
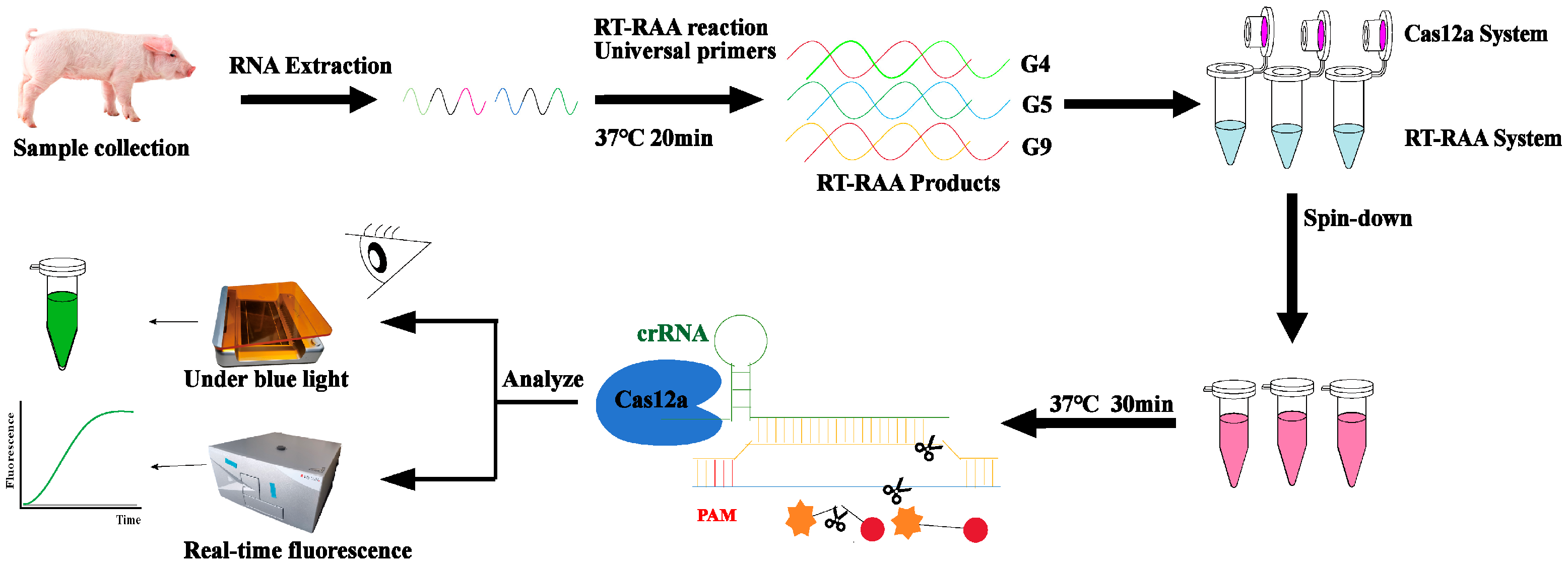
CRISPR/Cas12a-Based One-Tube RT-RAA Assay for PoRV Genotyping


Abstract
1. Introduction
2. Results
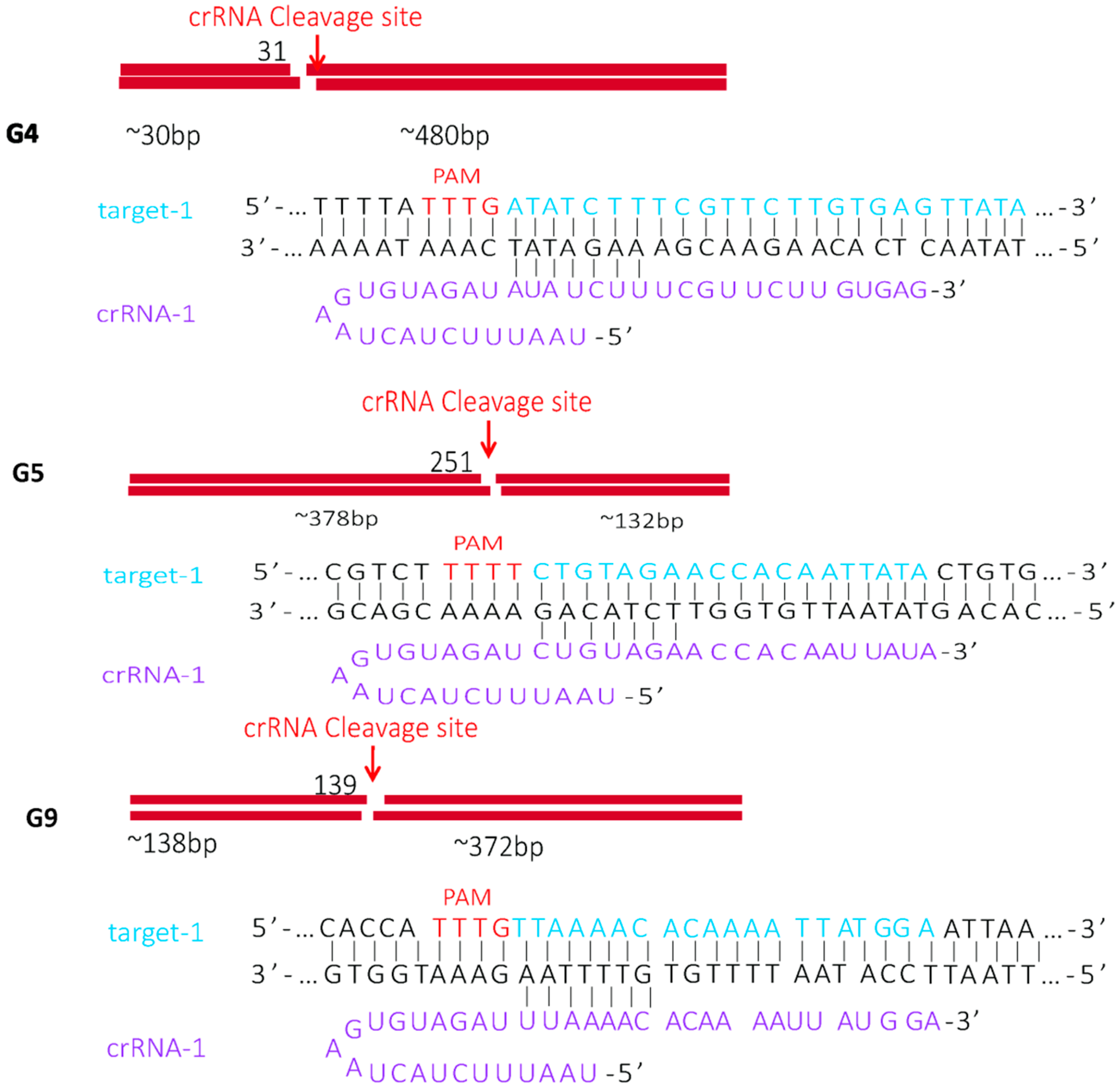
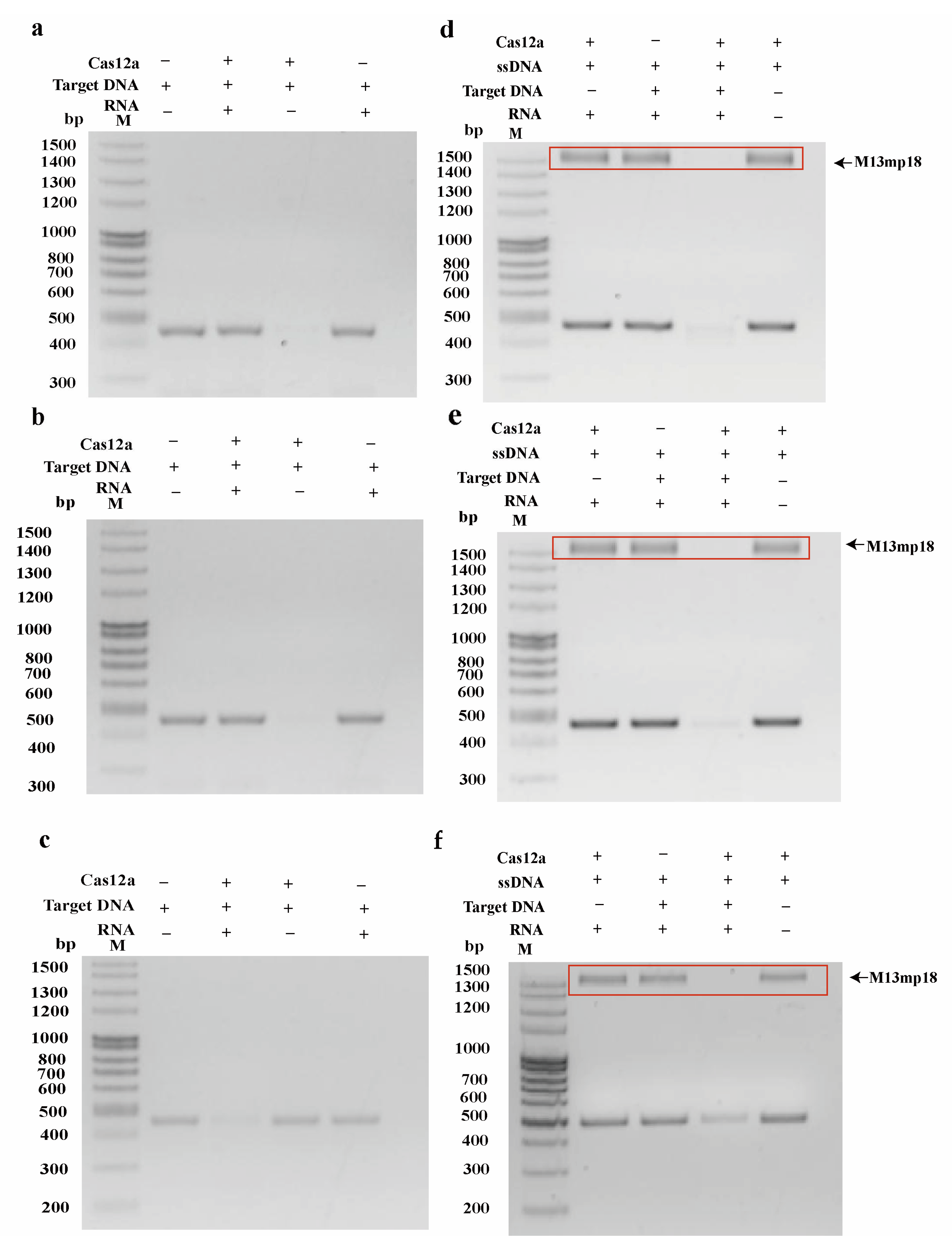
2.1. Identification of Cas12a Protein Cleavage Activity
2.2. Establishment and Optimization of RT-RAA System
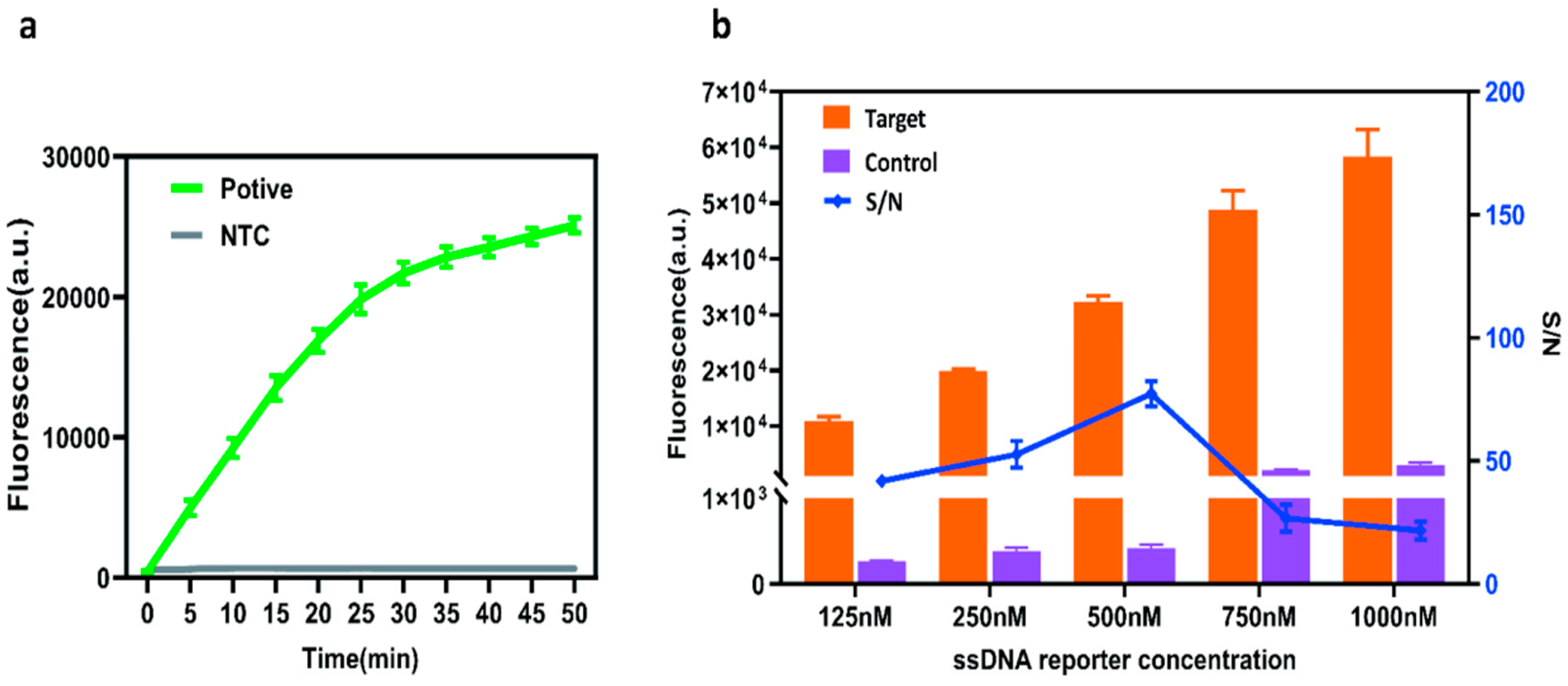
2.3. Optimization of a Single-Tube RT-RAA–CRISPR/Cas12a Assay System
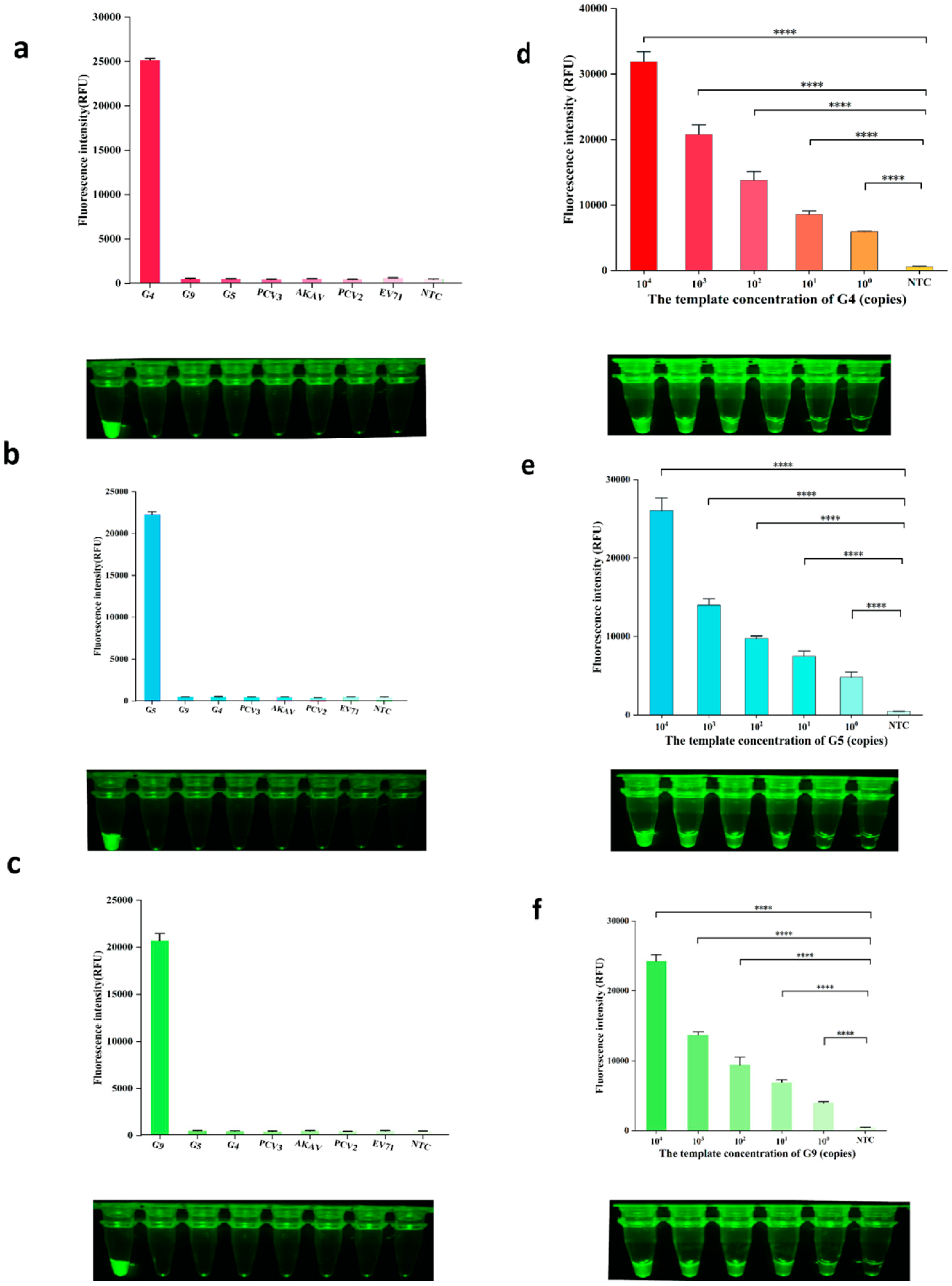
2.4. Sensitivity and Specificity of the Assay
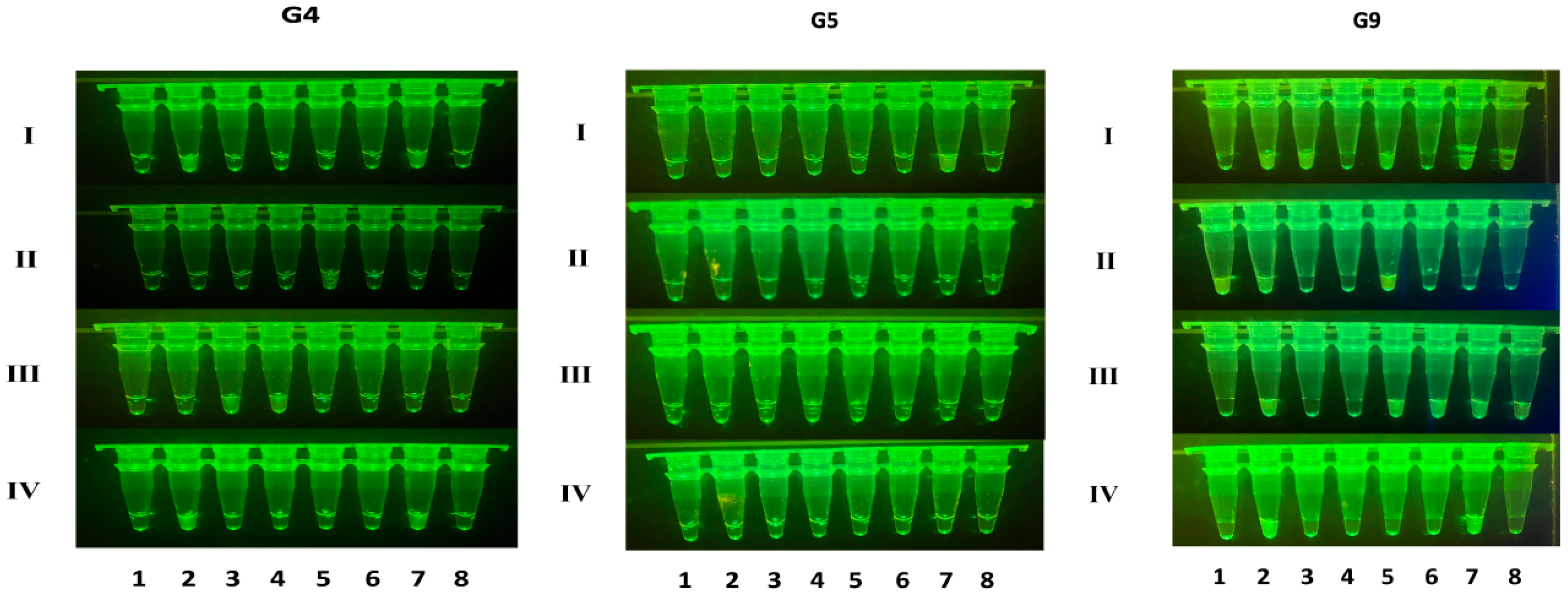
2.5. Clinical Sample Testing
3. Discussion
4. Materials and Methods
4.1. Design of Target DNA, crRNA, Primers, and PUC57 Plasmid
4.2. Expression and Purification of pET-28b-LbCas12a Protein
4.3. Sample Collection and Viral RNA Extraction
4.4. Optimization of RT-RAA Conditions
4.5. Sensitivity and Specificity Verification of the RT-RAA–CRISPR/Cas12a Genotyping Detection Method
4.6. Clinical Sample Validation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ren, J.; Zu, C.; Li, Y.; Li, M.; Gu, J.; Chen, F.; Li, X. Establishment and application of a TaqMan-based multiplex real-time PCR for simultaneous detection of three porcine diarrhea viruses. Front. Microbiol. 2024, 15, 1380849. [Google Scholar] [CrossRef]
- Hou, W.; Fan, M.; Zhu, Z.; Li, X. Establishment and Application of a Triplex Real-Time RT-PCR Assay for Differentiation of PEDV, PoRV, and PDCoV. Viruses 2023, 15, 1238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wu, C.; Chen, Y.; Li, Y.; Li, D.; Wang, W.; Wen, W.; Zhu, Z.; Li, X. Isolation, Genomic Characterization and Evolution of Six Porcine Rotavirus A Strains in a Pig Farming Group. Vet. Sci. 2024, 11, 436. [Google Scholar] [CrossRef] [PubMed]
