


TIM-3/Galectin-9 Immune Axis in Colorectal Cancer in Relation to KRAS, NRAS, BRAF, PIK3CA, AKT1 Mutations, MSI Status, and the Cytokine Milieu

 , , , and
, , , and
Abstract
1. Introduction
2. Results
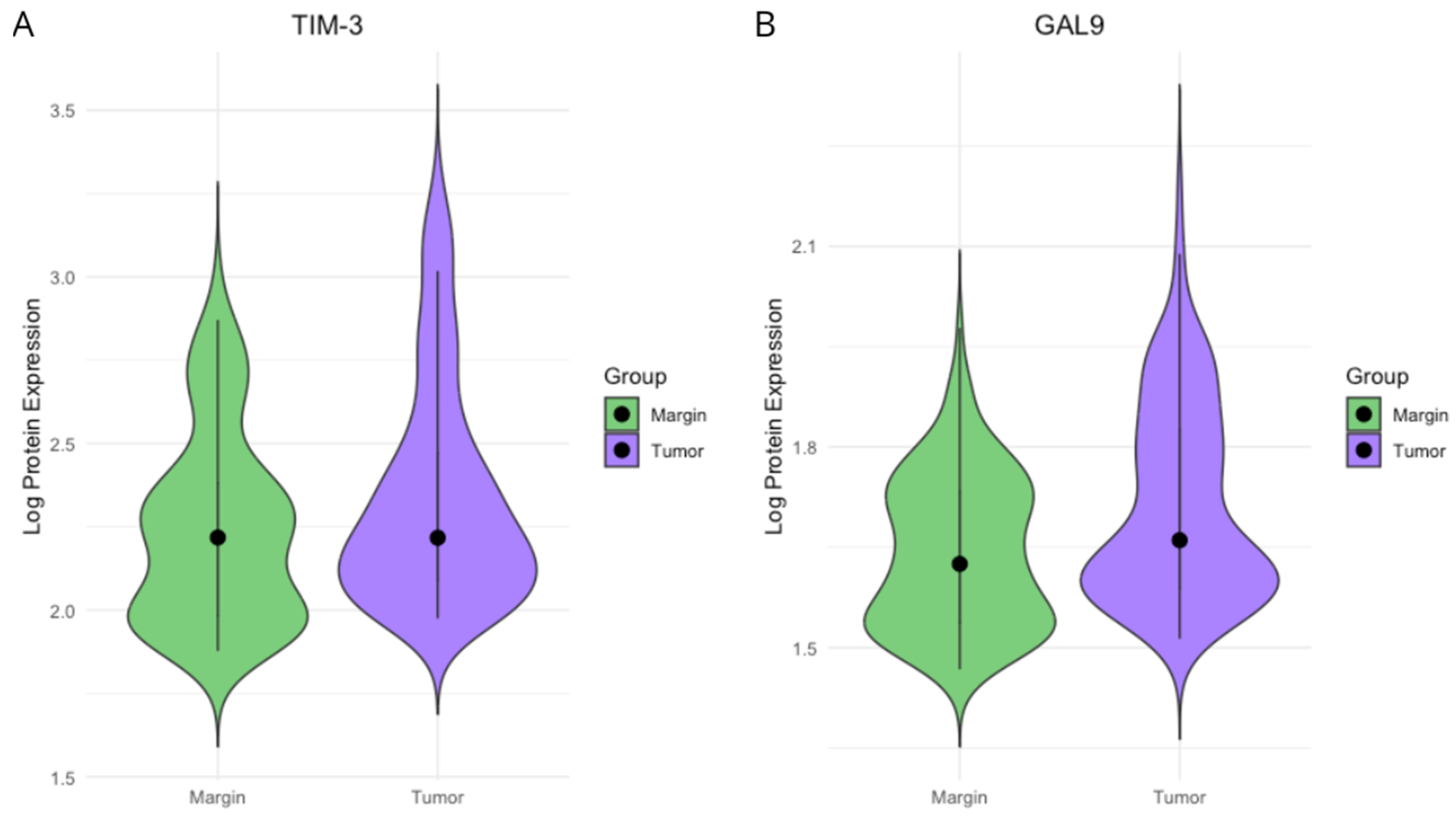
2.1. TIM3 and Gal-9 Protein Concentrations in CRC Tissue and Surgical Margin Tissue
2.2. Associations Between TIM-3 and Gal-9 Expression and KRAS, NRAS, BRAF, PIK3CA, and AKT1 Mutations
2.3. TIM-3 and Gal-9 Proteins Concentration and Selected Cytokine, Chemokine, and Growth Factor Profiles
2.4. TIM-3 and Gal-9 Protein Concentration on TNM Staging, Tumor Grade, Primary Tumor Localization, and MSI Status
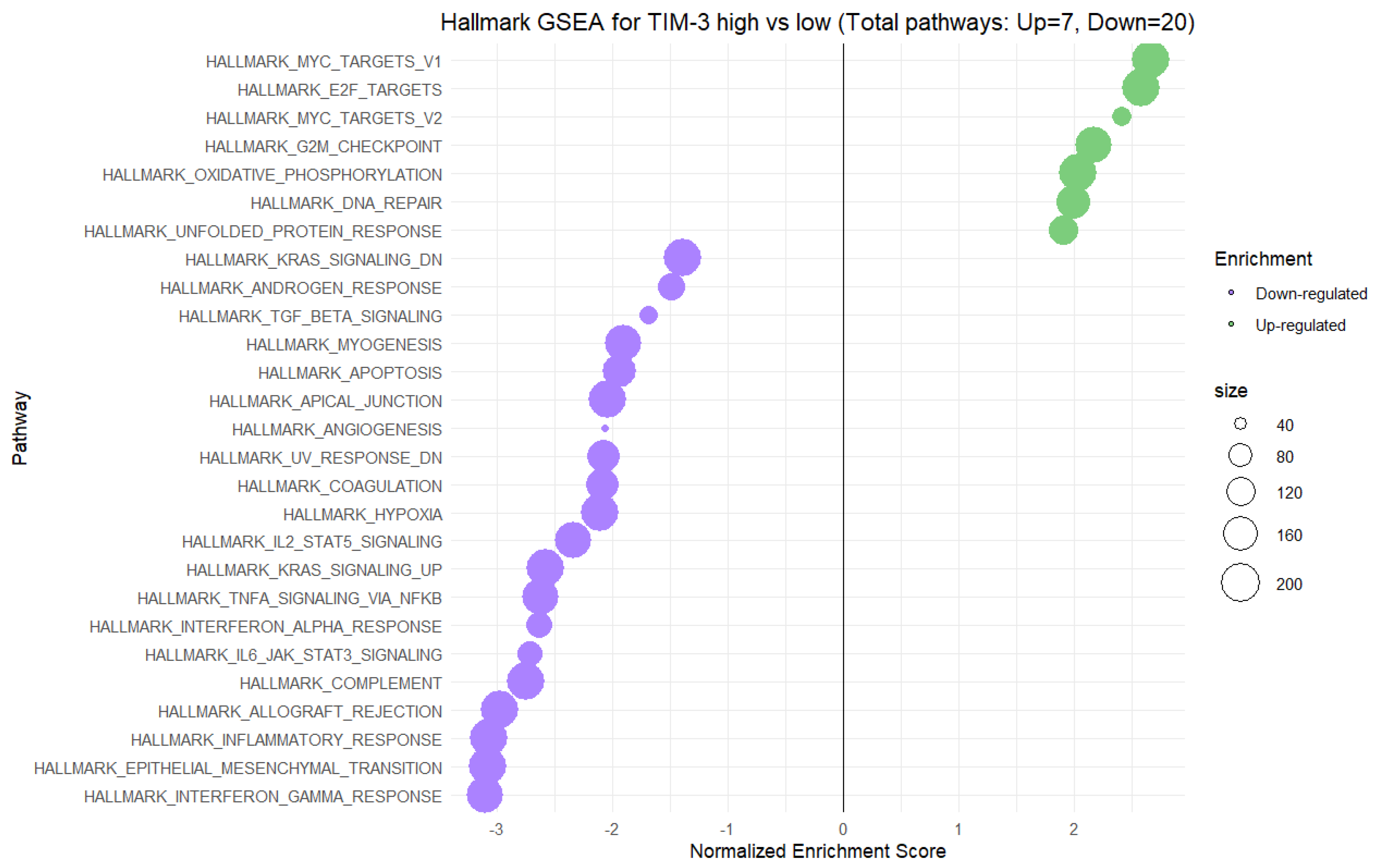
2.5. Gene Set Enrichment Analysis (GSEA) for High vs. Low Gene Expression of TIM-3 and Gal-9 in CRC Tumors
3. Discussion
4. Materials and Methods
4.1. Study Group Characteristics
4.2. Protein Quantification Protocol for TIM-3 and Gal-9
4.3. Assessment of KRAS, NRAS, BRAF, PIK3CA, and AKT Mutation Status
4.4. Microsatellite Instability (MSI) Evaluation
4.5. Principal Component Analysis (PCA) on Cytokine, Chemokine, and Growth Factor Concentration in CRC Homogenates
4.6. Gene Set Enrichment Analysis (GSEA) for TIM-3 and Gal-9 Gene Expression on CRC Data
4.7. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BasicFGF | Basic Fibroblast Growth Factor |
| CCL | Chemokine (C-C motif) ligand |
| CRC | Colorectal cancer |
| CSF | Colony-stimulating factor |
| CTACK | Cutaneous T Cell-Attracting Chemokine |
| CTLA-4 | Cytotoxic T lymphocyte antigen 4 |
| CXCL | Chemokine (C-X-C motif) ligand |
| EGFR | Epidermal growth factor receptor |
| Eotaxin | Eotaxin (CCL11) |
| FDR | False discovery rate |
| G-CSF | Granulocyte Colony-Stimulating Factor |
| Gal-9 | Galectin-9 |
| GM-CSF | Granulocyte-Macrophage Colony-Stimulating Factor |
| GO | Gene Ontology |
| GRO-α | Growth-Regulated Oncogene Alpha |
| GSEA | Gene Set Enrichment Analysis |
| HGF | Hepatocyte growth factor |
| ICB | Immune checkpoint blockade |
| IFN-γ | Interferon gamma |
| IL | Interleukin |
| IP-10 | Interferon Gamma-Induced Protein 10 |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KRAS | Kirsten rat sarcoma viral oncogene homolog |
| LAG-3 | Lymphocyte activation gene-3 |
| MAPK | Mitogen-activated protein kinase |
| MCP-1/MCP-3 | Monocyte Chemoattractant Protein 1/3 |
| MCSF | Macrophage Colony-Stimulating Factor |
| M-CSF | Macrophage colony-stimulating factor |
| MIF | Macrophage Migration Inhibitory Factor |
| MIG | Monokine Induced by Gamma Interferon |
| MIP-1α/MIP-1β | Macrophage Inflammatory Protein 1 Alpha/Beta |
| MMR | Mismatch repair |
| MSI | Microsatellite instability |
| MSS | Microsatellite stable |
| NRAS | Neuroblastoma RAS viral oncogene homolog |
| PCA | Principal Component Analysis |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed death-ligand 1 |
| PDGF-BB | Platelet-Derived Growth Factor BB |
| PI3K | Phosphoinositide 3-kinase |
| RANTES | Regulated on Activation, Normal T Cell Expressed and Secreted (CCL5) |
| SCF | Stem Cell Factor |
| SDF-1α | Stromal Cell-Derived Factor 1 Alpha |
| TAMs | Tumor-associated macrophages |
| TCGA | The Cancer Genome Atlas |
| TGF-β | Transforming growth factor beta |
| TILs | Tumor-infiltrating lymphocytes |
| TIM-3 | T cell immunoglobulin and mucin-domain containing-3 |
| TME | Tumor microenvironment |
| TNF-α | Tumor necrosis factor alpha |
| TNF-α/TNF-β | Tumor Necrosis Factor Alpha/Beta |
| TRAIL | TNF-related apoptosis-inducing ligand |
| VCAM-1 | Vascular cell adhesion protein 1 |
| VEGFA | Vascular Endothelial Growth Factor A |
References
- Fan, A.; Wang, B.; Wang, X.; Nie, Y.; Fan, D.; Zhao, X.; Lu, Y. Immunotherapy in Colorectal Cancer: Current Achievements and Future Perspective. Int. J. Biol. Sci. 2021, 17, 3837. [Google Scholar] [CrossRef] [PubMed]
