
Innovative Peptide Therapeutics in the Pipeline: Transforming Cancer Detection and Treatment



Abstract
1. Introduction
2. Peptide Classes
2.1. GnRH Analogues (Agonists and Antagonists)
2.2. Somatostatin (SST) Analogues
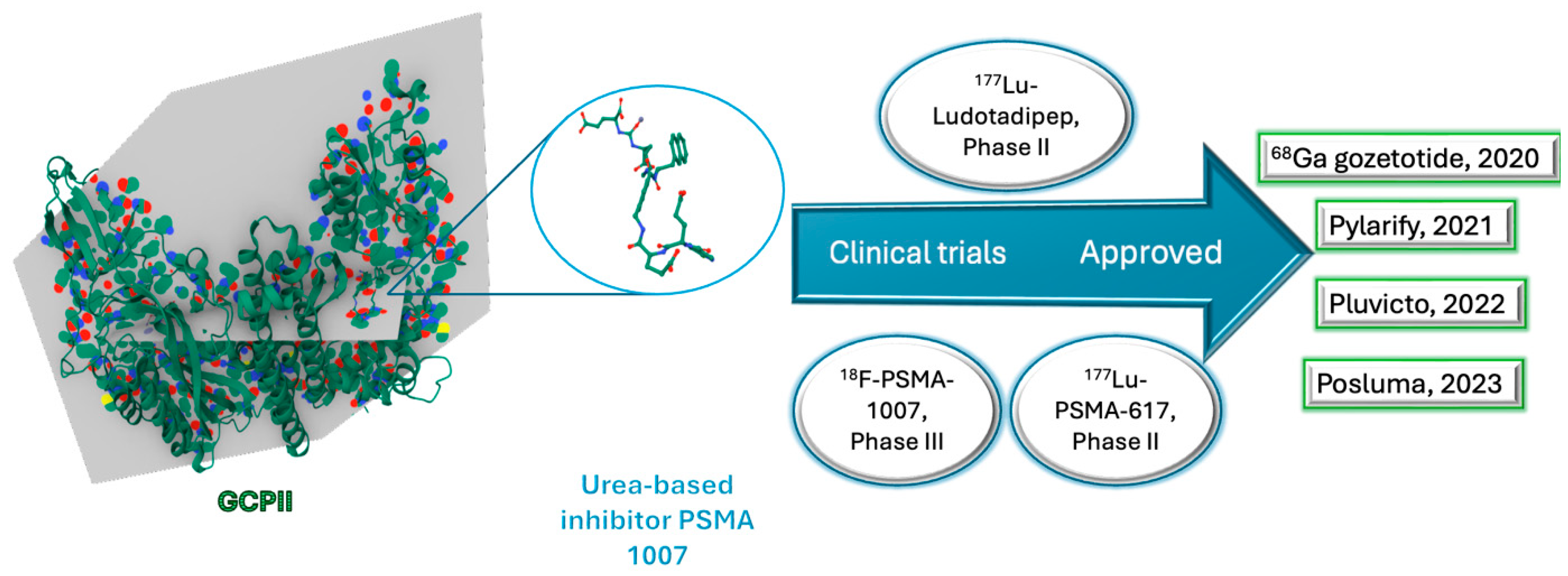
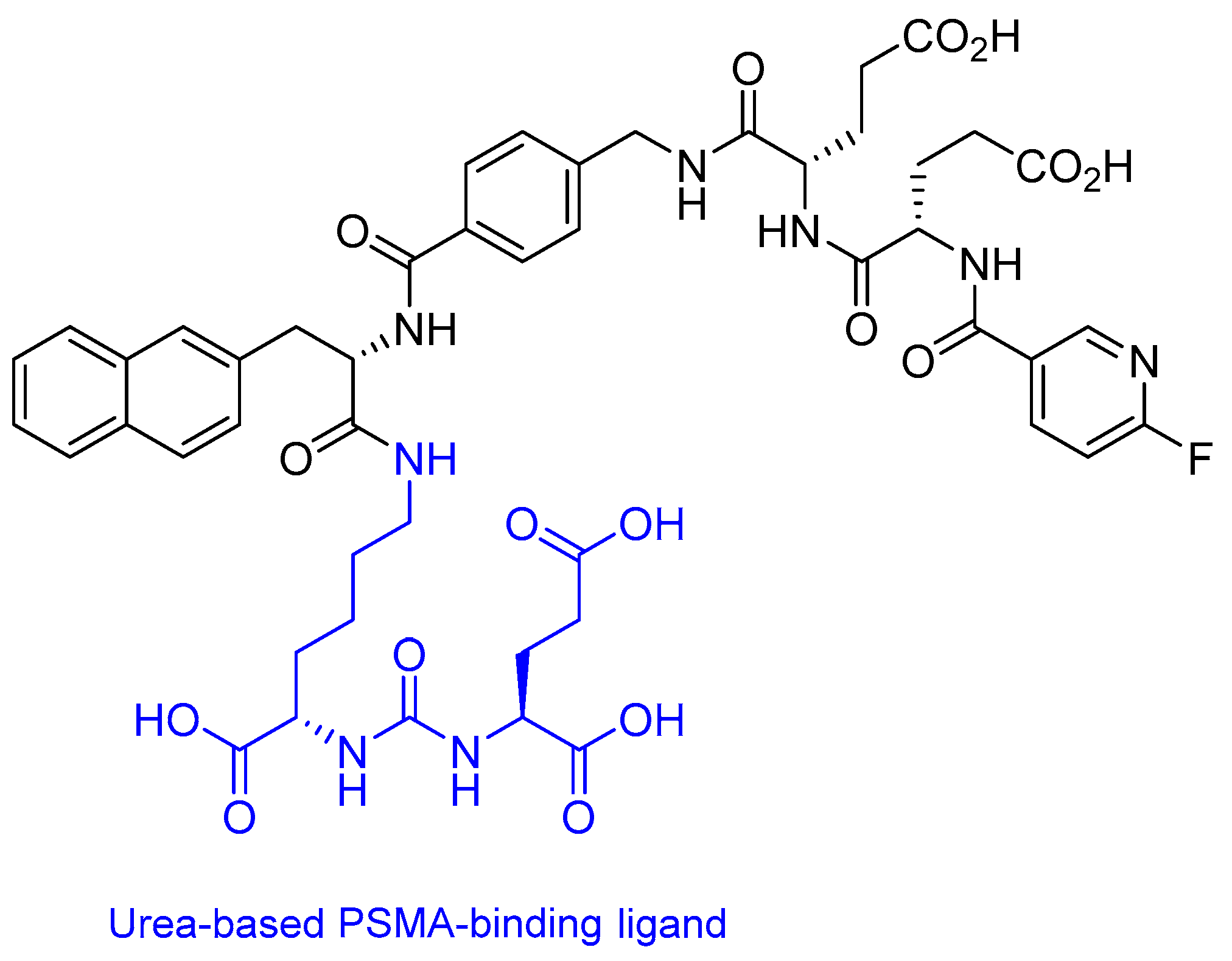
2.3. Prostate-Specific Membrane Antigen (PSMA) Peptide Antagonist
18Fluorine-PSMA-1007
2.4. Peptide Receptor Radionuclide Therapy (PRRT)
2.4.1. RAYZ-8009 (DOTA-RYZ-GPC3)
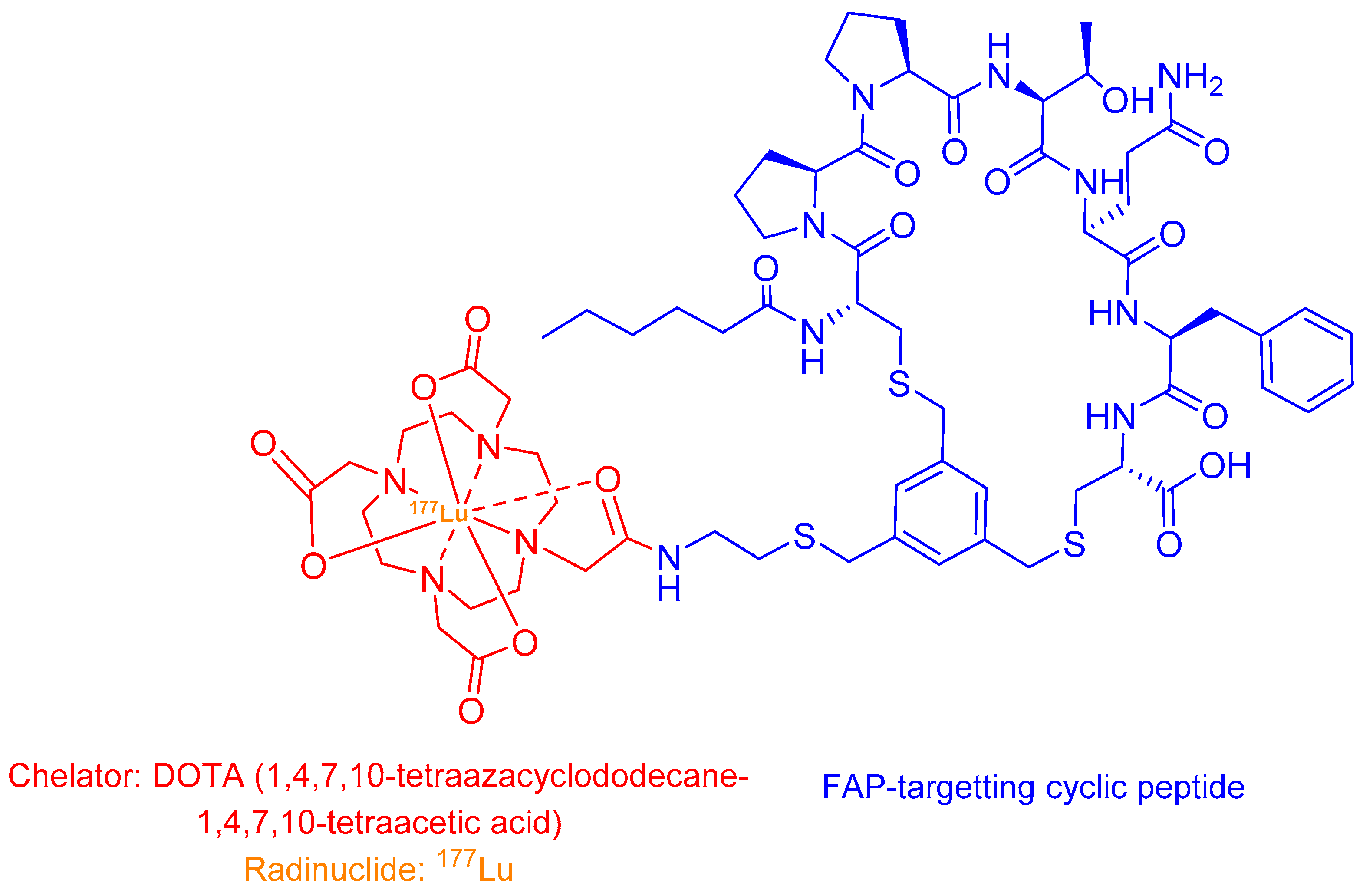
2.4.2. 177Lu-FAP-2286
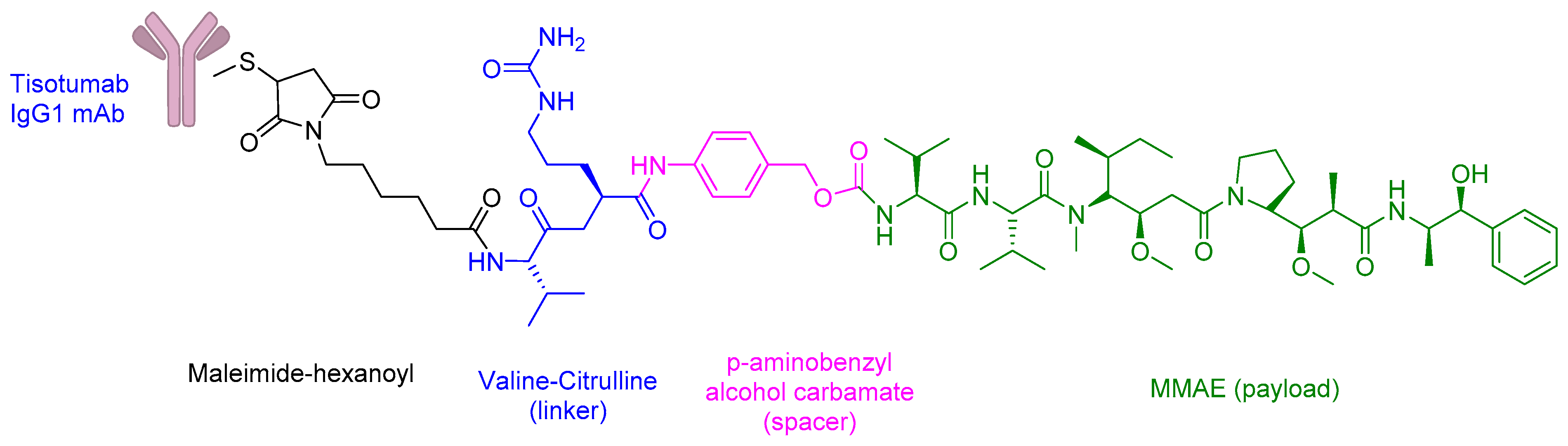
2.5. Antibody–Drug Conjugates (ADCs)
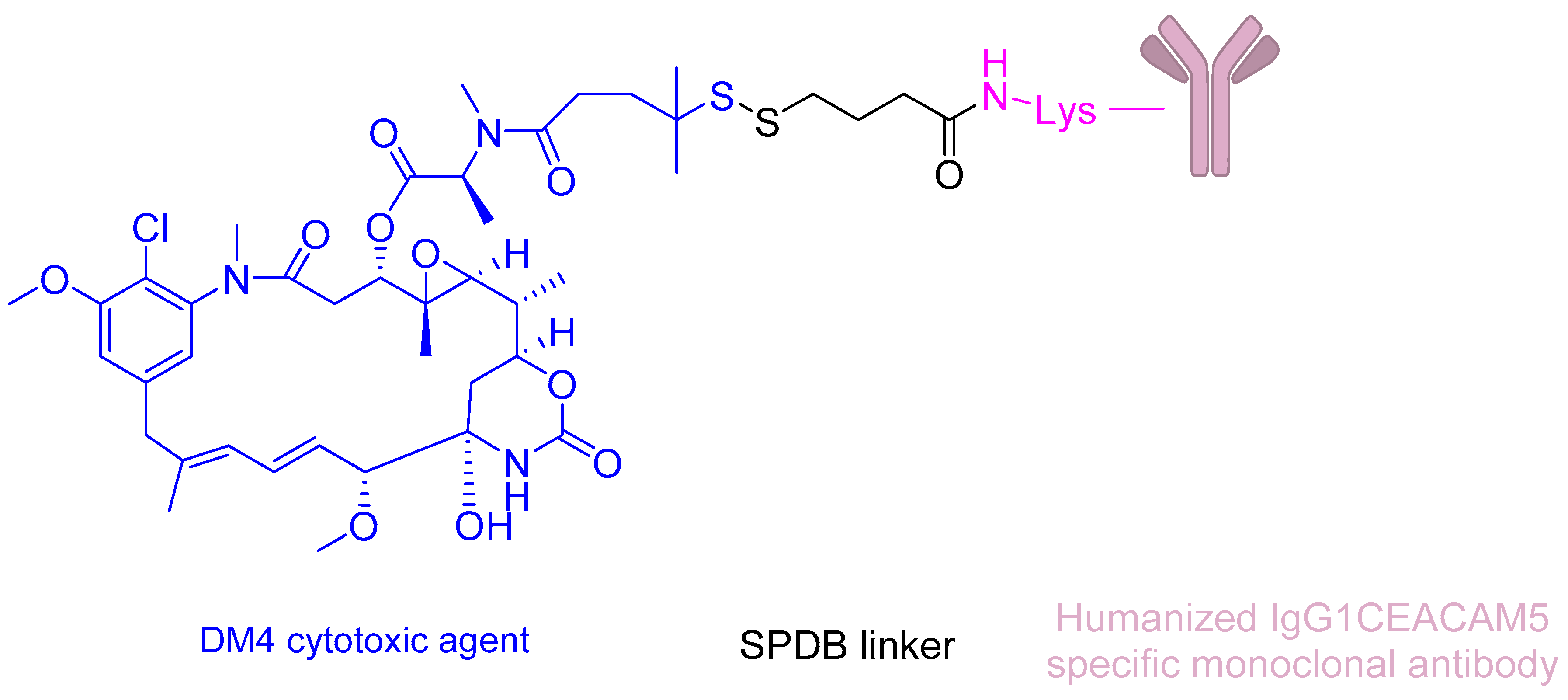
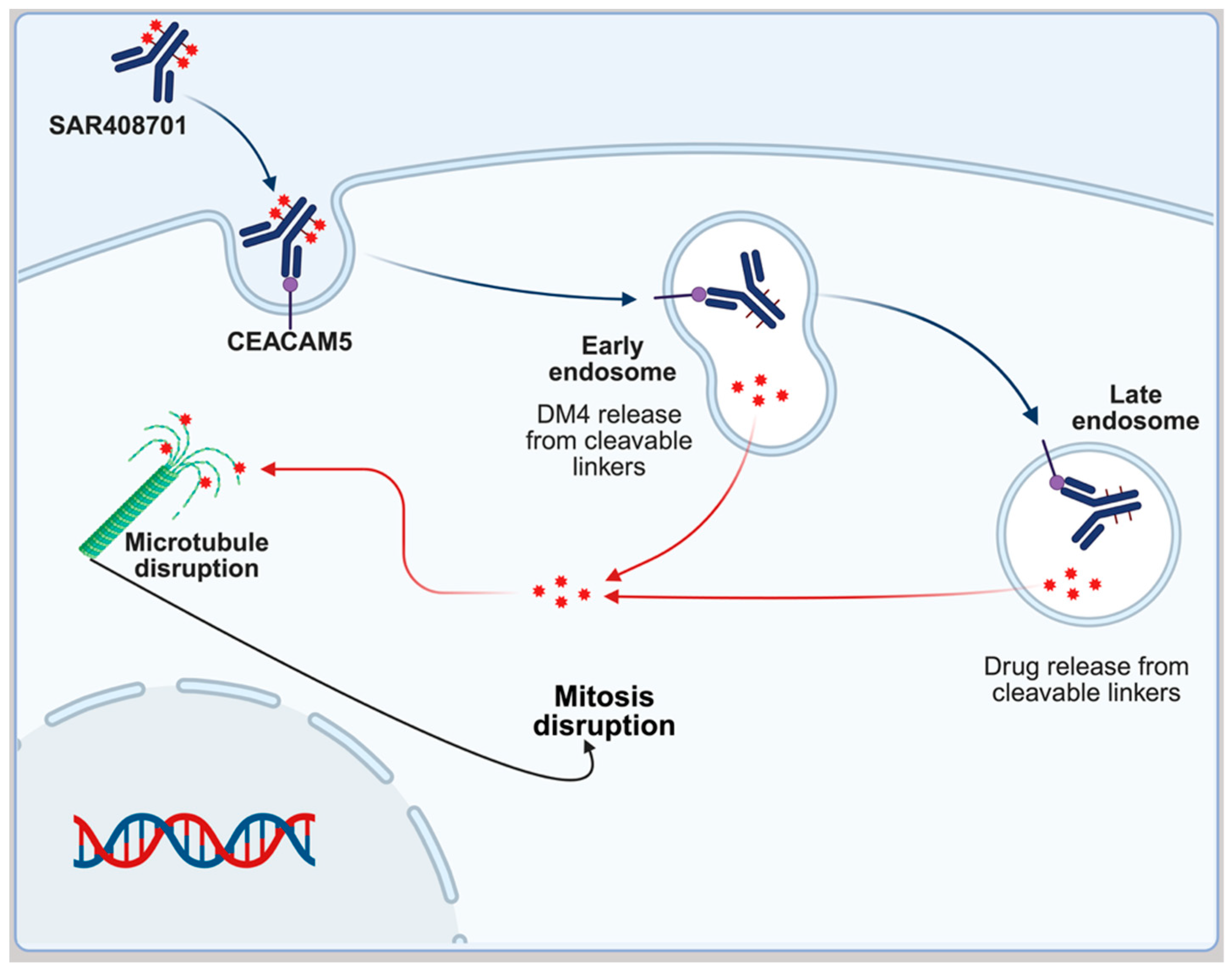
SAR408701
2.6. Peptide–Drug Conjugates (PDCs)
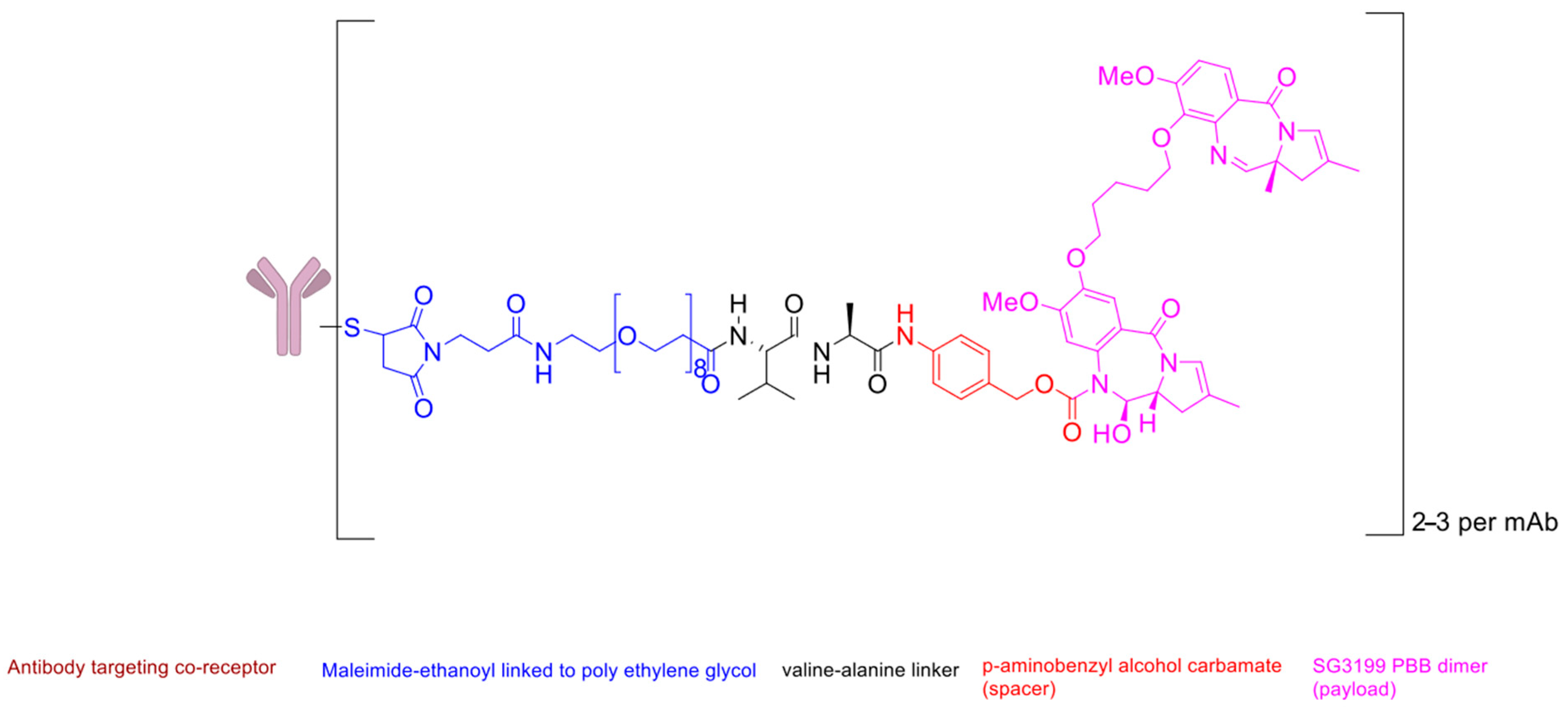
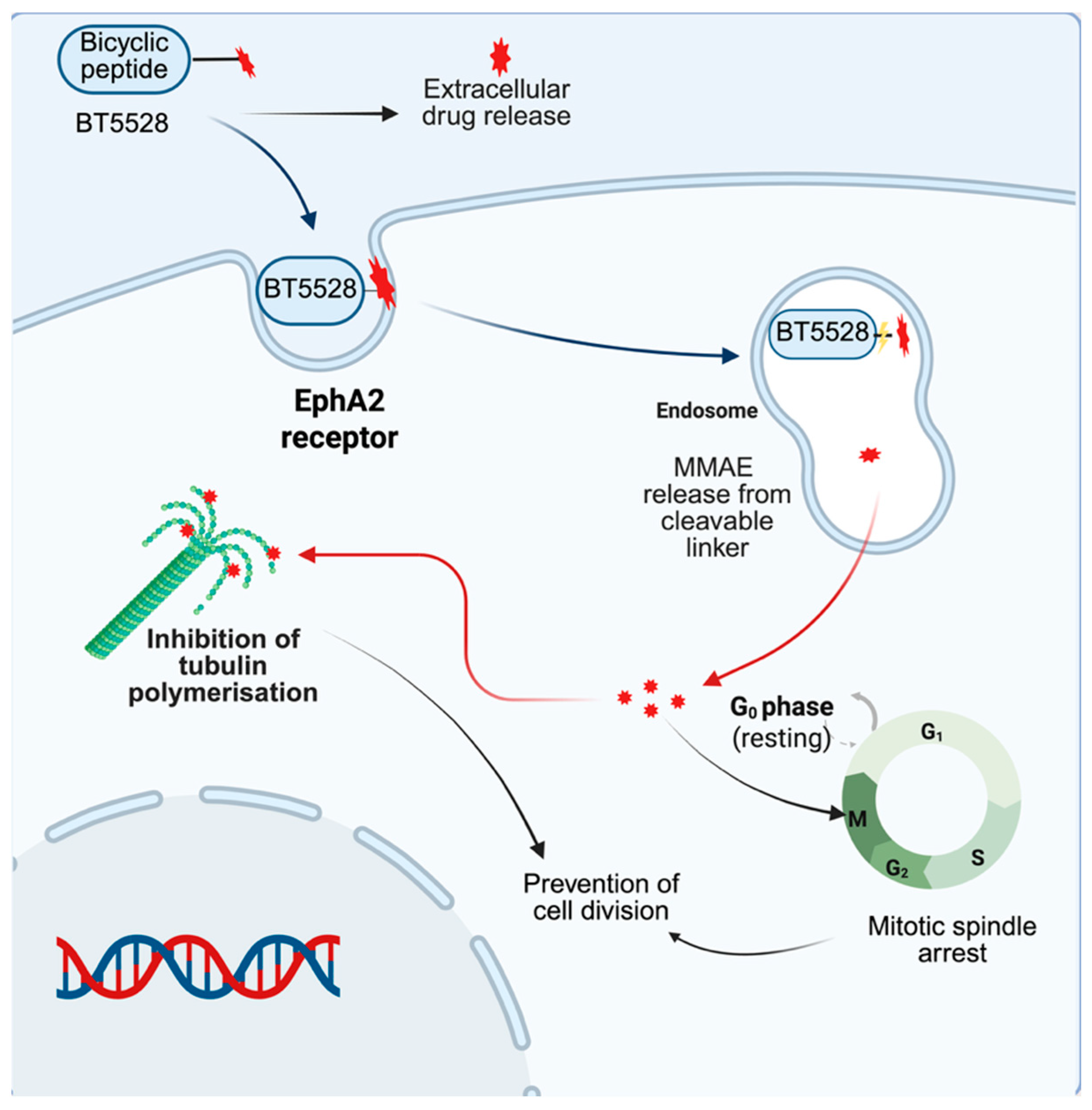
Bicycle Toxin Conjugate (BTC) BT5528
2.7. Other Classes
2.7.1. Lumisight (Pegulicianine) and Lumicell Direct Visualisation System (DVS)
2.7.2. CLP002 (TR3-M-NP) Peptide in Immunotherapy
3. Obstacles and Opportunities
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dickerson, H.; Diab, A.; Al Musaimi, O. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Cancer: Current Use and Future Prospects. Int. J. Mol. Sci. 2024, 25, 10008. [Google Scholar] [CrossRef]
- Karati, D.; Mahadik, K.R.; Trivedi, P.; Kumar, D. Alkylating Agents, the Road Less Traversed, Changing Anticancer Therapy. Anti-Cancer Agents Med. Chem. 2022, 22, 1478–1495. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.A.; Gernert, K.M.; Nettles, J.H.; Aneja, R. Drugs that target dynamic microtubules: A new molecular perspective. Med. Res. Rev. 2011, 31, 443–481. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.; Jaffee, E.M. Immunotherapy transforms cancer treatment. J. Clin. Investig. 2019, 129, 46–47. [Google Scholar] [CrossRef] [PubMed]
- Lamb, H.O.; Benfield, A.H.; Henriques, S.T. Peptides as innovative strategies to combat drug resistance in cancer therapy. Drug Discov. Today 2024, 29, 104206. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Lumisight Approval. 2024. Available online: https://lumicell.com/the-science-propelling-lumicell/ (accessed on 10 July 2025).