- Qiao, M.; Li, M.; Li, Y.; Wang, Z.; Hu, Z.; Qing, J.; Huang, J.; Jiang, J.; Jiang, Y.; Zhang, J.; et al. Recent Molecular Characterization of Porcine Rotaviruses Detected in China and Their Phylogenetic Relationships with Human Rotaviruses. Viruses 2024, 16, 453. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Huang, S.; Zhang, L.; Yang, Q.; Liu, S.; Wang, Z.; Chu, Q.; Tian, M.; Zhao, L.; Sun, Y.; et al. Virus-like Particles vaccine based on co-expression of G5 Porcine rotavirus VP2-VP6-VP7 induces a powerful immune protective response in mice. Vet. Microbiol. 2024, 298, 110241. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Zhang, D.; Liu, F.; Li, X.; Gao, M.; Cheng, M.; Bao, H.; Zhan, J.; Zeng, Y.; et al. Lactiplantibacillus plantarum surface-displayed VP6 (PoRV) protein can prevent PoRV infection in piglets. Int. Immunopharmacol. 2024, 133, 112079. [Google Scholar] [CrossRef]
- Das, S.; Das, P.J.; Handique, P.J. Molecular characterization of porcine group A rotavirus to contain piglet diarrhea for productivity enhancement in North East India. Virusdisease 2021, 32, 314–319. [Google Scholar] [CrossRef]
- Zhang, F.; Luo, Y.; Lin, C.; Tan, M.; Wan, P.; Xie, B.; Xiong, L.; Ji, H. Epidemiological monitoring and genetic variation analysis of pathogens associated with porcine viral diarrhea in southern China from 2021 to 2023. Front. Microbiol. 2024, 15, 1303915. [Google Scholar] [CrossRef]
- Park, J.G.; Alfajaro, M.M.; Cho, E.H.; Kim, J.Y.; Soliman, M.; Baek, Y.B.; Park, C.H.; Lee, J.H.; Son, K.Y.; Cho, K.O.; et al. Development of a live attenuated trivalent porcine rotavirus A vaccine against disease caused by recent strains most prevalent in South Korea. Vet. Res. 2019, 50, 2. [Google Scholar] [CrossRef]
