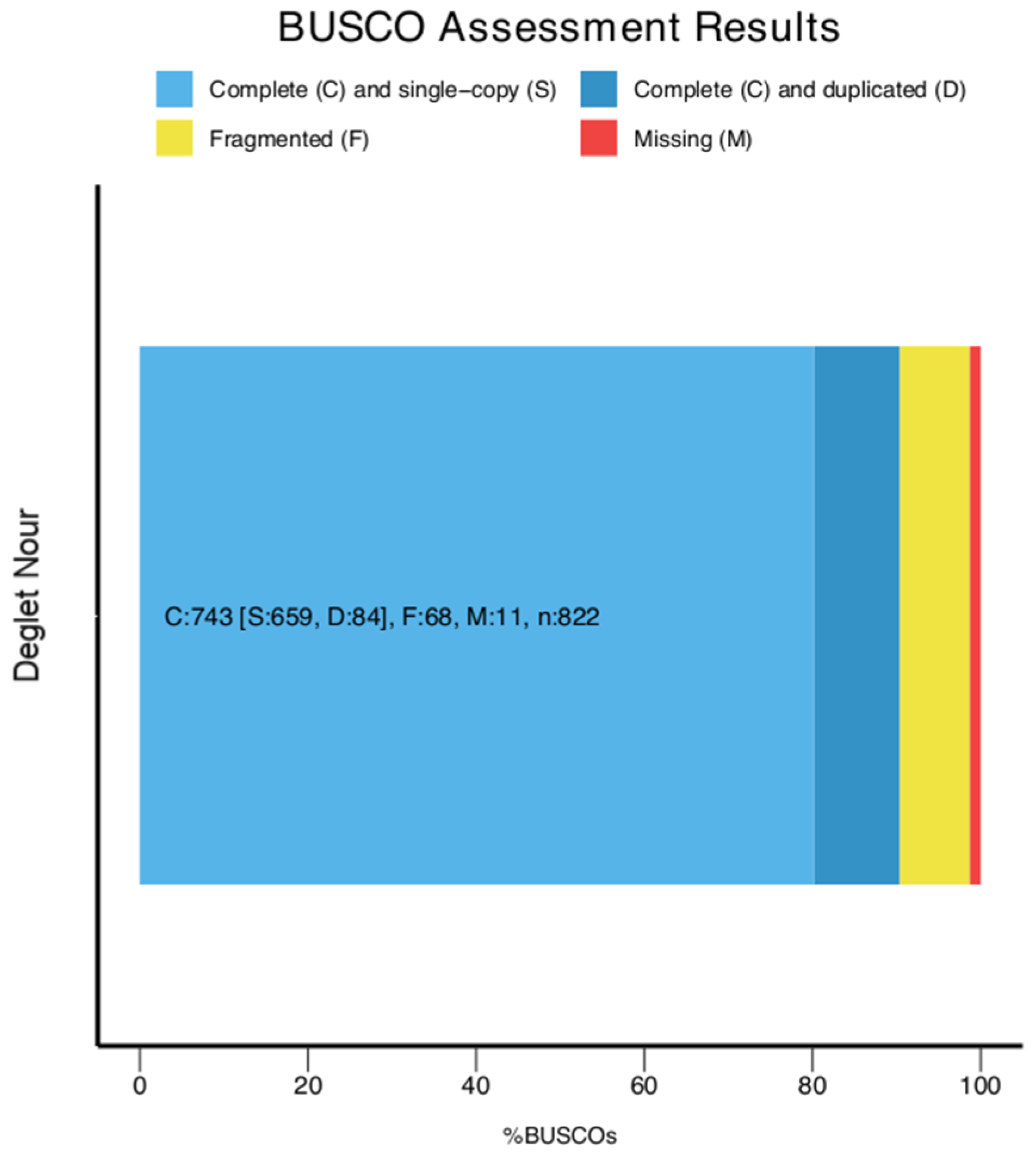
2.2. Genome Assembly Assessment
2.3. Transposable Elements
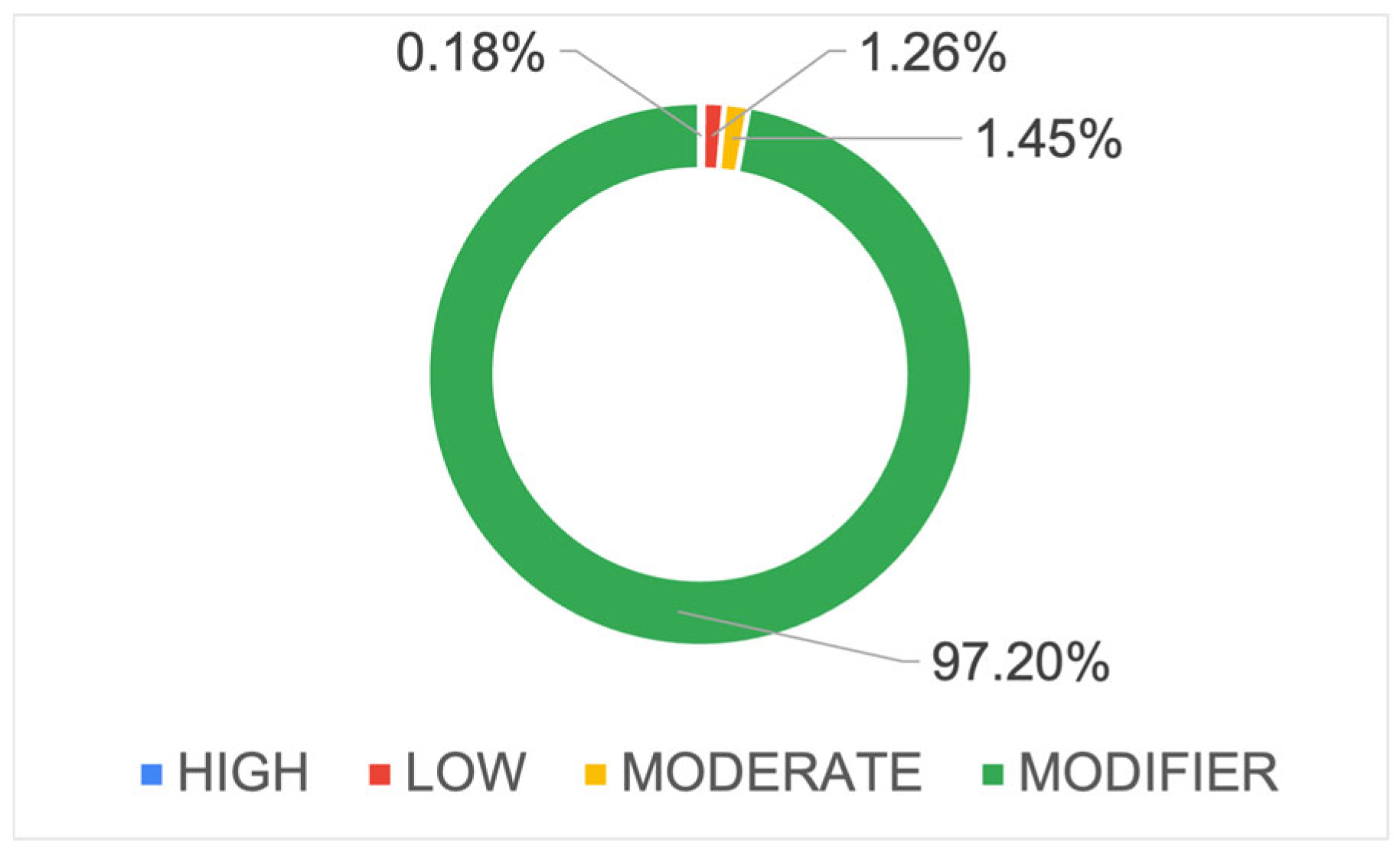
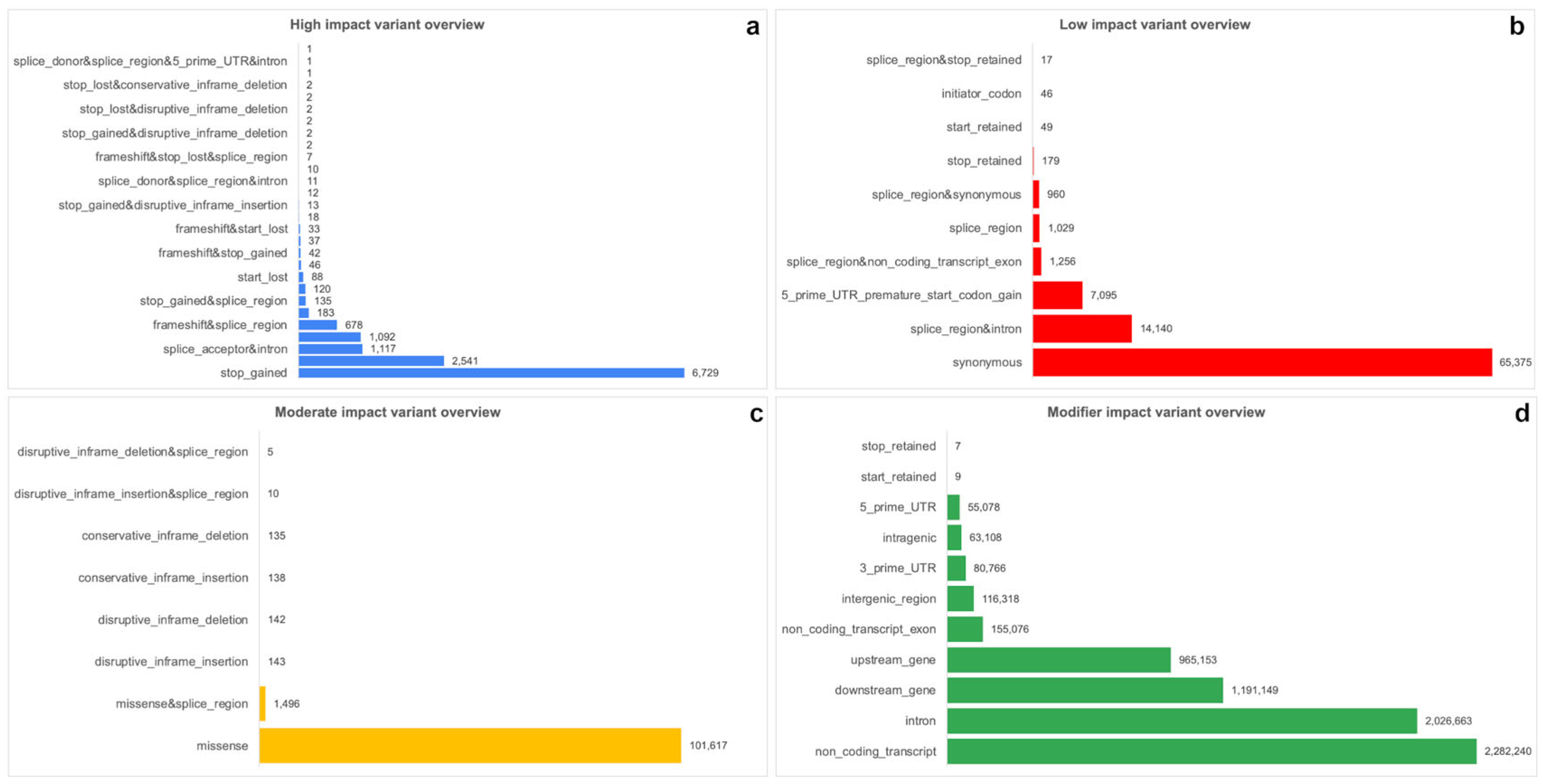
2.4. Annotation of Variants by SnpEff
3. Discussion
3.1. Genome Assembly and Basic Features
3.2. Transposable Element Composition
3.3. Genome-Wide Variant Detection
3.3.1. Variants in Sugar Metabolism Genes
3.3.2. Variants in Fruit Shape, Size, and Weight Genes
3.3.3. Variants in Fruit Firmness Genes
3.3.4. Implications for Breeding and Genetic Improvement
4. Materials and Methods
4.1. DNA Isolation and Sequencing
4.2. Pre-Processing of Raw Reads
4.3. Sequence Assembly and Annotation
4.4. Assessment of Sequence Assembly
4.5. Identification of Transposable Elements
4.6. SNP Identification
4.7. Prediction of Variants Impacts on Coding Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| bp | Base pair |
| CDS | Coding DNA sequence |
| Gbp | Giga base pair |
| GWAS | Genome-wide association study |
| INDELs | Insertions/deletions |
| Kb | Kilo base |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LINE | Long interspersed nuclear element |
| LTR | Long terminal repeat |
| Mb | Mega base |
| NCBI | National Center for Biotechnology Information |
| NGS | Next-generation sequencing |
| QTL | Quantitative trait loci |
| SINE | Short interspersed nuclear element |
| SNPs | Single nucleotide polymorphisms |
| TIR | Terminal inverted repeat |
References
- Lin, T.; Zhu, G.; Zhang, J.; Xu, X.; Yu, Q.; Zheng, Z.; Zhang, Z.; Lun, Y.; Li, S.; Wang, X.; et al. Genomic Analyses Provide Insights into the History of Tomato Breeding. Nat. Genet. 2014, 46, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Taranto, F.; D’Agostino, N.; Rodriguez, M.; Pavan, S.; Minervini, A.P.; Pecchioni, N.; Papa, R.; De Vita, P. Whole genome scan reveals molecular signatures of divergence and selection related to important traits in durum wheat germplasm. Front. Genet. 2020, 11, 217. [Google Scholar] [CrossRef] [PubMed]
- Al-Kilani, M.A.; Taranto, F.; D’Agostino, N.; Montemurro, C.; Belaj, A.; Ayoub, S.; Albdaiwi, R.; Hasan, S.; Al-Abdallat, A.M. Evaluation of Genetic Diversity among Olive Trees (Olea europaea L.) from Jordan. Front. Plant Sci. 2024, 15, 1437055. [Google Scholar] [CrossRef] [PubMed]
- Terracciano, I.; Cantarella, C.; Fasano, C.; Cardi, T.; Mennella, G.; D’Agostino, N. Liquid-phase sequence capture and targeted re-sequencing revealed novel polymorphisms in tomato genes belonging to the MEP carotenoid pathway. Sci. Rep. 2017, 7, 5616. [Google Scholar] [CrossRef] [PubMed]
- Crop Genomes and Beyond. Nat. Genet. 2020, 52, 865. [CrossRef] [PubMed]