- Thundimadathil, J. Cancer treatment using peptides: Current therapies and future prospects. J. Amino Acids 2012, 2012, 967347. [Google Scholar] [CrossRef] [PubMed]
- Al Musaimi, O. Peptide Therapeutics: Unveiling the Potential against Cancer—A Journey through 1989. Cancers 2024, 16, 1032. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr. Rev. 2003, 24, 389–427. [Google Scholar] [CrossRef]
- Hancock, R.E.; Haney, E.F.; Gill, E.E. The immunology of host defence peptides: Beyond antimicrobial activity. Nat. Rev. Immunol. 2016, 16, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.M.; Potts, G.K.; Ready, D.B.; Patterson, M.J. Specific MHC-I Peptides Are Induced Using PROTACs. Front. Immunol. 2018, 9, 2697. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2024: An Analysis of the FDA Drug Approvals from the Perspective of Molecules. Molecules 2025, 30, 482. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Sharma, K.K.; Sharma, A.; Jain, R. Peptide-based drug discovery: Current status and recent advances. Drug Discov. Today 2023, 28, 103464. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Al Musaimi, O. FDA’s stamp of approval: Unveiling peptide breakthroughs in cardiovascular diseases, ACE, HIV, CNS, and beyond. J. Pept. Sci. 2024, 30, e3627. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.; Coburn, F.; Nsereko, Y.; Al Musaimi, O. Peptide–Drug Conjugates: A New Hope for Cancer. J. Pept. Sci. 2025, 31, e70040. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yin, F.; Li, Z.; Zhang, Y.; Shi, C. Current progress and remaining challenges of peptide–drug conjugates (PDCs): Next generation of antibody-drug conjugates (ADCs)? J. Nanobiotechnol. 2025, 23, 305. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Liu, J.; Xia, M.; Yin, L.; Zhang, L.; Liu, X.; Cheng, Y. Peptide-drug conjugates: A new paradigm for targeted cancer therapy. Eur. J. Med. Chem. 2024, 265, 116119. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Yu, L.; Miao, Y.; Liu, X.; Yu, Z.; Wei, M. Peptide-drug conjugates (PDCs): A novel trend of research and development on targeted therapy, hype or hope? Acta Pharm. Sin. B 2023, 13, 498–516. [Google Scholar] [CrossRef] [PubMed]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody-Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef] [PubMed]
- Werner, R.A.; Derlin, T.; Lapa, C.; Sheikbahaei, S.; Higuchi, T.; Giesel, F.L.; Behr, S.; Drzezga, A.; Kimura, H.; Buck, A.K.; et al. (18)F-Labeled, PSMA-Targeted Radiotracers: Leveraging the Advantages of Radiofluorination for Prostate Cancer Molecular Imaging. Theranostics 2020, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- 68Ga-NTA-476 Imaging in Prostate Cancer. 2025. Available online: https://clin.larvol.com/trial-detail/ACTRN12623001157662 (accessed on 10 July 2025).
- Kelly, W.K.; Srinivas, S.; Maly, J.; Kilari, D.; Hussain, A.; Saraiya, B.; Siddiqui, B.A.; Monk, P.; Chatta, G.S.; Narayan, V.; et al. Phase 1/2 study of REGN4336 alone or in combination with cemiplimab or nezastomig in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2025, 43 (Suppl. 5), TPS295. [Google Scholar] [CrossRef]
- First Human Trial of Targeting MDM2/MDMX PET Imaging. 2024. Available online: https://clin.larvol.com/trial-detail/NCT06443762 (accessed on 10 July 2025).
- Kenny, L.M.; Gilbert, F.J.; Gopalakrishnan, G.; Aravind, P.; Barwick, T.; Patel, N.; Hiscock, D.R.; Boros, I.; Kealey, S.; Aigbirhio, F.I.; et al. The HERPET study: Imaging HER2 expression in breast cancer with the novel PET tracer [18F]GE-226, a first-in-patient study. J. Clin. Oncol. 2022, 40 (Suppl. 16), 3069. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.; Lian, X.; Zhang, C.; Feng, J.; Tao, H.; Wang, Z. Potential target within the tumor microenvironment—MT1-MMP. Front. Immunol. 2025, 16, 1517519. [Google Scholar] [CrossRef] [PubMed]
- de Valk, K.S.; Deken, M.M.; Handgraaf, H.J.M.; Bhairosingh, S.S.; Bijlstra, O.D.; van Esdonk, M.J.; Terwisscha van Scheltinga, A.G.T.; Valentijn, A.; March, T.L.; Vuijk, J.; et al. First-in-Human Assessment of cRGD-ZW800-1, a Zwitterionic, Integrin-Targeted, Near-Infrared Fluorescent Peptide in Colon Carcinoma. Clin. Cancer Res. 2020, 26, 3990–3998. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Fu, F.; Li, F.; Zhu, Z.; Yang, Y.; Chen, X.; Jia, B.; Zheng, S.; Huang, C.; Miao, W. Comparison of [(99m)Tc]3PRGD(2) Imaging and [(18)F]FDG PET/CT in Breast Cancer and Expression of Integrin α(v)β(3) in Breast Cancer Vascular Endothelial Cells. Mol. Imaging. Biol. 2018, 20, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Decary, S.; Berne, P.F.; Nicolazzi, C.; Lefebvre, A.M.; Dabdoubi, T.; Cameron, B.; Rival, P.; Devaud, C.; Prades, C.; Bouchard, H.; et al. Preclinical Activity of SAR408701: A Novel Anti-CEACAM5-maytansinoid Antibody-drug Conjugate for the Treatment of CEACAM5-positive Epithelial Tumors. Clin. Cancer Res. 2020, 26, 6589–6599. [Google Scholar] [CrossRef] [PubMed]
- Emons, G.; Gorchev, G.; Harter, P.; Wimberger, P.; Stähle, A.; Hanker, L.; Hilpert, F.; Beckmann, M.W.; Dall, P.; Gründker, C.; et al. Efficacy and safety of AEZS-108 (LHRH agonist linked to doxorubicin) in women with advanced or recurrent endometrial cancer expressing LHRH receptors: A multicenter phase 2 trial (AGO-GYN5). Int. J. Gynecol. Cancer 2014, 24, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.; Brown, A.; Mudd, G.; Huxley, P.; Van Rietschoten, K.; Pavan, S.; Chen, L.; Watcham, S.; Lahdenranta, J.; Keen, N. MMAE Delivery Using the Bicycle Toxin Conjugate BT5528. Mol. Cancer Ther. 2020, 19, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Kumthekar, P.; Tang, S.C.; Brenner, A.J.; Kesari, S.; Piccioni, D.E.; Anders, C.; Carrillo, J.; Chalasani, P.; Kabos, P.; Puhalla, S.; et al. ANG1005, a Brain-Penetrating Peptide-Drug Conjugate, Shows Activity in Patients with Breast Cancer with Leptomeningeal Carcinomatosis and Recurrent Brain Metastases. Clin. Cancer Res. 2020, 26, 2789–2799. [Google Scholar] [CrossRef] [PubMed]
- McKean, M.; Bendell, J.C.; Petrylak, D.P.; Powles, T.B.; Sonpavde, G.P.; Dickson, A.; Dosunmu, L.; Hennessy, M.G.; Jeffrey, P.; Rigby, M.; et al. 599TiP BT8009-100 phase I/II study of the safety, pharmacokinetics, & preliminary clinical activity of BT8009 in patients with Nectin-4 expressing advanced malignancies. Ann. Oncol. 2020, 31, S500–S501. [Google Scholar] [CrossRef]
- Lorusso, P.; Meric-Bernstam, F.; Hafez, N.; Rodriguez Rivera, I.I.; Tripathy, D.; Wilks, S.; Pearson, P.; Needle, M.N.; Tolcher, A.W. 669P CBX-12-101: Final results of a phase I study of CBX-12, a peptide drug conjugate (PDC) in patients (pts) with metastatic solid tumors. Ann. Oncol. 2024, 35, S525. [Google Scholar] [CrossRef]
- Gong, J.; Hu, X.; Zhang, J.; Du, Y.; Huang, R.; Teng, Y.; Tan, W.; Shen, L. Phase Ia study of CBP-1008, a bi-specific ligand drug conjugate targeting FRα and TRPV6, in patients with advanced solid tumors. J. Clin. Oncol. 2021, 39 (Suppl. 15), 3077. [Google Scholar] [CrossRef]
- Pagliaro, L.; Marchesini, M.; Roti, G. Targeting oncogenic Notch signaling with SERCA inhibitors. J. Hematol. Oncol. 2021, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Hartrampf, P.E.; Weinzierl, F.X.; Buck, A.K.; Rowe, S.P.; Higuchi, T.; Seitz, A.K.; Kübler, H.; Schirbel, A.; Essler, M.; Bundschuh, R.A.; et al. Matched-pair analysis of [(177)Lu]Lu-PSMA I&T and [(177)Lu]Lu-PSMA-617 in patients with metastatic castration-resistant prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 3269–3276. [Google Scholar] [CrossRef] [PubMed]
- Chavda, V.P.; Solanki, H.K.; Davidson, M.; Apostolopoulos, V.; Bojarska, J. Peptide-Drug Conjugates: A New Hope for Cancer Management. Molecules 2022, 27, 7232. [Google Scholar] [CrossRef] [PubMed]
- Gregorc, V.; Gaafar, R.M.; Favaretto, A.; Grossi, F.; Jassem, J.; Polychronis, A.; Bidoli, P.; Tiseo, M.; Shah, R.; Taylor, P.; et al. NGR-hTNF in combination with best investigator choice in previously treated malignant pleural mesothelioma (NGR015): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2018, 19, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.S.; Tang, K.; Lv, J. Peptide-drug conjugate-based novel molecular drug delivery system in cancer. Trends Pharmacol. Sci. 2021, 42, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Demeule, M.; Charfi, C.; Currie, J.C.; Zgheib, A.; Danalache, B.A.; Béliveau, R.; Marsolais, C.; Annabi, B. The TH1902 Docetaxel Peptide-Drug Conjugate Inhibits Xenografts Growth of Human SORT1-Positive Ovarian and Triple-Negative Breast Cancer Stem-like Cells. Pharmaceutics 2022, 14, 1910. [Google Scholar] [CrossRef] [PubMed]
- Charfi, C.; Demeule, M.; Currie, J.C.; Larocque, A.; Zgheib, A.; Danalache, B.A.; Ouanouki, A.; Béliveau, R.; Marsolais, C.; Annabi, B. New Peptide-Drug Conjugates for Precise Targeting of SORT1-Mediated Vasculogenic Mimicry in the Tumor Microenvironment of TNBC-Derived MDA-MB-231 Breast and Ovarian ES-2 Clear Cell Carcinoma Cells. Front. Oncol. 2021, 11, 760787. [Google Scholar] [CrossRef] [PubMed]
- Han, I.H.; Choi, I.; Choi, H.; Kim, S.; Jeong, C.; Yang, J.; Cao, Y.; Choi, J.; Lee, H.; Shin, J.S.; et al. Conformation-sensitive targeting of CD18 depletes M2-like tumor-associated macrophages resulting in inhibition of solid tumor progression. J. Immunother. Cancer 2025, 13, e011422. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhao, Z.; Fetse, J.; Mamani, U.F.; Guo, Y.; Li, Y.; Patel, P.; Liu, Y.; Lin, C.Y.; Li, Y.; Mustafa, B.; et al. Development of a peptide-based tumor-activated checkpoint inhibitor for cancer immunotherapy. Acta Biomater. 2025, 193, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Tanada, M.; Tamiya, M.; Matsuo, A.; Chiyoda, A.; Takano, K.; Ito, T.; Irie, M.; Kotake, T.; Takeyama, R.; Kawada, H.; et al. Development of Orally Bioavailable Peptides Targeting an Intracellular Protein: From a Hit to a Clinical KRAS Inhibitor. J. Am. Chem. Soc. 2023, 145, 16610–16620. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Harb, W.; Patnaik, A.; Ulahannan, S.; Mahdi, H.; Ahluwalia, M.; Patel, M.; Dowlati, A.; Bullock, A.; Wen, P.; et al. 374 A first-in-human Phase 1/2 open label trial evaluating the safety, pharmacology, and preliminary efficacy of VT1021 in subjects with advanced solid tumors. J. Immunother. Cancer 2020, 8 (Suppl. 3), A228. [Google Scholar] [CrossRef]