- Chiba, S.; Yokoyama, T.; Nakata, S.; Morita, Y.; Urasawa, T.; Taniguchi, K.; Urasawa, S.; Nakao, T. Protective effect of naturally acquired homotypic and heterotypic rotavirus antibodies. Lancet 1986, 328, 417–421. [Google Scholar] [CrossRef]
- Park, G.N.; Kim, D.I.; Choe, S.; Shin, J.; An, B.H.; Kim, K.S.; Hyun, B.H.; Lee, J.S.; An, D.J. Genetic Diversity of Porcine Group A Rotavirus Strains from Pigs in South Korea. Viruses 2022, 14, 2522. [Google Scholar] [CrossRef] [PubMed]
- Julius, L.A.; Saeed, M.M.; Kuijpers, T.; Sandu, S.; Henihan, G.; Dreo, T.; Schoen, C.D.; Mishra, R.; Dunne, N.J.; Carthy, E.; et al. Low-High-Low Rotationally Pulse-Actuated Serial Dissolvable Film Valves Applied to Solid Phase Extraction and LAMP Isothermal Amplification for Plant Pathogen Detection on a Lab-on-a-Disc. ACS Omega 2024, 9, 3262–3275. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, Y.; Luo, S.; Yang, X.; Liu, C.; Zhang, Q.; Liu, Y.; Zhang, X. Foldback-crRNA-Enhanced CRISPR/Cas13a System (FCECas13a) Enables Direct Detection of Ultrashort sncRNA. Anal. Chem. 2023, 95, 15606–15613. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef]
- He, Z.; Duan, X.; Zhao, Z.; Chen, Y.; Fu, C.; Zhang, F.; Wang, J.; Feng, J.; Lin, N.; Chen, H. Rapid on-site diagnosis of PEDV and PoRV co-infection by gold magnetic nanoparticles-based SERS immunochromatography. Talanta 2025, 285, 127428. [Google Scholar] [CrossRef]
- Arias, C.F.; López, S. Rotavirus cell entry: Not so simple after all. Curr. Opin. Virol. 2021, 48, 42–48. [Google Scholar] [CrossRef]
- Wang, Z.; Lv, C.; Xu, X.; Li, X.; Yao, Y.; Gao, X.; Sun, Z.; Wang, Y.; Sun, Y.; Xiao, Y.; et al. The dynamics of a Chinese porcine G9P [23] rotavirus production in MA-104 cells and intestines of 3-day-old piglets. J. Vet. Med. Sci. 2018, 80, 790–797. [Google Scholar] [CrossRef]