- Marks, R.A.; Hotaling, S.; Frandsen, P.B.; VanBuren, R. Representation and Participation across 20 Years of Plant Genome Sequencing. Nat. Plants 2021, 7, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Zehdi, S.; Trifi, M.; Billotte, N.; Marrakchi, M.; Christophe Pintaud, J. Genetic Diversity of Tunisian Date Palms (Phoenix dactylifera L.) Revealed by Nuclear Microsatellite Polymorphism: Genetic Diversity of Tunisian Date Palms. Hereditas 2005, 141, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Zehdi-Azouzi, S.; Cherif, E.; Moussouni, S.; Gros-Balthazard, M.; Abbas Naqvi, S.; Ludeña, B.; Castillo, K.; Chabrillange, N.; Bouguedoura, N.; Bennaceur, M.; et al. Genetic Structure of the Date Palm (Phoenix dactylifera) in the Old World Reveals a Strong Differentiation between Eastern and Western Populations. Ann. Bot. 2015, 116, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Cherif, E.; Zehdi, S.; Castillo, K.; Chabrillange, N.; Abdoulkader, S.; Pintaud, J.; Santoni, S.; Salhi-Hannachi, A.; Glémin, S.; Aberlenc-Bertossi, F. Male-Specific DNA Markers Provide Genetic Evidence of an XY Chromosome System, a Recombination Arrest and Allow the Tracing of Paternal Lineages in Date Palm. New Phytol. 2013, 197, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Al-Dous, E.K.; George, B.; Al-Mahmoud, M.E.; Al-Jaber, M.Y.; Wang, H.; Salameh, Y.M.; Al-Azwani, E.K.; Chaluvadi, S.; Pontaroli, A.C.; DeBarry, J.; et al. De Novo Genome Sequencing and Comparative Genomics of Date Palm (Phoenix dactylifera). Nat. Biotechnol. 2011, 29, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Al-Mssallem, I.S.; Hu, S.; Zhang, X.; Lin, Q.; Liu, W.; Tan, J.; Yu, X.; Liu, J.; Pan, L.; Zhang, T.; et al. Genome Sequence of the Date Palm Phoenix dactylifera L. Nat. Commun. 2013, 4, 2274. [Google Scholar] [CrossRef]
- Hazzouri, K.M.; Gros-Balthazard, M.; Flowers, J.M.; Copetti, D.; Lemansour, A.; Lebrun, M.; Masmoudi, K.; Ferrand, S.; Dhar, M.I.; Fresquez, Z.A.; et al. Genome-Wide Association Mapping of Date Palm Fruit Traits. Nat. Commun. 2019, 10, 4680. [Google Scholar] [CrossRef] [PubMed]
- Hazzouri, K.M.; Flowers, J.M.; Visser, H.J.; Khierallah, H.S.M.; Rosas, U.; Pham, G.M.; Meyer, R.S.; Johansen, C.K.; Fresquez, Z.A.; Masmoudi, K.; et al. Whole Genome Re-Sequencing of Date Palms Yields Insights into Diversification of a Fruit Tree Crop. Nat. Commun. 2015, 6, 8824. [Google Scholar] [CrossRef] [PubMed]
- Rhouma, A. Le Palmier Dattier en Tunisie I: Le Patrimoine Génétique; Arabesques: La Marsa, Tunisia, 1994. [Google Scholar]
- Rhouma, A. Le Palmier Dattier en Tunisie: I. Le Patrimoine Génétique—Volume 2; IPGRI: Rome, Italy, 2005; 255p, ISBN 978-92-9043-677-5/92-9043-677-8. [Google Scholar]