- Guerlavais, V.; Sawyer, T.K.; Carvajal, L.; Chang, Y.S.; Graves, B.; Ren, J.G.; Sutton, D.; Olson, K.A.; Packman, K.; Darlak, K.; et al. Discovery of Sulanemadlin (ALRN-6924), the First Cell-Permeating, Stabilized α-Helical Peptide in Clinical Development. J. Med. Chem. 2023, 66, 9401–9417. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Singh, M.; Sanz Santos, G.; Guerlavais, V.; Carvajal, L.A.; Aivado, M.; Zhan, Y.; Oliveira, M.M.S.; Westerberg, L.S.; Annis, D.A.; et al. Pharmacologic Activation of p53 Triggers Viral Mimicry Response Thereby Abolishing Tumor Immune Evasion and Promoting Antitumor Immunity. Cancer Discov. 2021, 11, 3090–3105. [Google Scholar] [CrossRef] [PubMed]
- Bekaii-Saab, T.; Wei, L.; Wesolowski, R.; Ahn, D.; Wu, C.; Lustberg, M.; Mortazavi, A.; Ramaswamy, B.; Overholser, J.; Kaumaya, P. A Phase Ib of a combination of two chimeric (Trastuzumab-like and Pertuzumab-like) HER-2 B cell peptide vaccine emulsified in ISA 720 and nor-MDP adjuvant in patients with advanced solid tumors [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2019; 2019 Mar 29-Apr 3; Atlanta, GA. Philadelphia (PA): AACR. Cancer Res. 2019, 79 (Suppl. 13), Abstract nr CT017. [Google Scholar]
- Hackenbruch, C.; Bauer, J.; Heitmann, J.S.; Maringer, Y.; Nelde, A.; Denk, M.; Zieschang, L.; Kammer, C.; Federmann, B.; Jung, S.; et al. FusionVAC22_01: A phase I clinical trial evaluating a DNAJB1-PRKACA fusion transcript-based peptide vaccine combined with immune checkpoint inhibition for fibrolamellar hepatocellular carcinoma and other tumor entities carrying the oncogenic driver fusion. Front. Oncol. 2024, 14, 1367450. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Chen, Z.; Wu, J.; Huang, Q.; He, Y.; Shang, H.; Xia, D.; Peng, E.; Wang, Z.; Liang, X.; et al. METTL3 promotes an immunosuppressive microenvironment in bladder cancer via m6A-dependent CXCL5/CCL5 regulation. J. Immunother. Cancer 2025, 13, e011108. [Google Scholar] [CrossRef] [PubMed]
- Ohio State University Comprehensive Cancer Center. BZLF1 Peptide Vaccine (OSU-2131) with QS-21 for the Prevention of Epstein-Barr Virus Related Cancer in Patients Awaiting Solid Organ Transplants. 2024. Available online: https://clin.larvol.com/trial-detail/NCT06741072 (accessed on 10 July 2025).
- Alsalloum, A.; Shevchenko, J.A.; Sennikov, S. NY-ESO-1 antigen: A promising frontier in cancer immunotherapy. Clin. Transl. Med. 2024, 14, e70020. [Google Scholar] [CrossRef] [PubMed]
- Schoen, R.E.; Boardman, L.A.; Cruz-Correa, M.; Bansal, A.; Kastenberg, D.; Hur, C.; Dzubinski, L.; Kaufman, S.F.; Rodriguez, L.M.; Richmond, E.; et al. Randomized, Double-Blind, Placebo-Controlled Trial of MUC1 Peptide Vaccine for Prevention of Recurrent Colorectal Adenoma. Clin. Cancer Res. 2023, 29, 1678–1688. [Google Scholar] [CrossRef] [PubMed]
- ProteinQure to Present Data on PQ203, a Novel Peptide-Drug Conjugate for Triple Negative Breast Cancer, at 2024 San Antonio Breast Cancer Symposium. 2024. Available online: https://www.biospace.com/press-releases/proteinqure-to-present-data-on-pq203-a-novel-peptide-drug-conjugate-for-triple-negative-breast-cancer-at-2024-san-antonio-breast-cancer-symposium (accessed on 10 July 2025).
- Dmello, C.; Brenner, A.; Piccioni, D.; Wen, P.Y.; Drappatz, J.; Mrugala, M.; Lewis, L.D.; Schiff, D.; Fadul, C.E.; Chamberlain, M.; et al. Phase II trial of blood–brain barrier permeable peptide-paclitaxel conjugate ANG1005 in patients with recurrent high-grade glioma. Neuro-Oncol. Adv. 2024, 6, vdae186. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Hirte, H.; Welch, S.; Ilenchuk, T.T.; Lutes, T.; Rice, C.; Fields, N.; Nemet, A.; Dugourd, D.; Piha-Paul, S.; et al. First-in-human phase I study of SOR-C13, a TRPV6 calcium channel inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Clift, R.; Ehara, T.; Yanagida, H.; Horton, S.; Noncovich, A.; Guest, M.; Kim, D.; Salvador, K.; Richardson, S.; et al. Peptide Binder to Glypican-3 as a Theranostic Agent for Hepatocellular Carcinoma. J. Nucl. Med. 2024, 65, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Memorial Sloan Kettering Cancer Center. Phase I, Open-Label Study of the Safety and Dosimetry of a 3-Dose Regimen of Escalating Doses of 177Lu-DOTA-EB-TATE in Adult Patients with Advanced, Well-Differentiated Neuroendocrine Tumors. Available online: https://www.mskcc.org/cancer-care/clinical-trials/21-362 (accessed on 10 July 2025).
- Novartis. A Study of 177Lu-FAP-2286 in Advanced Solid Tumors. 2025. Available online: https://www.novartis.com/clinicaltrials/study/nct04939610 (accessed on 10 July 2025).
- Zoptarelin Doxorubicin Fails to Improve Survival in Phase III Endometrial Cancer Trial. 2017. Available online: https://www.targetedonc.com/view/zoptarelin-doxorubicin-fails-to-improve-survival-in-phase-iii-endometrial-cancer-trial (accessed on 10 July 2025).
- Al Musaimi, O.; AlShaer, D.; de la Torre, B.G.; Albericio, F. 2024 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2025, 18, 291. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.K.; Nikooienejad, A.; Bray, R.; Cui, X.; Wilson, J.; Duffin, K.; Milicevic, Z.; Haupt, A.; Robins, D.A. Dual GIP and GLP-1 Receptor Agonist Tirzepatide Improves Beta-cell Function and Insulin Sensitivity in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2021, 106, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.E.; Zhernosekov, K. The evolution of PRRT for the treatment of neuroendocrine tumors; What comes next? Front. Endocrinol. 2022, 13, 941832. [Google Scholar] [CrossRef] [PubMed]
- Feijtel, D.; de Jong, M.; Nonnekens, J. Peptide Receptor Radionuclide Therapy: Looking Back, Looking Forward. Curr. Top. Med. Chem. 2020, 20, 2959–2969. [Google Scholar] [CrossRef] [PubMed]
- Burkett, B.J.; Bartlett, D.J.; McGarrah, P.W.; Lewis, A.R.; Johnson, D.R.; Berberoğlu, K.; Pandey, M.K.; Packard, A.T.; Halfdanarson, T.R.; Hruska, C.B.; et al. A Review of Theranostics: Perspectives on Emerging Approaches and Clinical Advancements. Radiol. Imaging Cancer 2023, 5, e220157. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Li, C.; Hong, H.; Zhou, Z.; Wu, Z. Macrocyclic RGD-Peptides with High Selectivity for αvβ3 Integrin in Cancer Imaging and Therapy. RSC Med. Chem. 2025. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Cui, C.; Wang, L.; Liu, H.; Cui, G. Novel Tetrapeptide, RGDF, Mediated Tumor Specific Liposomal Doxorubicin (DOX) Preparations. Mol. Pharm. 2011, 8, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Al Musaimi, O. Lasso Peptides Realm: Insights and Applications. Peptides 2024, 182, 171317. [Google Scholar] [CrossRef] [PubMed]
- Heh, E.; Allen, J.; Ramirez, F.; Lovasz, D.; Fernandez, L.; Hogg, T.; Riva, H.; Holland, N.; Chacon, J. Peptide Drug Conjugates and Their Role in Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 829. [Google Scholar] [CrossRef] [PubMed]
- Nhàn, N.T.T.; Yamada, T.; Yamada, K.H. Peptide-Based Agents for Cancer Treatment: Current Applications and Future Directions. Int. J. Mol. Sci. 2023, 24, 12931. [Google Scholar] [CrossRef] [PubMed]
- de Castro, G.V.; Worm, D.J.; Grabe, G.J.; Rowan, F.C.; Haggerty, L.; de la Lastra, A.L.; Popescu, O.; Helaine, S.; Barnard, A. Characterization of the Key Determinants of Phd Antitoxin Mediated Doc Toxin Inactivation in Salmonella. ACS Chem. Biol. 2022, 17, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dawber, R.S.; Zhang, P.; Walko, M.; Wilson, A.J.; Wang, X. Peptide-based inhibitors of protein-protein interactions: Biophysical, structural and cellular consequences of introducing a constraint. Chem. Sci. 2021, 12, 5977–5993. [Google Scholar] [CrossRef] [PubMed]
- Hurine, J.A.; Lambalk, C.B. Gonadotropin-releasing-hormone-receptor antagonists. Lancet 2001, 358, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Carolsfeld, J.; Powell, J.F.; Park, M.; Fischer, W.H.; Craig, A.G.; Chang, J.P.; Rivier, J.E.; Sherwood, N.M. Primary structure and function of three gonadotropin-releasing hormones, including a novel form, from an ancient teleost, herring. Endocrinology 2000, 141, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Olberg, D.E.; Hausner, S.H.; Bauer, N.; Klaveness, J.; Indrevoll, B.; Andressen, K.W.; Dahl, M.; Levy, F.O.; Sutcliffe, J.L.; Haraldsen, I. Radiosynthesis of high affinity fluorine-18 labeled GnRH peptide analogues: In vitro studies and in vivo assessment of brain uptake in rats. MedChemComm 2015, 6, 708–714. [Google Scholar] [CrossRef]
- Gründker, C.; Emons, G. The Role of Gonadotropin-Releasing Hormone in Cancer Cell Proliferation and Metastasis. Front. Endocrinol. 2017, 8, 187. [Google Scholar] [CrossRef] [PubMed]
- Hawryłkiewicz, A.; Ptaszyńska, N. Gemcitabine Peptide-Based Conjugates and Their Application in Targeted Tumor Therapy. Molecules 2021, 26, 364. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.K.; Leung, P.C. Molecular biology of gonadotropin-releasing hormone (GnRH)-I, GnRH-II, and their receptors in humans. Endocr. Rev. 2005, 26, 283–306. [Google Scholar] [CrossRef] [PubMed]
- Limonta, P.; Moretti, R.M.; Montagnani Marelli, M.; Motta, M. The biology of gonadotropin hormone-releasing hormone: Role in the control of tumor growth and progression in humans. Front. Neuroendocrinol. 2003, 24, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Olberg, D.E.; Bauer, N.; Andressen, K.W.; Hjørnevik, T.; Cumming, P.; Levy, F.O.; Klaveness, J.; Haraldsen, I.; Sutcliffe, J.L. Brain penetrant small molecule (18)F-GnRH receptor (GnRH-R) antagonists: Synthesis and preliminary positron emission tomography imaging in rats. Nucl. Med. Biol. 2016, 43, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Al-Inany, H.G.; Youssef, M.A.; Ayeleke, R.O.; Brown, J.; Lam, W.S.; Broekmans, F.J. Gonadotrophin-releasing hormone antagonists for assisted reproductive technology. Cochrane Database Syst. Rev. 2016, 4, Cd001750. [Google Scholar] [CrossRef] [PubMed]
- Markatos, C.; Biniari, G.; Chepurny, O.G.; Karageorgos, V.; Tsakalakis, N.; Komontachakis, G.; Vlata, Z.; Venihaki, M.; Holz, G.G.; Tselios, T.; et al. Cytotoxic Activity of Novel GnRH Analogs Conjugated with Mitoxantrone in Ovarian Cancer Cells. Molecules 2024, 29, 4127. [Google Scholar] [CrossRef] [PubMed]
- Degarelix Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2008/022201s000_Lbl.pdf (accessed on 10 July 2025).