- Zhu, J.; Yang, Q.; Cao, L.; Dou, X.; Zhao, J.; Zhu, W.; Ding, F.; Bu, R.E.; Suo, S.; Ren, Y.; et al. Development of porcine rotavirus vp6 protein based ELISA for differentiation of this virus and other viruses. Virol. J. 2013, 10, 91. [Google Scholar] [CrossRef]
- Sanekata, T.; Kishimoto, E.; Sato, K.; Honma, H.; Otsuki, K.; Tsubokura, M. Detection of porcine rotavirus in stools by a latex agglutination test. Vet. Microbiol. 1991, 27, 245–251. [Google Scholar] [CrossRef]
- Memon, A.M.; Chen, F.; Khan, S.B.; Guo, X.; Khan, R.; Khan, F.A.; Zhu, Y.; He, Q. Development and evaluation of polyclonal antibodies based antigen capture ELISA for detection of porcine rotavirus. Anim. Biotechnol. 2022, 34, 1807–1814. [Google Scholar] [CrossRef]
- Ding, G.; Fu, Y.; Li, B.; Chen, J.; Wang, J.; Yin, B.; Sha, W.; Liu, G. Development of a multiplex RT-PCR for the detection of major diarrhoeal viruses in pig herds in China. Transbound. Emerg. Dis. 2020, 67, 678–685. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, Z.; Zhou, Z.; Sun, J.; Yan, S.; Gao, W.; Shao, Y.; Bai, Y.; Wu, Y.; Yan, Z.; et al. A TaqMan Probe-Based Multiplex Real-Time PCR for Simultaneous Detection of Porcine Epidemic Diarrhea Virus Subtypes G1 and G2, and Porcine Rotavirus Groups A and C. Viruses 2022, 14, 1819. [Google Scholar] [CrossRef]
- Han, W.; Ma, Z.; Li, Z.; Chang, C.; Yuan, Y.; Li, Y.; Feng, R.; Zheng, C.; Shi, Z.; Tian, H.; et al. A novel double antibody sandwich quantitative ELISA for detecting porcine epidemic diarrhea virus infection. Appl. Microbiol. Biotechnol. 2024, 108, 482. [Google Scholar] [CrossRef]