- Yang, M.; Zhang, X.; Liu, G.; Yin, Y.; Chen, K.; Yun, Q.; Zhao, D.; Al-Mssallem, I.S.; Yu, J. The Complete Chloroplast Genome Sequence of Date Palm (Phoenix dactylifera L.). PLoS ONE 2010, 5, e12762. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Wu, H.; Zhang, T.; Yang, M.; Yin, Y.; Pan, L.; Yu, X.; Zhang, X.; Hu, S.; Al-Mssallem, I.S.; et al. A Complete Sequence and Transcriptomic Analyses of Date Palm (Phoenix dactylifera L.) Mitochondrial Genome. PLoS ONE 2012, 7, e37164. [Google Scholar] [CrossRef] [PubMed]
- Hamza, H.; Villa, S.; Torre, S.; Marchesini, A.; Benabderrahim, M.A.; Rejili, M.; Sebastiani, F. Whole Mitochondrial and Chloroplast Genome Sequencing of Tunisian Date Palm Cultivars: Diversity and Evolutionary Relationships. BMC Genom. 2023, 24, 772. [Google Scholar] [CrossRef] [PubMed]
- Henniges, M.C.; Johnston, E.; Pellicer, J.; Hidalgo, O.; Bennett, M.D.; Leitch, I.J. The plant DNA C-values database: A one-stop shop for plant genome size data. In Plant Genomic and Cytogenetic Databases; Springer: New York, NY, USA, 2023; pp. 111–122. [Google Scholar]
- Galindo-González, L.; Mhiri, C.; Deyholos, M.K.; Grandbastien, M.A. LTR-Retrotransposons in Plants: Engines of Evolution. Gene 2017, 626, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Vitte, C.; Panaud, O. LTR Retrotransposons and Flowering Plant Genome Size: Emergence of the Increase/Decrease Model. Cytogenet. Genome Res. 2005, 110, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Filho, J.A.F.; De Brito, L.S.; Leão, A.P.; Alves, A.A.; Formighieri, E.F.; Souza, M.T. In Silico Approach for Characterization and Comparison of Repeats in the Genomes of Oil and Date Palms. Bioinform. Biol. Insights 2017, 11, 117793221770238. [Google Scholar] [CrossRef] [PubMed]
- Beulé, T.; Agbessi, M.D.; Dussert, S.; Jaligot, E.; Guyot, R. Genome-Wide Analysis of LTR-Retrotransposons in Oil Palm. BMC Genom. 2015, 16, 795. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Xu, P.; Fan, H.; Baudouin, L.; Xia, W.; Bocs, S.; Xu, J.; Li, Q.; Guo, A.; Zhou, L.; et al. The Genome Draft of Coconut (Cocos nucifera). GigaScience 2017, 6, gix095. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Zhang, X.; Fang, Y.; Pan, L.; Sun, G.; Xin, C.; Ba Abdullah, M.M.; Yu, X.; Hu, S.; Al-Mssallem, I.S.; et al. High-Throughput Sequencing-Based Gene Profiling on Multi-Staged Fruit Development of Date Palm (Phoenix dactylifera L.). Plant Mol. Biol. 2012, 78, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Malek, J.A.; Mathew, S.; Mathew, L.S.; Younuskunju, S.; Mohamoud, Y.A.; Suhre, K. Deletion of Beta-fructofuranosidase (Invertase) Genes Is Associated with Sucrose Content in Date Palm Fruit. Plant Direct 2020, 4, e00214. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, A.; Moriguchi, T.; Koyama, K.; Omura, M.; Akihama, T. Analysis of Sucrose Synthase Genes in Citrus Suggests Different Roles and Phylogenetic Relationships. J. Exp. Bot. 2002, 53, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Pan, L.; Yin, Y.; Liu, W.; Huang, D.; Zhang, T.; Wang, L.; Xin, C.; Lin, Q.; Sun, G.; et al. Large-Scale Collection and Annotation of Gene Models for Date Palm (Phoenix dactylifera L.). Plant Mol. Biol. 2012, 79, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, B.; Keyhaninejad, N.; Rodríguez, G.R.; Kim, H.J.; Chakrabarti, M.; Illa-Berenguer, E.; Taitano, N.K.; Gonzalo, M.J.; Díaz, A.; et al. A Common Genetic Mechanism Underlies Morphological Diversity in Fruits and Other Plant Organs. Nat. Commun. 2018, 9, 4734. [Google Scholar] [CrossRef] [PubMed]
- Monforte, A.J.; Diaz, A.; Caño-Delgado, A.; Van Der Knaap, E. The Genetic Basis of Fruit Morphology in Horticultural Crops: Lessons from Tomato and Melon. J. Exp. Bot. 2013, 65, 4625–4637. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Van Eck, J.; Cong, B.; Tanksley, S.D. A New Class of Regulatory Genes Underlying the Cause of Pear-Shaped Tomato Fruit. Proc. Natl. Acad. Sci. USA 2002, 99, 13302–13306. [Google Scholar] [CrossRef] [PubMed]
- Ku, H.-M.; Doganlar, S.; Chen, K.-Y.; Tanksley, S.D. The Genetic Basis of Pear-Shaped Tomato Fruit. Theor. Appl. Genet. 1999, 99, 844–850. [Google Scholar] [CrossRef]
- Ku, H.-M.; Liu, J.; Doganlar, S.; Tanksley, S.D. Exploitation of Arabidopsis–Tomato Synteny to Construct a High-Resolution Map of the Ovate-containing Region in Tomato Chromosome 2. Genome 2001, 44, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Blas, A.L.; Yu, Q.; Veatch, O.J.; Paull, R.E.; Moore, P.H.; Ming, R. Genetic Mapping of Quantitative Trait Loci Controlling Fruit Size and Shape in Papaya. Mol. Breed. 2012, 29, 457–466. [Google Scholar] [CrossRef]
- Cong, B.; Liu, J.; Tanksley, S.D. Natural Alleles at a Tomato Fruit Size Quantitative Trait Locus Differ by Heterochronic Regulatory Mutations. Proc. Natl. Acad. Sci. USA 2002, 99, 13606–13611. [Google Scholar] [CrossRef] [PubMed]
- Alfred, J. Sizing up Developmental Timing. Nat. Rev. Genet. 2002, 3, 900. [Google Scholar] [CrossRef]
- Nesbitt, T.C.; Tanksley, S.D. Fw2.2 Directly Affects the Size of Developing Tomato Fruit, with Secondary Effects on Fruit Number and Photosynthate Distribution. Plant Physiol. 2001, 127, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Beauchet, A.; Gévaudant, F.; Gonzalez, N.; Chevalier, C. In Search of the Still Unknown Function of FW2.2/CELL NUMBER REGULATOR, a Major Regulator of Fruit Size in Tomato. J. Exp. Bot. 2021, 72, 5300–5311. [Google Scholar] [CrossRef] [PubMed]