- Luis, A.; Diaz, J.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; et al. Pembrolizumab versus Chemotherapy for Microsatellite Instability-High or Mismatch Repair-Deficient Metastatic Colorectal Cancer (KEYNOTE-177): Final Analysis of a Randomised, Open-Label, Phase 3 Study. Lancet Oncol. 2022, 23, 659. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit with Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Singh, M.; Morris, V.K.; Bandey, I.N.; Hong, D.S.; Kopetz, S. Advancements in Combining Targeted Therapy and Immunotherapy for Colorectal Cancer. Trends Cancer 2024, 10, 598–609. [Google Scholar] [CrossRef]
- Joller, N.; Anderson, A.C.; Kuchroo, V.K. LAG-3, TIM-3, and TIGIT: Distinct Functions in Immune Regulation. Immunity 2024, 57, 206–222. [Google Scholar] [CrossRef]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-Specific Cell Surface Protein Tim-3 Regulates Macrophage Activation and Severity of an Autoimmune Disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Golden-Mason, L.; Palmer, B.E.; Kassam, N.; Townshend-Bulson, L.; Livingston, S.; McMahon, B.J.; Castelblanco, N.; Kuchroo, V.; Gretch, D.R.; Rosen, H.R. Negative Immune Regulator Tim-3 Is Overexpressed on T Cells in Hepatitis C Virus Infection and Its Blockade Rescues Dysfunctional CD4+ and CD8+ T Cells. J. Virol. 2009, 83, 9122–9130. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 Expression Is Associated with Tumor Antigen-Specific CD8+ T Cell Dysfunction in Melanoma Patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Zhao, Y.; Shao, Q.; Peng, G. Exhaustion and Senescence: Two Crucial Dysfunctional States of T Cells in the Tumor Microenvironment. Cell. Mol. Immunol. 2019, 17, 27. [Google Scholar] [CrossRef]
- Tang, Z.-H.; Liang, S.; Potter, J.; Jiang, X.; Mao, H.-Q.; Li, Z. Tim-3/Galectin-9 Regulate the Homeostasis of Hepatic NKT Cells in a Murine Model of Nonalcoholic Fatty Liver Disease. J. Immunol. 2013, 190, 1788–1796. [Google Scholar] [CrossRef]
- Anderson, A.C.; Anderson, D.E.; Bregoli, L.; Hastings, W.D.; Kassam, N.; Lei, C.; Chandwaskar, R.; Karman, J.; Su, E.W.; Hirashima, M.; et al. Promotion of Tissue Inflammation by the Immune Receptor Tim-3 Expressed on Innate Immune Cells. Science 2007, 318, 1141–1143. [Google Scholar] [CrossRef]
- Chang, T.; Yang, J.; Deng, H.; Chen, D.; Yang, X.; Tang, Z.-H. Depletion and Dysfunction of Dendritic Cells: Understanding SARS-CoV-2 Infection. Front. Immunol. 2022, 13, 843342. [Google Scholar] [CrossRef] [PubMed]
- van de Weyer, P.S.; Muehlfeit, M.; Klose, C.; Bonventre, J.V.; Walz, G.; Kuehn, E.W. A Highly Conserved Tyrosine of Tim-3 Is Phosphorylated upon Stimulation by Its Ligand Galectin-9. Biochem. Biophys. Res. Commun. 2006, 351, 571–576. [Google Scholar] [CrossRef]
- Kandel, S.; Adhikary, P.; Li, G.; Cheng, K. The TIM3/Gal9 Signaling Pathway: An Emerging Target for Cancer Immunotherapy. Cancer Lett. 2021, 510, 67–78. [Google Scholar] [CrossRef]
- Freeman, G.J.; Casasnovas, J.M.; Umetsu, D.T.; DeKruyff, R.H. TIM Genes: A Family of Cell Surface Phosphatidylserine Receptors That Regulate Innate and Adaptive Immunity. Immunol. Rev. 2010, 235, 172–189. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.-S.; Melum, E.; Pertel, T.; et al. CEACAM1 Regulates TIM-3-Mediated Tolerance and Exhaustion. Nature 2015, 517, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-Infiltrating DCs Suppress Nucleic Acid–Mediated Innate Immune Responses through Interactions between the Receptor TIM-3 and the Alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Ma, X.; Ma, Y.; Du, Y.; Feng, J. A New Emerging Target in Cancer Immunotherapy: Galectin-9 (LGALS9). Genes Dis. 2022, 10, 2366. [Google Scholar] [CrossRef]
- Leal-Pinto, E.; Tao, W.; Rappaport, J.; Richardson, M.; Knorr, B.A.; Abramson, R.G. Molecular Cloning and Functional Reconstitution of a Urate Transporter/Channel. J. Biol. Chem. 1997, 272, 617–625. [Google Scholar] [CrossRef]
- Matsumoto, R.; Matsumoto, H.; Seki, M.; Hata, M.; Asano, Y.; Kanegasaki, S.; Stevens, R.L.; Hirashima, M. Human Ecalectin, a Variant of Human Galectin-9, Is a Novel Eosinophil Chemoattractant Produced by T Lymphocytes. J. Biol. Chem. 1998, 273, 16976–16984. [Google Scholar] [CrossRef]