- Huang, X.; Chen, J.; Yao, G.; Guo, Q.; Wang, J.; Liu, G. A TaqMan-probe-based multiplex real-time RT-qPCR for simultaneous detection of porcine enteric coronaviruses. Appl. Microbiol. Biotechnol. 2019, 103, 4943–4952. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Kellner, M.J.; Joung, J.; Collins, J.J.; Zhang, F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 2018, 360, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Cheng, Q.X.; Wang, J.M.; Li, X.Y.; Zhang, Z.L.; Gao, S.; Cao, R.B.; Zhao, G.P.; Wang, J. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 2018, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Du, L.; Liu, S.; Yang, Q.; Lei, C.; Wang, H.; Yang, L.; Yang, X. Development and Validation of RAA-CRISPR/Cas12a-Based Assay for Detecting Porcine Rotavirus. Animals 2024, 14, 3387. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ye, Q.; Chen, M.; Zhou, B.; Zhang, J.; Pang, R.; Xue, L.; Wang, J.; Zeng, H.; Wu, S.; et al. An ultrasensitive CRISPR/Cas12a based electrochemical biosensor for Listeria monocytogenes detection. Biosens. Bioelectron. 2021, 179, 113073. [Google Scholar] [CrossRef]
- Xu, J.; Jia, L.; Dang, C.; Liu, X.; Ding, Y. Effects of solid-liquid interaction and mixture concentration on wettability of nano-droplets: Molecular dynamics simulations. Aip Adv. 2022, 12, 105313. [Google Scholar] [CrossRef]
- Xiong, Y.; Cao, G.; Chen, X.; Yang, J.; Shi, M.; Wang, Y.; Nie, F.; Huo, D.; Hou, C. One-pot platform for rapid detecting virus utilizing recombinase polymerase amplification and CRISPR/Cas12a. Appl. Microbiol. Biotechnol. 2022, 106, 4607–4616. [Google Scholar] [CrossRef]