- Nantawan, U.; Kanchana-udomkan, C.; Bar, I.; Ford, R. Linkage Mapping and Quantitative Trait Loci Analysis of Sweetness and Other Fruit Quality Traits in Papaya. BMC Plant Biol. 2019, 19, 449. [Google Scholar] [CrossRef]
- Rose, J.K.C.; Lee, H.H.; Bennett, A.B. Expression of a Divergent Expansin Gene Is Fruit-Specific and Ripening-Regulated. Proc. Natl. Acad. Sci. USA 1997, 94, 5955–5960. [Google Scholar] [CrossRef] [PubMed]
- Harrison, E.P.; McQueen-Mason, S.J.; Manning, K. Expression of Six Expansin Genes in Relation to Extension Activity in Developing Strawberry Fruit. J. Exp. Bot. 2001, 52, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Lin, Y.; Wang, C.; Lu, J.; Liu, Z.; He, Z.; Shu, X.; Chen, W.; Wu, R.; Li, B.; et al. Expansin SlExp1 and Endoglucanase SlCel2 Synergistically Promote Fruit Softening and Cell Wall Disassembly in Tomato. Plant Cell 2024, 36, 709–726. [Google Scholar] [CrossRef] [PubMed]
- Hileman, L.C.; Sundstrom, J.F.; Litt, A.; Chen, M.; Shumba, T.; Irish, V.F. Molecular and Phylogenetic Analyses of the MADS-Box Gene Family in Tomato. Mol. Biol. Evol. 2006, 23, 2245–2258. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xu, H.; Ju, Z.; Cao, D.; Zhu, H.; Fu, D.; Grierson, D.; Qin, G.; Luo, Y.; Zhu, B. The RIN-MC Fusion of MADS-Box Transcription Factors Has Transcriptional Activity and Modulates Expression of Many Ripening Genes. Plant Physiol. 2018, 176, 891–909. [Google Scholar] [CrossRef] [PubMed]
- Gapper, N.E.; McQuinn, R.P.; Giovannoni, J.J. Molecular and Genetic Regulation of Fruit Ripening. Plant Mol. Biol. 2013, 82, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Manning, K.; Tör, M.; Poole, M.; Hong, Y.; Thompson, A.J.; King, G.J.; Giovannoni, J.J.; Seymour, G.B. A Naturally Occurring Epigenetic Mutation in a Gene Encoding an SBP-Box Transcription Factor Inhibits Tomato Fruit Ripening. Nat. Genet. 2006, 38, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Martel, C.; Vrebalov, J.; Tafelmeyer, P.; Giovannoni, J.J. The Tomato MADS-Box Transcription Factor RIPENING INHIBITOR Interacts with Promoters Involved in Numerous Ripening Processes in a COLORLESS NON RIPENING-Dependent Manner. Plant Physiol. 2011, 157, 1568–1579. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Hong, Y.; Yin, M.; Li, C.; Zhang, K.; Grierson, D. A Tomato HD-Zip Homeobox Protein, LeHB-1, Plays an Important Role in Floral Organogenesis and Ripening. Plant J. 2008, 55, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Fei, Z.; Chen, Y.