- Kageshita, T.; Kashio, Y.; Yamauchi, A.; Seki, M.; Abedin, M.J.; Nishi, N.; Shoji, H.; Nakamura, T.; Ono, T.; Hirashima, M. Possible Role of Galectin-9 in Cell Aggregation and Apoptosis of Human Melanoma Cell Lines and Its Clinical Significance. Int. J. Cancer 2002, 99, 809–816. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Mishra, R. Galectin-9: From Cell Biology to Complex Disease Dynamics. J. Biosci. 2016, 41, 507–534. [Google Scholar] [CrossRef] [PubMed]
- Kasamatsu, A.; Uzawa, K.; Nakashima, D.; Koike, H.; Shiiba, M.; Bukawa, H.; Yokoe, H.; Tanzawa, H. Galectin-9 as a Regulator of Cellular Adhesion in Human Oral Squamous Cell Carcinoma Cell Lines. Int. J. Mol. Med. 2005, 16, 269–273. [Google Scholar] [CrossRef]
- Irie, A.; Yamauchi, A.; Kontani, K.; Kihara, M.; Liu, D.; Shirato, Y.; Seki, M.; Nishi, N.; Nakamura, T.; Yokomise, H.; et al. Galectin-9 as a Prognostic Factor with Antimetastatic Potential in Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 2962–2968. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 Ligand Galectin-9 Negatively Regulates T Helper Type 1 Immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef]
- Enninga, E.A.L.; Chatzopoulos, K.; Butterfield, J.T.; Sutor, S.L.; Leontovich, A.A.; Nevala, W.K.; Flotte, T.J.; Markovic, S.N. CD206-Positive Myeloid Cells Bind Galectin-9 and Promote a Tumor-Supportive Microenvironment. J. Pathol. 2018, 245, 468–477. [Google Scholar] [CrossRef]
- Madireddi, S.; Eun, S.-Y.; Mehta, A.K.; Birta, A.; Zajonc, D.M.; Niki, T.; Hirashima, M.; Podack, E.R.; Schreiber, T.H.; Croft, M. Regulatory T Cell-Mediated Suppression of Inflammation Induced by DR3 Signaling Is Dependent on Galectin-9. J. Immunol. Baltim. Md 1950 2017, 199, 2721–2728. [Google Scholar] [CrossRef]
- Seki, M.; Oomizu, S.; Sakata, K.; Sakata, A.; Arikawa, T.; Watanabe, K.; Ito, K.; Takeshita, K.; Niki, T.; Saita, N.; et al. Galectin-9 Suppresses the Generation of Th17, Promotes the Induction of Regulatory T Cells, and Regulates Experimental Autoimmune Arthritis. Clin. Immunol. 2008, 127, 78–88. [Google Scholar] [CrossRef]
- Wang, Y.; Song, L.; Sun, J.; Sui, Y.; Li, D.; Li, G.; Liu, J.; Shu, Q. Expression of Galectin-9 and Correlation with Disease Activity and Vascular Endothelial Growth Factor in Rheumatoid Arthritis. Clin. Exp. Rheumatol. 2020, 38, 654–661. [Google Scholar]
- Yang, R.; Sun, L.; Li, C.-F.; Wang, Y.-H.; Yao, J.; Li, H.; Yan, M.; Chang, W.-C.; Hsu, J.-M.; Cha, J.-H.; et al. Galectin-9 Interacts with PD-1 and TIM-3 to Regulate T Cell Death and Is a Target for Cancer Immunotherapy. Nat. Commun. 2021, 12, 832. [Google Scholar] [CrossRef]
- Vaitaitis, G.M.; Wagner, D.H. Galectin-9 Controls CD40 Signaling through a Tim-3 Independent Mechanism and Redirects the Cytokine Profile of Pathogenic T Cells in Autoimmunity. PLoS ONE 2012, 7, e38708. [Google Scholar] [CrossRef]
- Yasinska, I.M.; Meyer, N.H.; Schlichtner, S.; Hussain, R.; Siligardi, G.; Casely-Hayford, M.; Fiedler, W.; Wellbrock, J.; Desmet, C.; Calzolai, L.; et al. Ligand-Receptor Interactions of Galectin-9 and VISTA Suppress Human T Lymphocyte Cytotoxic Activity. Front. Immunol. 2020, 11, 580557. [Google Scholar] [CrossRef]
- Madireddi, S.; Eun, S.-Y.; Lee, S.-W.; Nemčovičová, I.; Mehta, A.K.; Zajonc, D.M.; Nishi, N.; Niki, T.; Hirashima, M.; Croft, M. Galectin-9 Controls the Therapeutic Activity of 4-1BB-Targeting Antibodies. J. Exp. Med. 2014, 211, 1433–1448. [Google Scholar] [CrossRef] [PubMed]
- Daley, D.; Mani, V.R.; Mohan, N.; Akkad, N.; Ochi, A.; Heindel, D.W.; Lee, K.B.; Zambirinis, C.P.; Pandian, G.S.B.; Savadkar, S.; et al. Dectin 1 Activation on Macrophages by Galectin 9 Promotes Pancreatic Carcinoma and Peritumoral Immune Tolerance. Nat. Med. 2017, 23, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Arain, M.A.; Chen, Y.-J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Farkas, L.; et al. Colon Cancer, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 329–359. [Google Scholar] [CrossRef]