- Ma, L.; Zhu, M.; Meng, Q.; Wang, Y.; Wang, X. Real-time detection of Seneca Valley virus by one-tube RPA-CRISPR/Cas12a assay. Front. Cell. Infect. Microbiol. 2024, 13, 1305222. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Jiang, Q.; Yang, F.; Wu, Q.; Lyu, T.; Zhang, Q.; Wang, J.; Li, F.; Xu, D. Establishment and optimization of a system for the detection of Candida albicans based on enzymatic recombinase amplification and CRISPR/Cas12a system. Microbiol. Spectr. 2025, 13, e00268-25. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Bi, M.; Mo, X. CRISPR/Cas12a, combined with recombinase polymerase amplification (RPA) reaction for visual detection of Leishmania species. Microchem. J. 2024, 207, 112283. [Google Scholar] [CrossRef]
- Shi, H.; Chen, J.; Li, H.; Sun, D.; Wang, C.; Feng, L. Molecular characterization of a rare G9P[23] porcine rotavirus isolate from China. Arch. Virol. 2012, 157, 1897–1903. [Google Scholar] [CrossRef]
- Genz, B.; Gerszon, J.; Pollock, Y.; Gleeson, B.; Shankar, R.; Sellars, M.J.; Moser, R.J. Detection and genetic diversity of porcine rotavirus A, B and C in eastern Australian piggeries. Aust. Vet. J. 2023, 101, 153–163. [Google Scholar] [CrossRef]
- Eren, E.; Zamuda, K.; Patton, J.T. Modeling of the rotavirus group C capsid predicts a surface topology distinct from other rotavirus species. Virology 2016, 487, 150–162. [Google Scholar] [CrossRef]
- Liu, Y.; Han, X.; Zhang, X.; Liu, J.; Yao, L. Development of a droplet digital PCR assay for detection of group A porcine rotavirus. Front. Vet. Sci. 2023, 10, 1113537. [Google Scholar] [CrossRef]
- Aman, R.; Mahas, A.; Mahfouz, M. Nucleic Acid Detection Using CRISPR/Cas Biosensing Technologies. ACS Synth. Biol. 2020, 9, 1226–1233. [Google Scholar] [CrossRef]