-R.; Zheng, Y.; Huang, M.; Vrebalov, J.; McQuinn, R.; Gapper, N.; Liu, B.; Xiang, J.; et al. Single-Base Resolution Methylomes of Tomato Fruit Development Reveal Epigenome Modifications Associated with Ripening. Nat. Biotechnol. 2013, 31, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Seymour, G.B.; Ryder, C.D.; Cevik, V.; Hammond, J.P.; Popovich, A.; King, G.J.; Vrebalov, J.; Giovannoni, J.J.; Manning, K. A SEPALLATA Gene Is Involved in the Development and Ripening of Strawberry (Fragaria×ananassa Duch.) Fruit, a Non-Climacteric Tissue. J. Exp. Bot. 2011, 62, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Elitzur, T.; Vrebalov, J.; Giovannoni, J.J.; Goldschmidt, E.E.; Friedman, H. The Regulation of MADS-Box Gene Expression during Ripening of Banana and Their Regulatory Interaction with Ethylene. J. Exp. Bot. 2010, 61, 1523–1535. [Google Scholar] [CrossRef] [PubMed]
- Murayama, H.; Arikawa, M.; Sasaki, Y.; Dal Cin, V.; Mitsuhashi, W.; Toyomasu, T. Effect of Ethylene Treatment on Expression of Polyuronide-Modifying Genes and Solubilization of Polyuronides during Ripening in Two Peach Cultivars Having Different Softening Characteristics. Postharvest Biol. Technol. 2009, 52, 196–201. [Google Scholar] [CrossRef]
- Anees, M.; Gao, L.; Umer, M.J.; Yuan, P.; Zhu, H.; Lu, X.; He, N.; Gong, C.; Kaseb, M.O.; Zhao, S.; et al. Identification of Key Gene Networks Associated With Cell Wall Components Leading to Flesh Firmness in Watermelon. Front. Plant Sci. 2021, 12, 630243. [Google Scholar] [CrossRef] [PubMed]
- Salentijn, E.M.J.; Aharoni, A.; Schaart, J.G.; Boone, M.J.; Krens, F.A. Differential Gene Expression Analysis of Strawberry Cultivars That Differ in Fruit-firmness. Physiol. Plant. 2003, 118, 571–578. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, X.; Liu, X.; Ren, Y.; Jiang, D.; Shen, W.; Zhao, X.; Cao, L. Comparative Transcriptomic Profile of Two Mandarin Varieties during Maturation Reveals Pectinase Regulating Peelability. Sci. Hortic. 2024, 331, 113148. [Google Scholar] [CrossRef]
- Phan, T.D.; Bo, W.; West, G.; Lycett, G.W.; Tucker, G.A. Silencing of the Major Salt-Dependent Isoform of Pectinesterase in Tomato Alters Fruit Softening. Plant Physiol. 2007, 144, 1960–1967. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Zhang, F.; Wu, X.; Li, H. Characterization of the Tomato (Solanum lycopersicum) Pectin Methylesterases: Evolution, Activity of Isoforms and Expression During Fruit Ripening. Front. Plant Sci. 2020, 11, 238. [Google Scholar] [CrossRef] [PubMed]