- Ochman, B.; Kot, A.; Mielcarska, S.; Kula, A.; Dawidowicz, M.; Koszewska, D.; Hudy, D.; Szrot, M.; Piecuch, J.; Waniczek, D.; et al. Association of SIGLEC9 Expression with Cytokine Expression, Tumor Grading, KRAS, NRAS, BRAF, PIK3CA, AKT Gene Mutations, and MSI Status in Colorectal Cancer. Curr. Issues Mol. Biol. 2024, 46, 13617–13646. [Google Scholar] [CrossRef]
- Zhang, Y.; Deng, D.; Yin, W.; Luo, J.; Liu, J.; Xie, C.; Ji, X.; Ma, L.; Zhang, L.; Xia, X.; et al. Relationship Between Tim-3 and Galectin-9 Expression Levels, Clinical Pathological Characteristics, and Prognosis in Patients After Radical Resection of Colorectal Cancer. Sichuan Da Xue Xue Bao Yi Xue Ban 2024, 55, 375–382. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, J.; Ma, C.; Gao, W.; Song, B.; Xue, H.; Chen, W.; Chen, X.; Zhang, Y.; Shao, Q.; et al. Reduced Expression of Galectin-9 Contributes to a Poor Outcome in Colon Cancer by Inhibiting NK Cell Chemotaxis Partially through the Rho/ROCK1 Signaling Pathway. PLoS ONE 2016, 11, e0152599. [Google Scholar] [CrossRef]
- Sim, J.; Park, J.; Kim, S.; Hwang, S.; Sung, K.; Lee, J.-E.; Yang, S.; Cho, K.; Lee, S.; Moon, J.-S.; et al. Association of Tim-3/Gal-9 Axis with NLRC4 Inflammasome in Glioma Malignancy: Tim-3/Gal-9 Induce the NLRC4 Inflammasome. Int. J. Mol. Sci. 2022, 23, 2028. [Google Scholar] [CrossRef]
- Kim, H.-S.; Chang, C.Y.; Yoon, H.J.; Kim, K.S.; Koh, H.S.; Kim, S.S.; Lee, S.-J.; Kane, L.P.; Park, E.J. Glial TIM-3 Modulates Immune Responses in the Brain Tumor Microenvironment. Cancer Res. 2020, 80, 1833–1845. [Google Scholar] [CrossRef]
- Stempin, C.C.; Geysels, R.C.; Park, S.; Palacios, L.M.; Volpini, X.; Motran, C.C.; Rodríguez, E.V.A.; Nicola, J.P.; Cheng, S.; Pellizas, C.G.; et al. Secreted Factors by Anaplastic Thyroid Cancer Cells Induce Tumor-Promoting M2-like Macrophage Polarization through a TIM3-Dependent Mechanism. Cancers 2021, 13, 4821. [Google Scholar] [CrossRef]
- Su, H.; Xie, H.; Dai, C.; Ren, Y.; She, Y.; Xu, L.; Chen, D.; Xie, D.; Zhang, L.; Jiang, G.; et al. Characterization of TIM-3 Expression and Its Prognostic Value in Patients with Surgically Resected Lung Adenocarcinoma. Lung Cancer Amst. Neth. 2018, 121, 18–24. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, Y.; Li, G.; Huang, H.; Zhang, G.; Wang, F.; Sun, J.; Yang, Q.; Zhang, X.; Lu, B. TIM-3 Expression Characterizes Regulatory T Cells in Tumor Tissues and Is Associated with Lung Cancer Progression. PLoS ONE 2012, 7, e30676. [Google Scholar] [CrossRef]
- Yang, F.; Zeng, Z.; Li, J.; Ren, X.; Wei, F. TIM-3 and CEACAM1 Are Prognostic Factors in Head and Neck Squamous Cell Carcinoma. Front. Mol. Biosci. 2021, 8, 619765. [Google Scholar] [CrossRef]
- Veigas, F.; Mahmoud, Y.D.; Merlo, J.; Rinflerch, A.; Rabinovich, G.A.; Girotti, M.R. Immune Checkpoints Pathways in Head and Neck Squamous Cell Carcinoma. Cancers 2021, 13, 1018. [Google Scholar] [CrossRef]
- Cheng, G.; Li, M.; Wu, J.; Ji, M.; Fang, C.; Shi, H.; Zhu, D.; Chen, L.; Zhao, J.; Shi, L.; et al. Expression of Tim-3 in Gastric Cancer Tissue and Its Relationship with Prognosis. Int. J. Clin. Exp. Pathol. 2015, 8, 9452–9457. [Google Scholar]
- Burugu, S.; Gao, D.; Leung, S.; Chia, S.K.; Nielsen, T.O. TIM-3 Expression in Breast Cancer. Oncoimmunology 2018, 7, e1502128. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.; Li, Q.; Liang, D.; Wang, Q.; Liu, Q. Biomarkers of Response to Camrelizumab Combined with Apatinib: An Analysis from a Phase II Trial in Advanced Triple-Negative Breast Cancer Patients. Breast Cancer Res. Treat. 2021, 186, 687–697. [Google Scholar] [CrossRef]
- Yasinska, I.M.; Sakhnevych, S.S.; Pavlova, L.; Teo Hansen Selnø, A.; Teuscher Abeleira, A.M.; Benlaouer, O.; Gonçalves Silva, I.; Mosimann, M.; Varani, L.; Bardelli, M.; et al. The Tim-3-Galectin-9 Pathway and Its Regulatory Mechanisms in Human Breast Cancer. Front. Immunol. 2019, 10, 1594. [Google Scholar] [CrossRef]