- Guryanov, I.; Orlandin, A.; Viola, A.; Biondi, B.; Badocco, D.; Formaggio, F.; Ricci, A.; Cabri, W. Overcoming Chemical Challenges in the Solid-Phase Synthesis of High-Purity GnRH Antagonist Degarelix. Part 1. Org. Process Res. Dev. 2019, 23, 2746–2753. [Google Scholar] [CrossRef]
- Ali, M.; Raslan, M.; Ciebiera, M.; Zaręba, K.; Al-Hendy, A. Current approaches to overcome the side effects of GnRH analogs in the treatment of patients with uterine fibroids. Expert Opin. Drug Saf. 2022, 21, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Han, S.; Qiu, N.; Feng, W.; Lu, M.; Zhang, W.; Wang, M.; Zhou, Q.; Chen, S.; Xu, W.; et al. Structural insights into ligand recognition and selectivity of somatostatin receptors. Cell Res. 2022, 32, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Theodoropoulou, M.; Stalla, G.K. Somatostatin receptors: From signaling to clinical practice. Front. Neuroendocrinol. 2013, 34, 228–252. [Google Scholar] [CrossRef] [PubMed]
- Patel, Y.C. Molecular pharmacology of somatostatin receptor subtypes. J. Endocrinol. Investig. 1997, 20, 348–367. [Google Scholar] [CrossRef] [PubMed]
- Bergsma, H.; van Vliet, E.I.; Teunissen, J.J.; Kam, B.L.; de Herder, W.W.; Peeters, R.P.; Krenning, E.P.; Kwekkeboom, D.J. Peptide receptor radionuclide therapy (PRRT) for GEP-NETs. Best Pract. Res. Clin. Gastroenterol. 2012, 26, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Octreotide Approval Letter and Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/98/021008a_appltr_prntlbl.pdf (accessed on 10 July 2025).
- Mycapssa Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/208232Orig1s000AdminCorres.pdf (accessed on 10 July 2025).
- McCafferty, R.; Fawzy, R. Chapter 38—Miscellaneous Hormones. In Side Effects of Drugs Annual; Ray, S.D., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 447–455. [Google Scholar]
- Ladrière, T.; Faudemer, J.; Levigoureux, E.; Peyronnet, D.; Desmonts, C.; Vigne, J. Safety and Therapeutic Optimization of Lutetium-177 Based Radiopharmaceuticals. Pharmaceutics 2023, 15, 1240. [Google Scholar] [CrossRef] [PubMed]
- Stueven, A.K.; Kayser, A.; Wetz, C.; Amthauer, H.; Wree, A.; Tacke, F.; Wiedenmann, B.; Roderburg, C.; Jann, H. Somatostatin Analogues in the Treatment of Neuroendocrine Tumors: Past, Present and Future. Int. J. Mol. Sci. 2019, 20, 3049. [Google Scholar] [CrossRef] [PubMed]
- Lanreotide Approval Letter. 2007. Chrome-Extension. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2007/022074s000_Approv.pdf (accessed on 10 July 2025).
- Lanreotide Drug Label. 2007. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215395s000lbl.pdf (accessed on 10 July 2025).
- Zhao, J.; Wang, S.; Markison, S.; Kim, S.H.; Han, S.; Chen, M.; Kusnetzow, A.K.; Rico-Bautista, E.; Johns, M.; Luo, R.; et al. Discovery of Paltusotine (CRN00808), a Potent, Selective, and Orally Bioavailable Non-peptide SST2 Agonist. ACS Med. Chem. Lett. 2023, 14, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Juliana, C.A.; Chai, J.; Arroyo, P.; Rico-Bautista, E.; Betz, S.F.; De León, D.D. A selective nonpeptide somatostatin receptor 5 agonist effectively decreases insulin secretion in hyperinsulinism. J. Biol. Chem. 2023, 299, 104816. [Google Scholar] [CrossRef] [PubMed]
- Rogoza, O.; Megnis, K.; Kudrjavceva, M.; Gerina-Berzina, A.; Rovite, V. Role of Somatostatin Signalling in Neuroendocrine Tumours. Int. J. Mol. Sci. 2022, 23, 1447. [Google Scholar] [CrossRef] [PubMed]
- Gourni, E.; Henriksen, G. Metal-Based PSMA Radioligands. Molecules 2017, 22, 523. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Heston, W.D. Tumor target prostate specific membrane antigen (PSMA) and its regulation in prostate cancer. J. Cell Biochem. 2004, 91, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Singh, P.; Topaloglu, O.; Isaacs, J.T.; Denmeade, S.R. A dimeric peptide that binds selectively to prostate-specific membrane antigen and inhibits its enzymatic activity. Cancer Res. 2006, 66, 9171–9177. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Bennett, M.J.; Thomas, L.M.; Bjorkman, P.J. Crystal structure of prostate-specific membrane antigen, a tumor marker and peptidase. Proc. Natl. Acad. Sci. USA 2005, 102, 5981–5986. [Google Scholar] [CrossRef] [PubMed]
- Barinka, C.; Hlouchova, K.; Rovenska, M.; Majer, P.; Dauter, M.; Hin, N.; Ko, Y.S.; Tsukamoto, T.; Slusher, B.S.; Konvalinka, J.; et al. Structural basis of interactions between human glutamate carboxypeptidase II and its substrate analogs. J. Mol. Biol. 2008, 376, 1438–1450. [Google Scholar] [CrossRef] [PubMed]
- Mesters, J.R.; Barinka, C.; Li, W.; Tsukamoto, T.; Majer, P.; Slusher, B.S.; Konvalinka, J.; Hilgenfeld, R. Structure of glutamate carboxypeptidase II, a drug target in neuronal damage and prostate cancer. EMBO J. 2006, 25, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Barinka, C.; Starkova, J.; Konvalinka, J.; Lubkowski, J. A high-resolution structure of ligand-free human glutamate carboxypeptidase II. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2007, 63 Pt 3, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Barinka, C.; Byun, Y.; Dusich, C.L.; Banerjee, S.R.; Chen, Y.; Castanares, M.; Kozikowski, A.P.; Mease, R.C.; Pomper, M.G.; Lubkowski, J. Interactions between human glutamate carboxypeptidase II and urea-based inhibitors: Structural characterization. J. Med. Chem. 2008, 51, 7737–7743. [Google Scholar] [CrossRef] [PubMed]
- Pluvicto Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215833s000lbl.pdf (accessed on 10 July 2025).
- Foley, R.W.; Redman, S.L.; Graham, R.N.; Loughborough, W.W.; Little, D. Fluorine-18 labelled prostate-specific membrane antigen (PSMA)-1007 positron-emission tomography-computed tomography: Normal patterns, pearls, and pitfalls. Clin. Radiol. 2020, 75, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Olivier, P.; Giraudet, A.L.; Skanjeti, A.; Merlin, C.; Weinmann, P.; Rudolph, I.; Hoepping, A.; Gauthé, M. Phase III Study of (18)F-PSMA-1007 Versus (18)F-Fluorocholine PET/CT for Localization of Prostate Cancer Biochemical Recurrence: A Prospective, Randomized, Crossover Multicenter Study. J. Nucl. Med. 2023, 64, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Parghane, R.V.; Kamaldeep Chakrabarty, S. Peptide Receptor Radionuclide Therapy of Neuroendocrine Tumors. Semin. Nucl. Med. 2020, 50, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Rosar, F.; Kochems, N.; Bartholomä, M.; Maus, S.; Stemler, T.; Linxweiler, J.; Khreish, F.; Ezziddin, S. Renal Safety of [(177)Lu]Lu-PSMA-617 Radioligand Therapy in Patients with Compromised Baseline Kidney Function. Cancers 2021, 13, 3095. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Schar, J.C.; Waser, B.; Wenger, S.; Heppeler, A.; Schmitt, J.S.; Macke, H.R. Affinity profiles for human somatostatin receptor subtypes SST1-SST5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. Eur. J. Nucl. Med. 2000, 27, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Detectnet Drug Label. 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213227s000lbl.pdf (accessed on 16 January 2021).
- PeptiDream Announces Initial Public Offering of RayzeBio and Update on GPC3 Program. 2023. Available online: https://uk.marketscreener.com/quote/stock/PEPTIDREAM-INC-13370541/news/PeptiDream-Announces-Initial-Public-Offering-of-RayzeBio-and-Update-on-GPC3-Program-44886778/ (accessed on 10 July 2025).
- de la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2022: An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2023, 28, 1038. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.; Huang, Y.; Simms, A.E.; Mazur, A. Fibroblast activation protein-α: A key modulator of the microenvironment in multiple pathologies. Int. Rev. Cell Mol. Biol. 2012, 297, 83–116. [Google Scholar] [CrossRef] [PubMed]
- A Study of 177Lu-FAP-2286 in Advanced Solid Tumors (LuMIERE) Study Details Tab Study Overview. 2023. Available online: https://clinicaltrials.gov/about-site/disclaimer (accessed on 10 July 2025).
- McConathy, J.; Dhawan, M.; Goenka, A.; Moy, R.; Menda, Y.; Chasen, B.; Khushman, M.; Mintz, A.; Zakharia, Y.; Sunderland, J.; et al. 177Lu-FAP-2286 in patients with advanced or metastatic solid tumors: Initial data from a phase 1/2 study investigating safety, pharmacokinetics, dosimetry, and preliminary antitumor activity (LuMIERE). J. Nucl. Med. 2022, 63 (Suppl. 2), 2271. [Google Scholar] [CrossRef]
- Banihashemian, S.S.; Akbari, M.E.; Pirayesh, E.; Divband, G.; Abolhosseini Shahrnoy, A.; Nami, R.; Mazidi, S.M.; Nasiri, M. Feasibility and therapeutic potential of [(177)Lu]Lu-FAPI-2286 in patients with advanced metastatic sarcoma. Eur. J. Nucl. Med. Mol. Imaging 2024, 52, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Clovis Oncology Highlights Updated LuMIERE Phase 1 Data of Targeted Radiotherapy Candidate FAP-2286 at the 35th Annual EANM Congress. 2022. Available online: https://www.businesswire.com/news/home/20221017005267/en/Clovis-Oncology-Highlights-Updated-LuMIERE-Phase-1-Data-of-Targeted-Radiotherapy-Candidate-FAP-2286-at-the-35th-Annual-EANM-Congress (accessed on 10 July 2025).
- Dorywalska, M.; Dushin, R.; Moine, L.; Farias, S.E.; Zhou, D.; Navaratnam, T.; Lui, V.; Hasa-Moreno, A.; Casas, M.G.; Tran, T.T.; et al. Molecular Basis of Valine-Citrulline-PABC Linker Instability in Site-Specific ADCs and Its Mitigation by Linker Design. Mol. Cancer Ther. 2016, 15, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Shirpour, A.; Hadadi, A.; Zolghadri, S.; Vosoughi, S.; Rajabifar, S. Preclinical evaluation of [13xLa]La-FAP-2286 as a novel theranostic agent for tumors expressing fibroblast activation protein. Sci. Rep. 2025, 15, 7475. [Google Scholar] [CrossRef] [PubMed]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates--an emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Dal Corso, A.; Cazzamalli, S.; Gebleux, R.; Mattarella, M.; Neri, D. Protease-Cleavable Linkers Modulate the Anticancer Activity of Noninternalizing Antibody-Drug Conjugates. Bioconjugate Chem. 2017, 28, 1826–1833. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Tisotumab Vedotin: First Approval. Drugs 2021, 81, 2141–2147. [Google Scholar] [CrossRef] [PubMed]
- TIVDAK Drug Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761208Orig1s000lbledt.pdf (accessed on 10 July 2025).