- Castillejo, C.; de la Fuente, J.I.; Iannetta, P.; Botella, M.Á.; Valpuesta, V. Pectin Esterase Gene Family in Strawberry Fruit: Study of FaPE1, a Ripening-specific Isoform. J. Exp. Bot. 2004, 55, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Rahman, H.; Vikram, P.; Hammami, Z.; Singh, R.K. Recent Advances in Date Palm Genomics: A Comprehensive Review. Front. Genet. 2022, 13, 959266. [Google Scholar] [CrossRef] [PubMed]
- Frédérique, A.-B.; Pintaud Jean-, C.; Chabrillange, N.; Cherif, E.; Astillo-Perez, K.; Zehdi, S. Marqueur Moléculaire Et Méthodes Pour L’identification des Génotypes de Palmier Dattier 2015. European Patent WO2014080034A1, 30 September 2015. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Chikhi, R.; Medvedev, P. Informed and Automated K-Mer Size Selection for Genome Assembly. Bioinformatics 2014, 30, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Jackman, S.D.; Vandervalk, B.P.; Mohamadi, H.; Chu, J.; Yeo, S.; Hammond, S.A.; Jahesh, G.; Khan, H.; Coombe, L.; Warren, R.L.; et al. ABySS 2.0: Resource-Efficient Assembly of Large Genomes Using a Bloom Filter. Genome Res. 2017, 27, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Coombe, L.; Nikolić, V.; Chu, J.; Birol, I.; Warren, R.L. ntJoin: Fast and Lightweight Assembly-Guided Scaffolding Using Minimizer Graphs. Bioinformatics 2020, 36, 3885–3887. [Google Scholar] [CrossRef] [PubMed]
- Paulino, D.; Warren, R.L.; Vandervalk, B.P.; Raymond, A.; Jackman, S.D.; Birol, I. Sealer: A Scalable Gap-Closing Application for Finishing Draft Genomes. BMC Bioinform. 2015, 16, 230. [Google Scholar] [CrossRef] [PubMed]
- Bao, E.; Jiang, T.; Girke, T. AlignGraph: Algorithm for Secondary de Novo Genome Assembly Guided by Closely Related References. Bioinformatics 2014, 30, i319–i328. [Google Scholar] [CrossRef] [PubMed]
- Shumate, A.; Salzberg, S.L. Liftoff: Accurate Mapping of Gene Annotations. Bioinformatics 2021, 37, 1639–1643. [Google Scholar] [CrossRef] [PubMed]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile Genome Assembly Evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing Genomic Data Quality and Beyond. Curr. Protoc. 2021, 1, e323. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A Toolkit for Detection and Evolutionary Analysis of Gene Synteny and Collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Hoštáková, N.; Neumann, P.; Macas, J. DANTE and DANTE_LTR: Lineage-Centric Annotation Pipelines for Long Terminal Repeat Retrotransposons in Plant Genomes. NAR Genom. Bioinform. 2024, 6, lqae113. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wu, S.; Lin, Q.; Gao, S.; Ding, F.; Zhang, X.; Aljohi, H.A.; Yu, J.; Hu, S. RGAAT: A Reference-Based Genome Assembly and Annotation Tool for New Genomes and Upgrade of Known Genomes. Genom. Proteom. Bioinform. 2018, 16, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Raw Reads | Raw Read Q30 (%) | Paired Processed Reads | Processed Read Q30 (%) | Surviving Single Reads |