- Chen, Z.; Dong, D.; Zhu, Y.; Pang, N.; Ding, J. The Role of Tim-3/Galectin-9 Pathway in T-Cell Function and Prognosis of Patients with Human Papilloma Virus-Associated Cervical Carcinoma. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21401. [Google Scholar] [CrossRef]
- Seifert, A.M.; Reiche, C.; Heiduk, M.; Tannert, A.; Meinecke, A.-C.; Baier, S.; von Renesse, J.; Kahlert, C.; Distler, M.; Welsch, T.; et al. Detection of Pancreatic Ductal Adenocarcinoma with Galectin-9 Serum Levels. Oncogene 2020, 39, 3102–3113. [Google Scholar] [CrossRef]
- Yang, J.; Zhu, L.; Cai, Y.; Suo, J.; Jin, J. Role of Downregulation of Galectin-9 in the Tumorigenesis of Gastric Cancer. Int. J. Oncol. 2014, 45, 1313–1320. [Google Scholar] [CrossRef]
- Lahm, H.; André, S.; Hoeflich, A.; Fischer, J.R.; Sordat, B.; Kaltner, H.; Wolf, E.; Gabius, H.J. Comprehensive Galectin Fingerprinting in a Panel of 61 Human Tumor Cell Lines by RT-PCR and Its Implications for Diagnostic and Therapeutic Procedures. J. Cancer Res. Clin. Oncol. 2001, 127, 375–386. [Google Scholar] [CrossRef]
- Hou, N.; Ma, J.; Li, W.; Zhao, L.; Gao, Q.; Mai, L. T-Cell Immunoglobulin and Mucin Domain-Containing Protein-3 and Galectin-9 Protein Expression: Potential Prognostic Significance in Esophageal Squamous Cell Carcinoma for Chinese Patients. Oncol. Lett. 2017, 14, 8007–8013. [Google Scholar] [CrossRef]
- Boughdady, I.S.; Kinsella, A.R.; Haboubi, N.Y.; Schofield, P.F. K-Ras Gene Mutations in Adenomas and Carcinomas of the Colon. Surg. Oncol. 1992, 1, 275–282. [Google Scholar] [CrossRef]
- Bos, J.L.; Fearon, E.R.; Hamilton, S.R.; Verlaan-de Vries, M.; van Boom, J.H.; van der Eb, A.J.; Vogelstein, B. Prevalence of Ras Gene Mutations in Human Colorectal Cancers. Nature 1987, 327, 293–297. [Google Scholar] [CrossRef]
- Xu, M.; Zhao, X.; Wen, T.; Qu, X. Unveiling the Role of KRAS in Tumor Immune Microenvironment. Biomed. Pharmacother. 2024, 171, 116058. [Google Scholar] [CrossRef]
- Scheffler, M.; Ihle, M.A.; Hein, R.; Merkelbach-Bruse, S.; Scheel, A.H.; Siemanowski, J.; Brägelmann, J.; Kron, A.; Abedpour, N.; Ueckeroth, F.; et al. K-Ras Mutation Subtypes in NSCLC and Associated Co-Occuring Mutations in Other Oncogenic Pathways. J. Thorac. Oncol. 2019, 14, 606–616. [Google Scholar] [CrossRef]
- Fu, X.; Wang, X.; Duanmu, J.; Li, T.; Jiang, Q. KRAS Mutations Are Negatively Correlated with Immunity in Colon Cancer. Aging 2020, 13, 750–768. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, H.S.; Kim, B.J. Prognostic Value of KRAS Mutation in Advanced Non-Small-Cell Lung Cancer Treated with Immune Checkpoint Inhibitors: A Meta-Analysis and Review. Oncotarget 2017, 8, 48248–48252. [Google Scholar] [CrossRef]
- Liu, H.; Liang, Z.; Zhou, C.; Zeng, Z.; Wang, F.; Hu, T.; He, X.; Wu, X.; Wu, X.; Lan, P. Mutant KRAS Triggers Functional Reprogramming of Tumor-Associated Macrophages in Colorectal Cancer. Signal Transduct. Target. Ther. 2021, 6, 144. [Google Scholar] [CrossRef]
- Afrăsânie, V.-A.; Marinca, M.V.; Alexa-Stratulat, T.; Gafton, B.; Păduraru, M.; Adavidoaiei, A.M.; Miron, L.; Rusu, C. KRAS, NRAS, BRAF, HER2 and Microsatellite Instability in Metastatic Colorectal Cancer–Practical Implications for the Clinician. Radiol. Oncol. 2019, 53, 265–274. [Google Scholar] [CrossRef]
- Mei, Z.B.; Duan, C.Y.; Li, C.B.; Cui, L.; Ogino, S. Prognostic Role of Tumor PIK3CA Mutation in Colorectal Cancer: A Systematic Review and Meta-Analysis. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 1836–1848. [Google Scholar] [CrossRef]
- Wang, C.; Pan, D. Mutation Patterns and Prognostic Analysis of BRAF/KRAS/PIK3CA in Colorectal Cancer. J. Clin. Lab. Anal. 2022, 36, e24444. [Google Scholar] [CrossRef]
- De Roock, W.; De Vriendt, V.; Normanno, N.; Ciardiello, F.; Tejpar, S. KRAS, BRAF, PIK3CA, and PTEN Mutations: Implications for Targeted Therapies in Metastatic Colorectal Cancer. Lancet Oncol. 2011, 12, 594–603. [Google Scholar] [CrossRef]