- Bogani, G.; Coleman, R.L.; Vergote, I.; Raspagliesi, F.; Lorusso, D.; Monk, B.J. Tisotumab vedotin in recurrent or metastatic cervical cancer. Curr. Probl. Cancer 2023, 47, 100952. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Concin, N.; Hong, D.S.; Thistlethwaite, F.C.; Machiels, J.P.; Arkenau, H.T.; Plummer, R.; Jones, R.H.; Nielsen, D.; Windfeld, K.; et al. Tisotumab vedotin in patients with advanced or metastatic solid tumours (InnovaTV 201): A first-in-human, multicentre, phase 1-2 trial. Lancet Oncol. 2019, 20, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Alley, S.C.; Harris, J.R.; Cao, A.; Heuvel EG-vd Velayudhan, J.; Satijn, D.; Verploegen, S.; Dominguez, T.; Breij, E.C. Abstract 221: Tisotumab vedotin induces anti-tumor activity through MMAE-mediated, Fc-mediated, and Fab-mediated effector functions in vitro. Cancer Res. 2019, 79 (Suppl. 13), 221. [Google Scholar] [CrossRef]
- de Goeij, B.E.; Satijn, D.; Freitag, C.M.; Wubbolts, R.; Bleeker, W.K.; Khasanov, A.; Zhu, T.; Chen, G.; Miao, D.; van Berkel, P.H.; et al. High turnover of tissue factor enables efficient intracellular delivery of antibody-drug conjugates. Mol. Cancer Ther. 2015, 14, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Harris, J.R.; Burm, S.M.; Vanderstichele, A.; Houtkamp, M.A.; Aarass, S.; Riisnaes, R.; Figueiredo, I.; Nava Rodrigues, D.; Christova, R.; et al. Systematic study of tissue factor expression in solid tumors. Cancer Rep. 2023, 6, e1699. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fan, S.; Zhong, W.; Zhou, X.; Li, S. Development and Properties of Valine-Alanine based Antibody-Drug Conjugates with Monomethyl Auristatin E as the Potent Payload. Int. J. Mol. Sci. 2017, 18, 1860. [Google Scholar] [CrossRef] [PubMed]
- CEACAM5-Directed Tusamitamab Ravtansine Progresses to Phase 3 Trial in NSCLC. 2023. Available online: https://www.onclive.com/view/ceacam5-directed-tusamitamab-ravtansine-progresses-to-phase-3-trial-in-nsclc (accessed on 10 July 2025).
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide-Drug Conjugates and Their Targets in Advanced Cancer Therapies. Front. Chem. 2020, 8, 571. [Google Scholar] [CrossRef] [PubMed]
- Hennrich, U.; Kopka, K. Lutathera®: The First FDA- and EMA-Approved Radiopharmaceutical for Peptide Receptor Radionuclide Therapy. Pharmaceuticals 2019, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Olivier, T.; Prasad, V. The approval and withdrawal of melphalan flufenamide (melflufen): Implications for the state of the FDA. Transl. Oncol. 2022, 18, 101374. [Google Scholar] [CrossRef] [PubMed]
- Pepaxti Drug Approval Letter. 2022. Available online: https://www.pharmaceutical-technology.com/news/mhra-oncopeptides-multiple-myeloma-drug/#:~:text=MHRA%20grants%20marketing%20authorisation%20for,or%20following%20the%20last%20treatment (accessed on 10 July 2025).
- Bashir, B.; Wang, J.S.; Falchook, G.; Fontana, E.; Arkenau, H.T.; Carter, L.; Galot, R.; Basu, B.; Greystoke, A.; Subbiah, V.; et al. Results From First-in-Human Phase I Dose-Escalation Study of a Novel Bicycle Toxin Conjugate Targeting EphA2 (BT5528) in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2024, 42, 3443–3452. [Google Scholar] [CrossRef] [PubMed]
- Al Musaimi, O.; Lombardi, L.; Williams, D.R.; Albericio, F. Strategies for Improving Peptide Stability and Delivery. Pharmaceuticals 2022, 15, 1283. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, C.; Pazgier, M.; Li, C.; Mao, Y.; Lv, Y.; Gu, B.; Wei, G.; Yuan, W.; Zhan, C.; et al. D-peptide inhibitors of the p53-MDM2 interaction for targeted molecular therapy of malignant neoplasms. Proc. Natl. Acad. Sci. USA 2010, 107, 14321–14326. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. Advances in oral peptide therapeutics. Nat. Rev. Drug Discov. 2020, 19, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Andreadis, P.; Karagiannis, T.; Malandris, K.; Avgerinos, I.; Liakos, A.; Manolopoulos, A.; Bekiari, E.; Matthews, D.R.; Tsapas, A. Semaglutide for type 2 diabetes mellitus: A systematic review and meta-analysis. Diabetes Obes. Metab. 2018, 20, 2255–2263. [Google Scholar] [CrossRef] [PubMed]
- Tuvia, S.; Pelled, D.; Marom, K.; Salama, P.; Levin-Arama, M.; Karmeli, I.; Idelson, G.H.; Landau, I.; Mamluk, R. A novel suspension formulation enhances intestinal absorption of macromolecules via transient and reversible transport mechanisms. Pharm. Res. 2014, 31, 2010–2021. [Google Scholar] [CrossRef] [PubMed]
- Tuvia, S.; Atsmon, J.; Teichman, S.L.; Katz, S.; Salama, P.; Pelled, D.; Landau, I.; Karmeli, I.; Bidlingmaier, M.; Strasburger, C.J.; et al. Oral octreotide absorption in human subjects: Comparable pharmacokinetics to parenteral octreotide and effective growth hormone suppression. J. Clin. Endocrinol. Metab. 2012, 97, 2362–2369. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, X.; Xie, Z.; Xie, C.; Zhan, C.; Hu, X.; Shen, Q.; Wei, X.; Su, B.; Wang, J.; et al. D-Peptides as Recognition Molecules and Therapeutic Agents. Chem. Rec. 2016, 16, 1772–1786. [Google Scholar] [CrossRef] [PubMed]