|---|---|---|---|---|---|
| DN_R1 | 172,691,117 | 93.95 | 157,872,866 | 90.40 | 9,766,454 |
| DN_R2 | 172,691,117 | 94.81 | 157,872,866 | 94.81 | 2,324,394 |
| Genomes | Size (Mb) | Number of Scaffolds | N50 (Kb) | Length of Sequences Anchored to LGs (Mb) |
|---|---|---|---|---|
| Al-Dous et al. [10] * | 381 | 57,277 | 30.5 | 0 |
| Al-Mssallem et al. [11] ** | 558 | 82,354 | 330.0 | 0 |
| Hazzouri et al. [12] *** | 772 | 2706 | 897.2 | 385.6 |
| Present study | 431 | 16,167 | 12.2 | 0 |
| Variant Calling | Genome Deglet Nour |
|---|---|
| SNPs | 1,062,681 |
| Insertions | 63,274 |
| Deletions | 51,033 |
| Total | 1,176,998 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zarkouna, R.; Hachef, A.; Fruggiero, C.; Aufiero, G.; D’Angelo, D.; Bourguiba, H.; Mezghani-Khemakhem, M.; D’Agostino, N.; Zehdi-Azouzi, S. Comparative Genomics and Draft Genome Assembly of the Elite Tunisian Date Palm Cultivar Deglet Nour: Insights into the Genetic Variations Linked to Fruit Ripening and Quality Traits. Int. J. Mol. Sci. 2025, 26, 6844. https://doi.org/10.3390/ijms26146844
Zarkouna R, Hachef A, Fruggiero C, Aufiero G, D’Angelo D, Bourguiba H, Mezghani-Khemakhem M, D’Agostino N, Zehdi-Azouzi S. Comparative Genomics and Draft Genome Assembly of the Elite Tunisian Date Palm Cultivar Deglet Nour: Insights into the Genetic Variations Linked to Fruit Ripening and Quality Traits. International Journal of Molecular Sciences. 2025; 26(14):6844. https://doi.org/10.3390/ijms26146844
Chicago/Turabian StyleZarkouna, Rahma, Afifa Hachef, Carmine Fruggiero, Gaetano Aufiero, Davide D’Angelo, Hedia Bourguiba, Maha Mezghani-Khemakhem, Nunzio D’Agostino, and Salwa Zehdi-Azouzi. 2025. "Comparative Genomics and Draft Genome Assembly of the Elite Tunisian Date Palm Cultivar Deglet Nour: Insights into the Genetic Variations Linked to Fruit Ripening and Quality Traits" International Journal of Molecular Sciences 26, no. 14: 6844. https://doi.org/10.3390/ijms26146844
APA StyleZarkouna, R., Hachef, A., Fruggiero, C., Aufiero, G., D’Angelo, D., Bourguiba, H., Mezghani-Khemakhem, M., D’Agostino, N., & Zehdi-Azouzi, S. (2025). Comparative Genomics and Draft Genome Assembly of the Elite Tunisian Date Palm Cultivar Deglet Nour: Insights into the Genetic Variations Linked to Fruit Ripening and Quality Traits. International Journal of Molecular Sciences, 26(14), 6844. https://doi.org/10.3390/ijms26146844