- Arroyo-Olarte, R.D.; Bravo Rodríguez, R.; Morales-Ríos, E. Genome Editing in Bacteria: CRISPR-Cas and Beyond. Microorganisms 2021, 9, 844. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR-Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef]
- Ji, S.; Wang, X.; Wang, Y.; Sun, Y.; Su, Y.; Lv, X.; Song, X. Advances in Cas12a-Based Amplification-Free Nucleic Acid Detection. Cris. J. 2023, 6, 405–418. [Google Scholar] [CrossRef]
- Huang, Z.; Liu, S.; Pei, X.; Li, S.; He, Y.; Tong, Y.; Liu, G. Fluorescence Signal-Readout of CRISPR/Cas Biosensors for Nucleic Acid Detection. Biosensors 2022, 12, 779. [Google Scholar] [CrossRef]
- Ahamed, M.A.; Politza, A.J.; Liu, T.; Khalid, M.A.U.; Zhang, H.; Guan, W. CRISPR-based strategies for sample-to-answer monkeypox detection: Current status and emerging opportunities. Nanotechnology 2024, 36, 042001. [Google Scholar] [CrossRef]
- Dong, J.; Feng, W.; Lin, M.; Chen, S.; Liu, X.; Wang, X.; Chen, Q. Comparative Evaluation of PCR-Based, LAMP and RPA-CRISPR/Cas12a Assays for the Rapid Detection of Diaporthe aspalathi. Int. J. Mol. Sci. 2024, 25, 5773. [Google Scholar] [CrossRef]
- Arshad, F.; Abdillah, A.N.; Shivanand, P.; Ahmed, M.U. CeO2 nanozyme mediated RPA/CRISPR-Cas12a dual-mode biosensor for detection of invA gene in Salmonella. Biosens. Bioelectron. 2024, 247, 115940. [Google Scholar] [CrossRef]
- Jiang, G.; Yang, X.; Li, Z.; Mao, J.; Zeng, P.; Wang, D.; Wu, Z.; Liu, C.; Qiu, Y.; Cui, Y.; et al. Recombinant Polymerase Amplification Coupled with CRISPR/Cas12a Detection System for Rapid Visual Detection of Porcine Circovirus 3. Animals 2024, 14, 2527. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequences (5′-3′) | Product Length |
|---|---|---|
| RT-RAA-1F | ATGTATGGTATTGAATATACCACAGTTCT | 510 bp |
| RT-RAA-1R | TATATCCATTGGATTACATAACCATTCATT | |