- Zeissig, M.N.; Ashwood, L.M.; Kondrashova, O.; Sutherland, K.D. Next Batter up! Targeting Cancers with KRAS-G12D Mutations. Trends Cancer 2023, 9, 955–967. [Google Scholar] [CrossRef]
- Mahdi, Y.; Khmou, M.; Souadka, A.; Agouri, H.E.; Ech-Charif, S.; Mounjid, C.; Khannoussi, B.E. Correlation between KRAS and NRAS Mutational Status and Clinicopathological Features in 414 Cases of Metastatic Colorectal Cancer in Morocco: The Largest North African Case Series. BMC Gastroenterol. 2023, 23, 193. [Google Scholar] [CrossRef]
- Benmokhtar, S.; Laraqui, A.; Hilali, F.; Bajjou, T.; El Zaitouni, S.; Jafari, M.; Baba, W.; Elannaz, H.; Lahlou, I.A.; Hafsa, C.; et al. RAS/RAF/MAPK Pathway Mutations as Predictive Biomarkers in Middle Eastern Colorectal Cancer: A Systematic Review. Clin. Med. Insights Oncol. 2024, 18, 11795549241255651. [Google Scholar] [CrossRef]
- Koulouridi, A.; Karagianni, M.; Messaritakis, I.; Sfakianaki, M.; Voutsina, A.; Trypaki, M.; Bachlitzanaki, M.; Koustas, E.; Karamouzis, M.V.; Ntavatzikos, A.; et al. Prognostic Value of KRAS Mutations in Colorectal Cancer Patients. Cancers 2022, 14, 3320. [Google Scholar] [CrossRef] [PubMed]
- Ucar, G.; Ergun, Y.; Aktürk Esen, S.; Acikgoz, Y.; Dirikoc, M.; Esen, İ.; Bal, Ö.; Uncu, D. Prognostic and Predictive Value of KRAS Mutation Number in Metastatic Colorectal Cancer. Medicine 2020, 99, e22407. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Fan, W.; Li, J.; Wu, G.; Wu, H. KRAS/NRAS Mutations Associated with Distant Metastasis and BRAF/PIK3CA Mutations Associated with Poor Tumor Differentiation in Colorectal Cancer. Int. J. Gen. Med. 2023, 16, 4109–4120. [Google Scholar] [CrossRef]
- Benmokhtar, S.; Laraqui, A.; El Boukhrissi, F.; Hilali, F.; Bajjou, T.; Jafari, M.; Elzaitouni, S.; Baba, W.; El Mchichi, B.; Elannaz, H.; et al. Clinical Significance of Somatic Mutations in RAS/RAF/MAPK Signaling Pathway in Moroccan and North African Colorectal Cancer Patients. Asian Pac. J. Cancer Prev. APJCP 2022, 23, 3725–3733. [Google Scholar] [CrossRef]
- Poulsen, T.S.; de Oliveira, D.V.N.P.; Espersen, M.L.M.; Klarskov, L.L.; Skovrider-Ruminski, W.; Hogdall, E. Frequency and Coexistence of KRAS, NRAS, BRAF and PIK3CA Mutations and Occurrence of MMR Deficiency in Danish Colorectal Cancer Patients. APMIS 2021, 129, 61–69. [Google Scholar] [CrossRef]
- Tol, J.; Nagtegaal, I.D.; Punt, C.J.A. BRAF Mutation in Metastatic Colorectal Cancer. N. Engl. J. Med. 2009, 361, 98–99. [Google Scholar] [CrossRef]
- Roth, A.D.; Tejpar, S.; Delorenzi, M.; Yan, P.; Fiocca, R.; Klingbiel, D.; Dietrich, D.; Biesmans, B.; Bodoky, G.; Barone, C.; et al. Prognostic Role of KRAS and BRAF in Stage II and III Resected Colon Cancer: Results of the Translational Study on the PETACC-3, EORTC 40993, SAKK 60-00 Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 466–474. [Google Scholar] [CrossRef]
- Liao, X.; Morikawa, T.; Lochhead, P.; Imamura, Y.; Kuchiba, A.; Yamauchi, M.; Nosho, K.; Qian, Z.R.; Nishihara, R.; Meyerhardt, J.A.; et al. Prognostic Role of PIK3CA Mutation in Colorectal Cancer: Cohort Study and Literature Review. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 2257–2268. [Google Scholar] [CrossRef]
- Papadatos-Pastos, D.; Rabbie, R.; Ross, P.; Sarker, D. The Role of the PI3K Pathway in Colorectal Cancer. Crit. Rev. Oncol. Hematol. 2015, 94, 18–30. [Google Scholar] [CrossRef]
- Crane, C.A.; Panner, A.; Murray, J.C.; Wilson, S.P.; Xu, H.; Chen, L.; Simko, J.P.; Waldman, F.M.; Pieper, R.O.; Parsa, A.T. PI(3) Kinase Is Associated with a Mechanism of Immunoresistance in Breast and Prostate Cancer. Oncogene 2009, 28, 306–312. [Google Scholar] [CrossRef]
- Ugai, T.; Zhao, M.; Shimizu, T.; Akimoto, N.; Shi, S.; Takashima, Y.; Zhong, R.; Lau, M.C.; Haruki, K.; Arima, K.; et al. Association of PIK3CA Mutation and PTEN Loss with Expression of CD274 (PD-L1) in Colorectal Carcinoma. Oncoimmunology 2021, 10, 1956173. [Google Scholar] [CrossRef]