- Welch, B.D.; Francis, J.N.; Redman, J.S.; Paul, S.; Weinstock, M.T.; Reeves, J.D.; Lie, Y.S.; Whitby, F.G.; Eckert, D.M.; Hill, C.P.; et al. Design of a potent D-peptide HIV-1 entry inhibitor with a strong barrier to resistance. J. Virol. 2010, 84, 11235–11244. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Pazgier, M.; Li, C.; Yuan, W.; Li, C.; Lu, W. A left-handed solution to peptide inhibition of the p53-MDM2 interaction. Angew. Chem. Int. Ed. Engl. 2010, 49, 3649–3652. [Google Scholar] [CrossRef] [PubMed]
- Maxian, T.; Gerlitz, L.; Riedl, S.; Rinner, B.; Zweytick, D. Effect of L- to D-Amino Acid Substitution on Stability and Activity of Antitumor Peptide RDP215 against Human Melanoma and Glioblastoma. Int. J. Mol. Sci. 2021, 22, 8469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Sun, X.; Liu, W.; Wu, J.; Wu, Y.; Jiang, S.; Wang, X.; Gao, X.; Zuo, Q.; Zhang, H.; et al. Determining the Multivalent Effects of d-Peptide-Based Radiotracers. Chem. Biomed. Imaging 2025, 3, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, Y.; Tan, Y.; Hu, R.; Niu, M.-M. An NRP1/MDM2-Targeted D-Peptide Supramolecular Nanomedicine for High-Efficacy and Low-Toxic Liver Cancer Therapy. Adv. Healthc. Mater. 2021, 10, 2002197. [Google Scholar] [CrossRef] [PubMed]

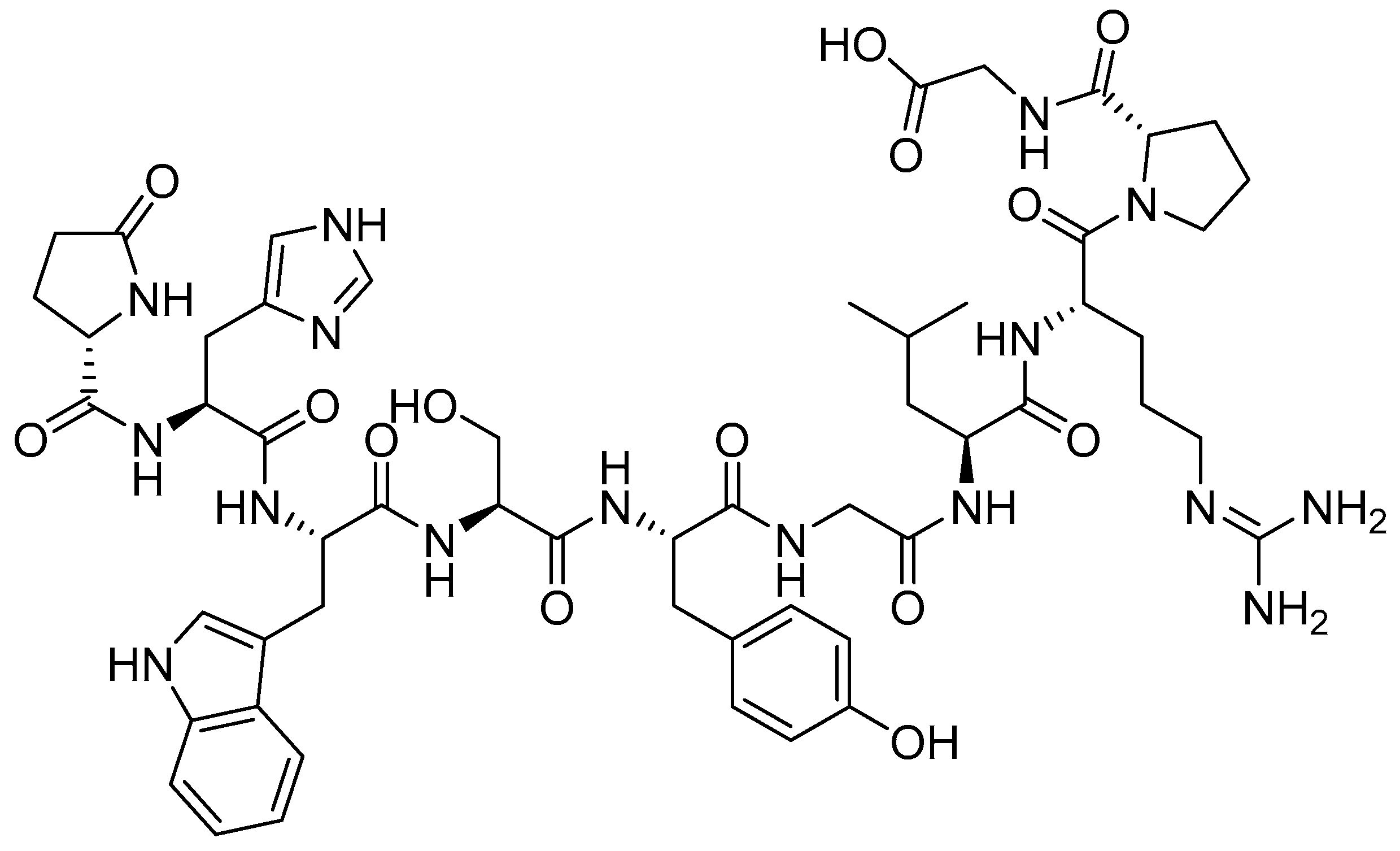

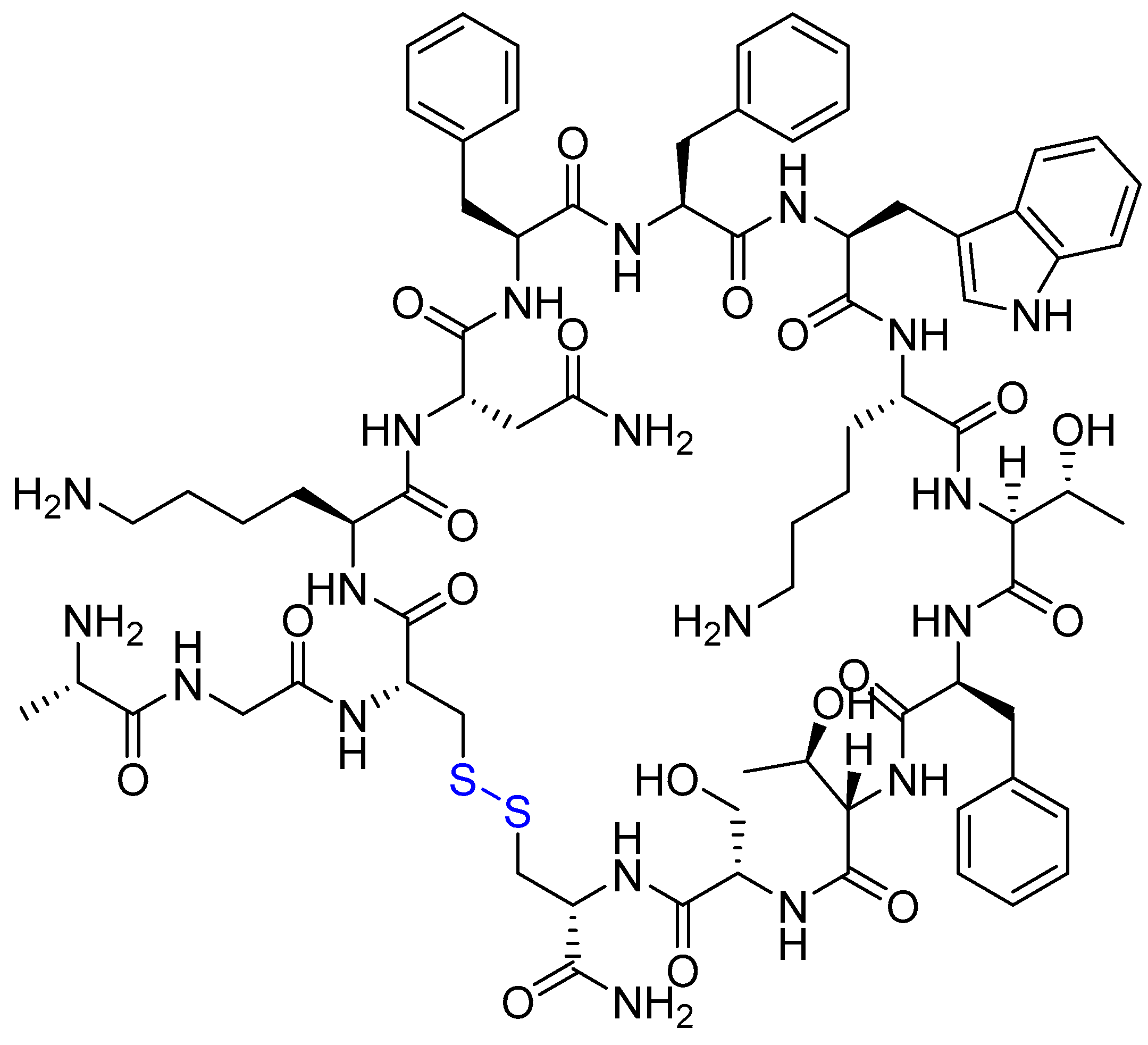
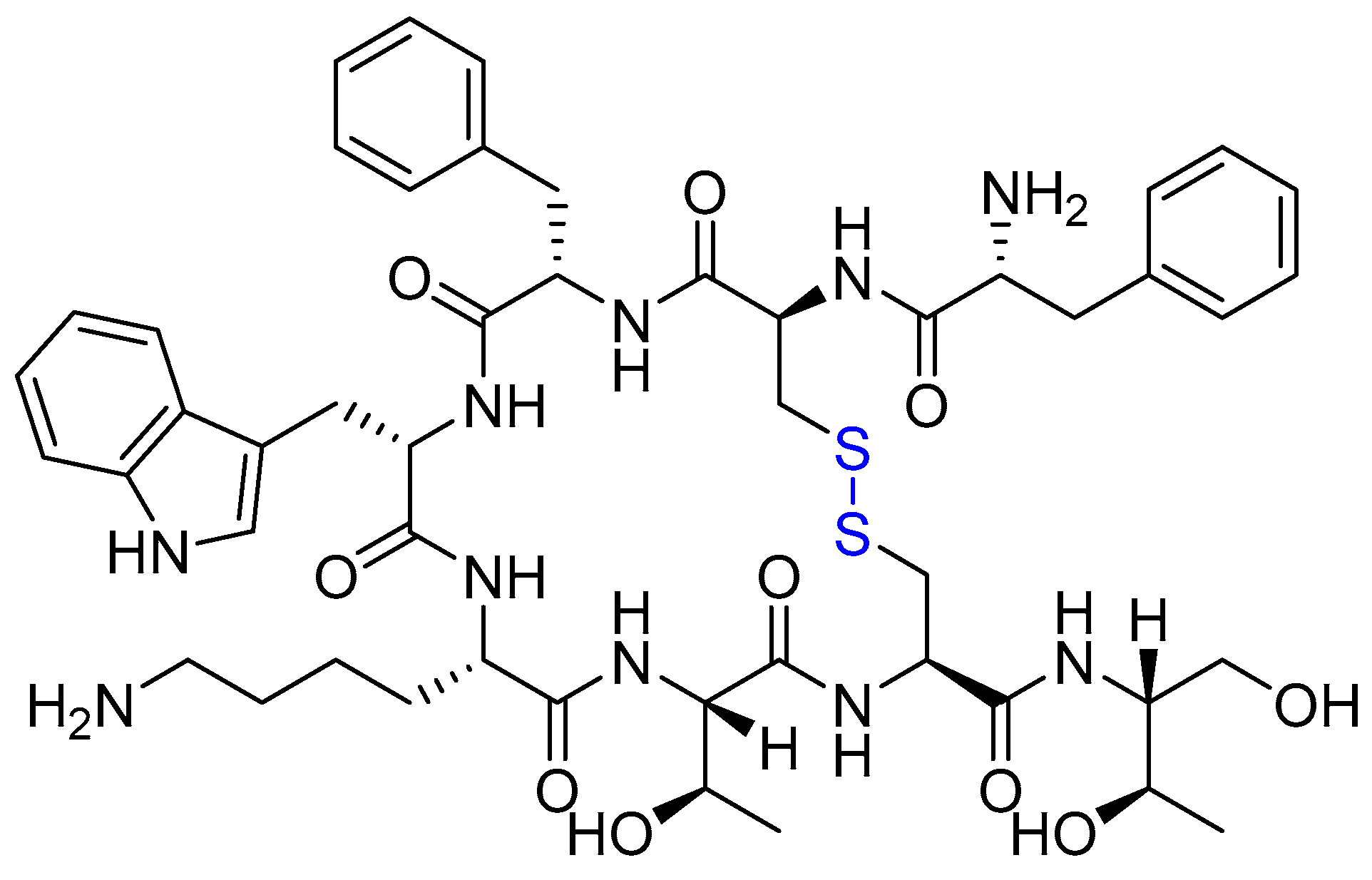
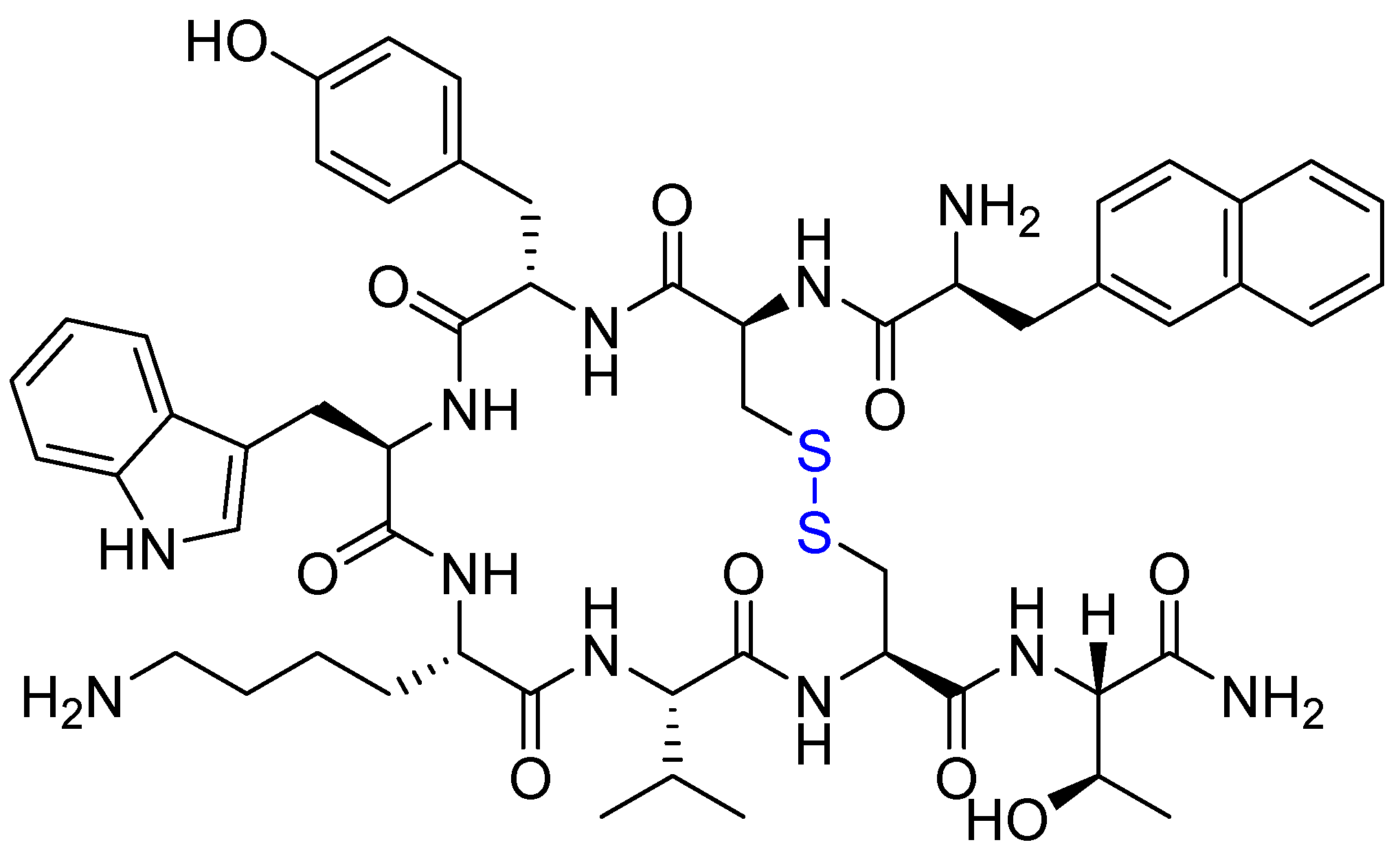


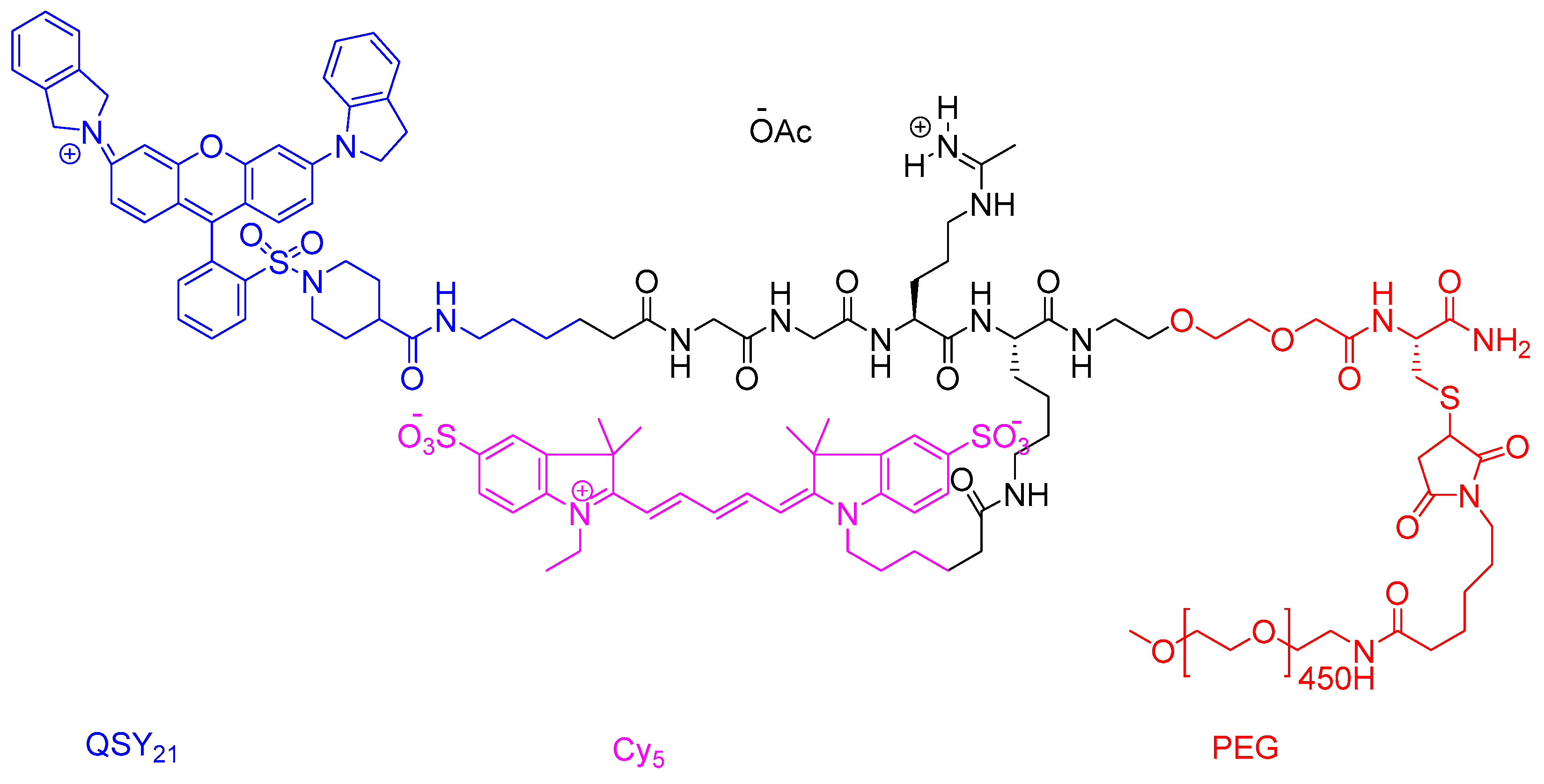
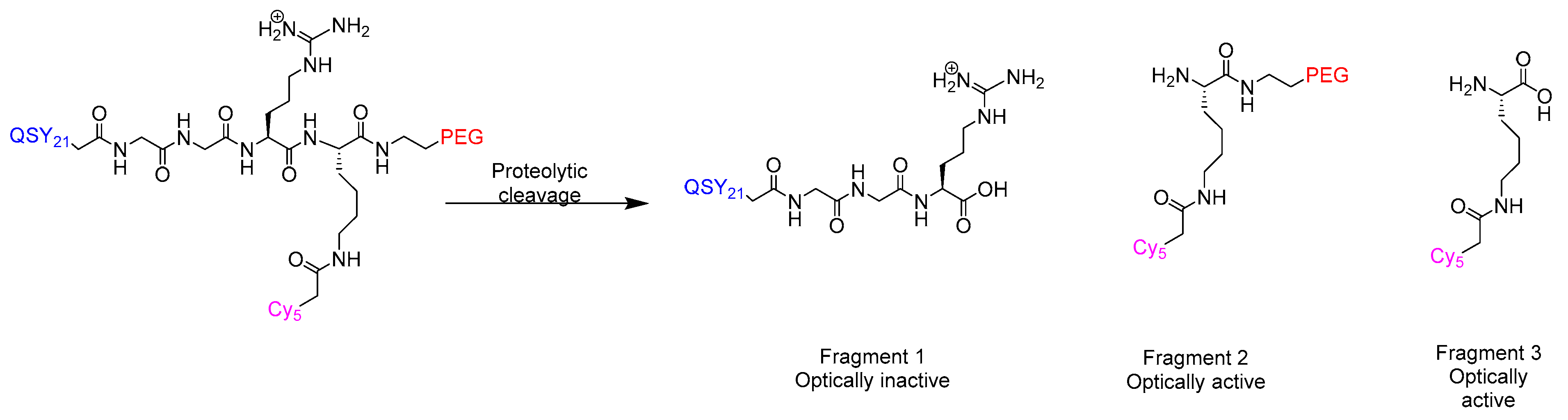
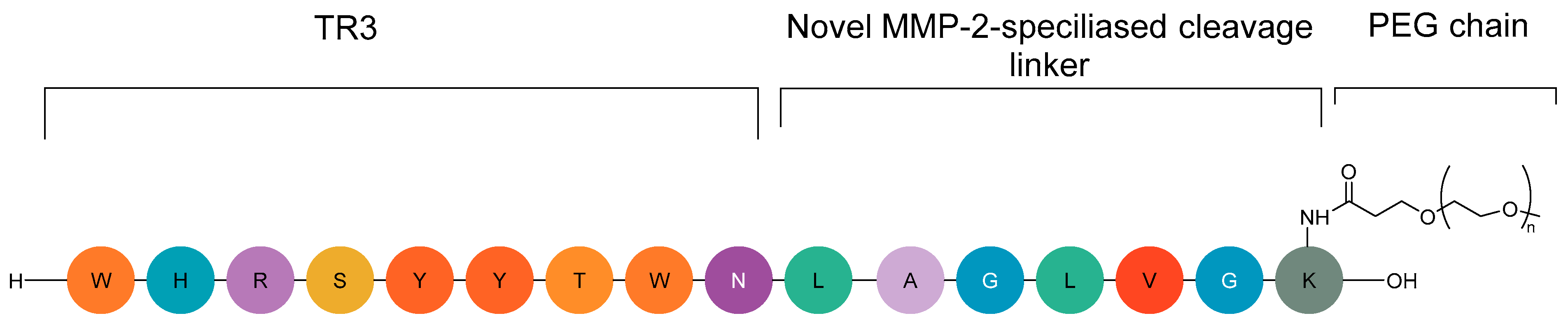

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Clinical Trials ID | Drug | Indications | Therapeutic Target | Phase | Ref |
|---|---|---|---|---|---|---|
| Diagnostic | ||||||
| 1 | NCT06723665 | 18F-PSMA-1007 | Prostate cancer | PSMA | III | [23] |
| 2 | NCT06520449 | 68Ga-NTA-476 | Prostate cancer | PSMA | I | [24] |
| 3 | NCT05125016 | REGN4336 | Metastatic castration-resistant prostate cancer (mCRPC) | PSMA/CD3 | I/II | [25] |
| 4 | NCT06443762 | [68Ga] MDM2/MDMX | Positron emission tomography (PET) imaging | MDM2/MDMX | I | [26] |
| 5 | NCT03827317 | [18F]GE-226 | HER2 in breast cancer | HER2 | N/A | [27] |
| 6 | NCT06719856 | MMP14 (MT1-MMP) Targeting Bicyclic Peptide Probe | PET imaging in solid tumours | MMP14 (MT1-MMP) | N/A | [28] |
| 7 | NCT05518071 | FLUOPANC | Fluorescence-guided surgery of pancreatic and bileduct tumours | Integrin | II | [29] |
| 8 | NCT02742168 | 99mTc-3PRGD2 | PET in breast cancer | Integrin | N/A | [30] |
| Therapeutic | ||||||
| 9 | NCT02187848 | SAR408701 | NSCLC | CEACAM5 | II | [31] |
| 10 | NCT00569257 * | AEZS-108 | Endometrial and ovarian cancer | LHRH receptor | Terminated | [32] |
| 11 | NCT04180371 | BT5528 | Solid tumours | EphA2 receptor | I/II | [33] |
| 12 | NCT02048059 | ANG1005 | Brain metastases from breast cancer | LRP1 | III | [34] |
| 13 | NCT03486730 | BT1718 | Solid tumours | MT1-MMP protein | I/II | [21] |
| 14 | NCT06225596 | BT8009 | Advanced or metastatic urothelial cancer | Nectin-4 | II/III | [35] |
| 15 | NCT06730100 | CBX-12 | Solid tumours, Platinum-resistant and refractory ovarian cancer | Not applicable | II | [36] |
| 16 | NCT04740398 | CBP-1008 | Solid tumour | FRα, TRPV6 | I | [37] |
| 17 | NCT04928612 | CBP-1018 | Lung tumour | FOLR1, PSMA | I | [21] |
| 18 | NCT02082691 | G-202 | Hepatocellular carcinoma | PSMA | II | [38] |
| 19 | NCT06972628 | 177Lu-PSMA-617 | Prostate cancer | PSMA | II | [39] |
| 20 | NCT05579184 | 177Lu-Ludotadipep | Prostate cancer | PSMA | II | [21] |
| 21 | NCT05465590 ** | MB1707 | Solid tumour | CXCR4 | Withdrawn | [40] |
| 22 | NCT01098266 | NGR015 | Malignant pleural mesothelioma | pAPN | III | [41] |
| 23 | NCT02936323 | PEN-221 | Lung cancer | SSTR2 | II | [42] |
| 24 | NCT04706962 | TH1902 | Triple negative breast cancer | SORT1 | I | [43,44] |
| 25 | NCT06400160 | TB511 | Advanced solid tumours | M2 macrophages | I/II | [45] |
| 26 | NCT05597917 | tTF-NGR | Soft tissue sarcoma | CD13, αvβ3 integrin | III | [21] |
| 27 | N/A | CLP002 | Different cancer types | PD-L1 | II | [46] |
| 28 | NCT05012618 | LUNA18 | Solid tumours | GTPases like KRAS | I | [47] |
| 29 | NCT03364400 | VT1021 | Solid tumours | CD36 and CD47 | I/II | [48] |
| 30 | NCT02264613 | Sulanemadlin (ALRN-6924) | Chemoprotection agent | MDM2 and MDM4 | N/A | [49,50] |
| 31 | NCT06949410 | HER2 Vaccine | Locally advanced breast cancer | HER2 | I | [51] |
| 32 | NCT06789198 | Peptide Vaccine | For fibrolamellar hepatocellular carcinoma patients and other tumour entities carrying the driver fusion DNAJB1-PRKACA (FusionVAC22_02) | DNAJB1-PRKACA fusion transcript | I | [52] |
| 33 | NCT06762925 | METTL3 Peptide Inhibitors | Enhancing anti-tumour immune response by reshaping the TME | CXCL5/CCL5 | N/A | [53] |
| 34 | NCT06741072 | BZLF1 Peptide Vaccine | Prevention of Epstein–Barr virus related cancer in patients awaiting solid organ transplants | The promoters of early EBV lytic genes | Ib | [54] |
| 35 | NCT05479045 | New York oesophageal squamous cell carcinoma 1 (NY-ESO-1) peptide vaccine | Anti-PD1 resistance in patients with platinum-refractory stage III/IV ovarian cancer (OC) | NY-ESO-1 | II | [55] |