| RT-RAA-2F | ATGTATGGTATTGAATATACCACAGTTCT | 545 bp |
| RT-RAA-2R | TTCGCTTCATCTGTTTGCTGATAATAATA | |
| RT-RAA-3F | ATGTATGGTATTGAATATACCACAGTTCT | 564 bp |
| RT-RAA-3R | TCCCATCGATATCCATTTATTCGCTTC | |
| RT-RAA-4F | ATGTATGGTATTGAATATACCACAGTTCT | 476 bp |
| RT-RAA-4R | ATCAAATCAGCCAATTCAGACATATCTAGCT | |
| crRNA-G4 | UAAUUUCUACUCUUGUAGAUGAUAUCUUUCGUUCUUGUGAG | |
| crRNA-G5 | UAAUUUCUACUCUUGUAGAUCUGUAGAACCACAAUUAUA | |
| crRNA-G9 | UAAUUUCUACUCUUGUAGAUUUAAAACACAAAAUUAUGGA | |
| FQ-ssDNA reporter | 5′-FAM-TTTTTTTTTTT-BHQ-1′ | |
| RT-qPCR-F-G4 | CCTATGCTAATTCAACGCAAAATG | 102 bp |
| RT-qPCR-R-G4 | GGCCATCCTTTAGTTAGAAACAGTT | |
| RT-qPCR-F-G5 | AGTTACTAGAACAATGGACTTTATC | 157 bp |
| RT-qPCR-R-G5 | CAAAAATGTTTCACTTGTAGTTGA | |
| RT-qPCR-F-G9 | TTGATGTAACTACAAGTACCTGTACAA | 139 bp |
| RT-qPCR-R-G9 | TATCTAACACTTCTGAGCCACC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bi, M.; Wang, Z.; Li, K.; Ren, Y.; Ma, D.; Mo, X. CRISPR/Cas12a-Based One-Tube RT-RAA Assay for PoRV Genotyping. Int. J. Mol. Sci. 2025, 26, 6846. https://doi.org/10.3390/ijms26146846
Bi M, Wang Z, Li K, Ren Y, Ma D, Mo X. CRISPR/Cas12a-Based One-Tube RT-RAA Assay for PoRV Genotyping. International Journal of Molecular Sciences. 2025; 26(14):6846. https://doi.org/10.3390/ijms26146846
Chicago/Turabian StyleBi, Mingfang, Zunbao Wang, Kaijie Li, Yuhe Ren, Dan Ma, and Xiaobing Mo. 2025. "CRISPR/Cas12a-Based One-Tube RT-RAA Assay for PoRV Genotyping" International Journal of Molecular Sciences 26, no. 14: 6846. https://doi.org/10.3390/ijms26146846
APA StyleBi, M., Wang, Z., Li, K., Ren, Y., Ma, D., & Mo, X. (2025). CRISPR/Cas12a-Based One-Tube RT-RAA Assay for PoRV Genotyping. International Journal of Molecular Sciences, 26(14), 6846. https://doi.org/10.3390/ijms26146846