- He, S.; Lin, Q.; Chen, J.; Ma, C.; Liu, Z.; Sun, Y.; Mao, W.; Shen, D.; Wang, J. Differential Expression of Tim3 Protein in Colorectal Cancer Associated with MSI and Braf Mutation. Histol. Histopathol. 2022, 37, 441–448. [Google Scholar] [CrossRef]
- Cen, S.; Liu, K.; Zheng, Y.; Shan, J.; Jing, C.; Gao, J.; Pan, H.; Bai, Z.; Liu, Z. BRAF Mutation as a Potential Therapeutic Target for Checkpoint Inhibitors: A Comprehensive Analysis of Immune Microenvironment in BRAF Mutated Colon Cancer. Front. Cell Dev. Biol. 2021, 9, 705060. [Google Scholar] [CrossRef]
- Yan, Y.; Gao, Z.; Han, H.; Zhao, Y.; Zhang, Y.; Ma, X.; Chen, H. NRAS Expression Is Associated with Prognosis and Tumor Immune Microenvironment in Lung Adenocarcinoma. J. Cancer Res. Clin. Oncol. 2022, 148, 565–575. [Google Scholar] [CrossRef]
- De’ Angelis, G.L.; Bottarelli, L.; Azzoni, C.; De’ Angelis, N.; Leandro, G.; Di Mario, F.; Gaiani, F.; Negri, F. Microsatellite Instability in Colorectal Cancer. Acta Bio-Medica Atenei Parm. 2018, 89, 97–101. [Google Scholar] [CrossRef]
- Yamamoto, H.; Watanabe, Y.; Arai, H.; Umemoto, K.; Tateishi, K.; Sunakawa, Y. Microsatellite Instability: A 2024 Update. Cancer Sci. 2024, 115, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Li, W.; Do, H.; Kwon, H.Y.; Kim, B.; Kim, K.; Kim, Y.; Cho, M.-Y. The Expression Patterns of Immune Checkpoint Molecules in Colorectal Cancer: An Analysis Based on Microsatellite Status. Biomedicines 2024, 12, 752. [Google Scholar] [CrossRef]
- Katagata, M.; Okayama, H.; Nakajima, S.; Saito, K.; Sato, T.; Sakuma, M.; Fukai, S.; Endo, E.; Sakamoto, W.; Saito, M.; et al. TIM-3 Expression and M2 Polarization of Macrophages in the TGFβ-Activated Tumor Microenvironment in Colorectal Cancer. Cancers 2023, 15, 4943. [Google Scholar] [CrossRef] [PubMed]
- Gossink, E.M.; Coffer, P.J.; Cutilli, A.; Lindemans, C.A. Immunomodulation by Galectin-9: Distinct Role in T Cell Populations, Current Therapeutic Avenues and Future Potential. Cell. Immunol. 2025, 407, 104890. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Wu, Y.-F.; Chou, F.-C.; Wu, Y.-H.; Yeh, L.-T.; Lin, K.-I.; Liu, F.-T.; Sytwu, H.-K. Intracellular Galectin-9 Enhances Proximal TCR Signaling and Potentiates Autoimmune Diseases. J. Immunol. 2020, 204, 1158–1172. [Google Scholar] [CrossRef] [PubMed]
- Kashio, Y.; Nakamura, K.; Abedin, M.J.; Seki, M.; Nishi, N.; Yoshida, N.; Nakamura, T.; Hirashima, M. Galectin-9 Induces Apoptosis Through the Calcium-Calpain-Caspase-1 Pathway1. J. Immunol. 2003, 170, 3631–3636. [Google Scholar] [CrossRef]
- Gooden, M.J.M.; Wiersma, V.R.; Samplonius, D.F.; Gerssen, J.; van Ginkel, R.J.; Nijman, H.W.; Hirashima, M.; Niki, T.; Eggleton, P.; Helfrich, W.; et al. Galectin-9 Activates and Expands Human T-Helper 1 Cells. PLoS ONE 2013, 8, e65616. [Google Scholar] [CrossRef]
- Lhuillier, C.; Barjon, C.; Niki, T.; Gelin, A.; Praz, F.; Morales, O.; Souquere, S.; Hirashima, M.; Wei, M.; Dellis, O.; et al. Impact of Exogenous Galectin-9 on Human T Cells: CONTRIBUTION OF THE T CELL RECEPTOR COMPLEX TO ANTIGEN-INDEPENDENT ACTIVATION BUT NOT TO APOPTOSIS INDUCTION. J. Biol. Chem. 2015, 290, 16797–16811. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, R.; Zhang, Y.; Guo, Y.; Hui, Z.; Wang, P.; Sun, Y. Galectin-9 Expression Clinically Associated with Mature Dendritic Cells Infiltration and T Cell Immune Response in Colorectal Cancer. BMC Cancer 2022, 22, 1319. [Google Scholar] [CrossRef]
- Rahmati, A.; Bigam, S.; Elahi, S. Galectin-9 Promotes Natural Killer Cells Activity via Interaction with CD44. Front. Immunol. 2023, 14, 1131379. [Google Scholar] [CrossRef] [PubMed]
- Golden-Mason, L.; McMahan, R.H.; Strong, M.; Reisdorph, R.; Mahaffey, S.; Palmer, B.E.; Cheng, L.; Kulesza, C.; Hirashima, M.; Niki, T.; et al. Galectin-9 Functionally Impairs Natural Killer Cells in Humans and Mice. J. Virol. 2013, 87, 4835–4845. [Google Scholar] [CrossRef]