| 36 | NCT01720836 | MUC1 peptide vaccine | NSCLC | Cell surface mucin | I/II | [56] |
| 37 | N/A | PQ203 proteinqure | Triple negative breast cancer (TNBC) | sortillin receptor | Ia/b | [57] |
| 38 | NCT01967810 | ANG1005 | High-grade glioma (HGG) | Microtubules | II | [58] |
| 39 | NCT03784677 | SOR-C13 | Advanced solid tumours | Microtubules | I | [59] |
| Theranostic | ||||||
| 40 | NCT06726161 | RAYZ-8009/811 | Hepatocellular carcinoma | GPC3 | I/1b | [60] |
| 41 | NCT06991738 | 177Lu-DOTA-EB-TATE | Adult patients with metastatic, radioactive iodine non-responsive oncocytic (Hurthle cell) thyroid cancer | neuroendocrine tumours (NETs) | I/II | [61] |
| 42 | NCT04939610 | 177Lu-FAP-2286 | Advanced metastatic cancer | FAP | I/II | [62] |
| GnRH Analogues | Indication | Approval Year |
|---|---|---|
| Agonists | ||
| Goserelin (Zoladex) | Localised prostate cancer | 1989 |
| Leuprolide (Lupron) | Advanced prostate cancer and central precocious puberty | 1995 |
| Nafarelin (Synarel) | Endometriosis | 1998 |
| Trelstar (triptorelin) | Prostate cancer | 2000 |
| Histrelin (Supprelin LA) | Advanced prostate cancer and central precocious puberty (CPP) in children | 2007 |
| Antagonist | ||
| Ganirelix (Antagon) | Prevent premature LH surges or ovulation in women undergoing fertility treatment of controlled ovarian hyperstimulation | 1999 |
| Cetrorelix (cetrolide) | Prevent premature ovulation as part of controlled ovarian stimulation treatment | 2000 |
| Abarelix (Plenaxis) | Palliative treatment of advanced prostate cancer | 2003 |
| Degarelix (Firmagon) | Advanced prostate cancer | 2008 |
| Somatostatin Analogues | Indication | Approval Year |
|---|---|---|
| Octreotide (Sandostatin) | To treat acromegaly and alleviate symptoms associated with metastatic carcinoid tumours | 1998 |
| Lanreotide (Somatuline) | NETs | 2007 |
| PSMA Antagonist | Indication | Approval Year |
|---|---|---|
| 68Ga-PSMA-11 (68Ga gozetotide) | Diagnosis of prostate cancer | 2020 |
| Piflufolastat F-18 (Pylarify) | To detect and monitor prostate cancer | 2021 |
| Lutetium 177Lu vipivotide tetraxetan (Pluvicto) | To treat adults with a specific type of advanced prostate cancer called PSMA-positive mCRPC | 2022 |
| Flotufolastat F-18 (Posluma) | To reveal a more precise image of prostate cancer | 2023 |
| PRRT | Indication | Approval Year |
|---|---|---|
| Depreotide (Neotect) | To identify SSTR-bearing pulmonary masses in patients presenting with pulmonary lesions on computed tomography and/or chest x-ray who have known malignancy or who are highly suspect for malignancy | 1999 |
| 68Ga-DOTATATE (Netspot) | For diagnosing and staging NETs | 2016 |
| 177Lu-DOTATATE (Lutathera) | To treat NETs | 2018 |
| 68Ga-DOTATOC | Diagnosis and staging of NETs | 2019 |
| 64Cu-DOTATATE (Detectnet) | To aid locating and identifying SSTR-positive NETs in adult patients | 2020 |
| ADCs | Indication | Approval Year |
|---|---|---|
| Enfortumab Vedotin-Ejfv (Padcev) | Treating adults with urothelial cancer | 2019 |
| Polatuzumab vedotin-piiq (Polivy) | To treat adults with diffuse large B-cell lymphoma (DLBCL), a blood cancer affecting white blood cells, in patients whose cancer has not been treated before | 2019 |
| Fam-trastuzumab deruxtecan-nxki (Enhertu) | Treating adults with HER2-positive breast cancer that is metastatic (has spread to other parts of the body) or cannot be removed by surgery | 2019 |
| Belantamab Mafodotin-Blmf (Blenrep) | Treatment for multiple myeloma, a cancer of the bone marrow | 2020 |
| Tisotumab Vedotin-Tftv (TIVDAK) | To treat adults with cervical cancer when the disease has worsened during or after previous systemic (whole-body) treatment | 2021 |
| Loncastuximab Tesirine-Lpyl (Zynlonta) | Treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) and high-grade B-cell lymphoma (HGBL), after two or more lines of systemic therapy | 2021 |
| Telisotuzumab vedotin-tllv (Emrelis) | To treat locally advanced or metastatic, non-squamous non-small cell lung cancer (NSCLC) with high c-Met protein overexpression after prior systemic therapy | 2025 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nsereko, Y.; Armstrong, A.; Coburn, F.; Al Musaimi, O. Innovative Peptide Therapeutics in the Pipeline: Transforming Cancer Detection and Treatment. Int. J. Mol. Sci. 2025, 26, 6815. https://doi.org/10.3390/ijms26146815
Nsereko Y, Armstrong A, Coburn F, Al Musaimi O. Innovative Peptide Therapeutics in the Pipeline: Transforming Cancer Detection and Treatment. International Journal of Molecular Sciences. 2025; 26(14):6815. https://doi.org/10.3390/ijms26146815
Chicago/Turabian StyleNsereko, Yanyamba, Amy Armstrong, Fleur Coburn, and Othman Al Musaimi. 2025. "Innovative Peptide Therapeutics in the Pipeline: Transforming Cancer Detection and Treatment" International Journal of Molecular Sciences 26, no. 14: 6815. https://doi.org/10.3390/ijms26146815
APA StyleNsereko, Y., Armstrong, A., Coburn, F., & Al Musaimi, O. (2025). Innovative Peptide Therapeutics in the Pipeline: Transforming Cancer Detection and Treatment. International Journal of Molecular Sciences, 26(14), 6815. https://doi.org/10.3390/ijms26146815