- Nobumoto, A.; Oomizu, S.; Arikawa, T.; Katoh, S.; Nagahara, K.; Miyake, M.; Nishi, N.; Takeshita, K.; Niki, T.; Yamauchi, A.; et al. Galectin-9 Expands Unique Macrophages Exhibiting Plasmacytoid Dendritic Cell-like Phenotypes That Activate NK Cells in Tumor-Bearing Mice. Clin. Immunol. 2009, 130, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, M.F.; Bohner, P.; Pieraerts, C.; Lhermitte, B.; Gourmaud, J.; Nobile, A.; Rotman, S.; Cesson, V.; Martin, V.; Legris, A.-S.; et al. Immunoregulation of Dendritic Cell Subsets by Inhibitory Receptors in Urothelial Cancer. Eur. Urol. 2017, 71, 854–857. [Google Scholar] [CrossRef]
- Sakuma, M.; Katagata, M.; Okayama, H.; Nakajima, S.; Saito, K.; Sato, T.; Fukai, S.; Tsumuraya, H.; Onozawa, H.; Sakamoto, W.; et al. TIM-3 Expression on Dendritic Cells in Colorectal Cancer. Cancers 2024, 16, 1888. [Google Scholar] [CrossRef]
- González-Reyes, S.; Marín, L.; González, L.; González, L.O.; del Casar, J.M.; Lamelas, M.L.; González-Quintana, J.M.; Vizoso, F.J. Study of TLR3, TLR4 and TLR9 in Breast Carcinomas and Their Association with Metastasis. BMC Cancer 2010, 10, 665. [Google Scholar] [CrossRef]
- Ndhlovu, L.C.; Lopez-Vergès, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 Marks Human Natural Killer Cell Maturation and Suppresses Cell-Mediated Cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef]
- Gleason, M.K.; Lenvik, T.R.; McCullar, V.; Felices, M.; O’Brien, M.S.; Cooley, S.A.; Verneris, M.R.; Cichocki, F.; Holman, C.J.; Panoskaltsis-Mortari, A.; et al. Tim-3 Is an Inducible Human Natural Killer Cell Receptor That Enhances Interferon Gamma Production in Response to Galectin-9. Blood 2012, 119, 3064–3072. [Google Scholar] [CrossRef]
- Wang, Z.; Yin, N.; Zhang, Z.; Zhang, Y.; Zhang, G.; Chen, W. Upregulation of T-Cell Immunoglobulin and Mucin-Domain Containing-3 (Tim-3) in Monocytes/Macrophages Associates with Gastric Cancer Progression. Immunol. Invest. 2017, 46, 134–148. [Google Scholar] [CrossRef]
- Galassi, C.; Chan, T.A.; Vitale, I.; Galluzzi, L. The Hallmarks of Cancer Immune Evasion. Cancer Cell 2024, 42, 1825–1863. [Google Scholar] [CrossRef] [PubMed]
- Ochman, B.; Mielcarska, S.; Kula, A.; Dawidowicz, M.; Robotycka, J.; Piecuch, J.; Szrot, M.; Dzięgielewska-Gęsiak, S.; Muc-Wierzgoń, M.; Waniczek, D.; et al. Do Elevated YKL-40 Levels Drive the Immunosuppressive Tumor Microenvironment in Colorectal Cancer? Assessment of the Association of the Expression of YKL-40, MMP-8, IL17A, and PD-L1 with Coexisting Type 2 Diabetes, Obesity, and Active Smoking. Curr. Issues Mol. Biol. 2023, 45, 2781–2797. [Google Scholar] [CrossRef]
- Mielcarska, S.; Dawidowicz, M.; Kula, A.; Kiczmer, P.; Skiba, H.; Krygier, M.; Chrabańska, M.; Piecuch, J.; Szrot, M.; Ochman, B.; et al. B7H3 Role in Reshaping Immunosuppressive Landscape in MSI and MSS Colorectal Cancer Tumours. Cancers 2023, 15, 3136. [Google Scholar] [CrossRef] [PubMed]
- Dawidowicz, M.; Kula, A.; Mielcarska, S.; Kiczmer, P.; Skiba, H.; Krygier, M.; Chrabańska, M.; Piecuch, J.; Szrot, M.; Robotycka, J.; et al. B7H4 Expression Is More Frequent in MSS Status Colorectal Cancer and Is Negatively Associated with Tumour Infiltrating Lymphocytes. Cells 2023, 12, 861. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for Taxonomy-Based Analysis of Pathways and Genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Dampier, C.H.; Devall, M.; Jennelle, L.T.; Díez-Obrero, V.; Plummer, S.J.; Moreno, V.; Casey, G. Oncogenic Features in Histologically Normal Mucosa: Novel Insights into Field Effect from a Mega-Analysis of Colorectal Transcriptomes. Clin. Transl. Gastroenterol. 2020, 11, e00210. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Mutation Status | Percent (%) | ||
|---|---|---|---|---|
| n = 104 | Wild-Type | Mutant | Percent Among Mutation of the One Gene | Percent Among All Group |
| KRAS | 66 | 38 | 36.53% | |
| KRAS-117-STATUS | 100 | 4 | 10.52% | 3.84% |
| KRAS-12/13-STATUS | 75 | 29 | 76.31% | 27.88% |
| KRAS-59-STATUS | 101 | 3 | 7.89% | 2.88% |
| KRAS-146-STATUS | 102 | 2 | 5.26% | 1.92% |
| KRAS-61-STATUS | 101 | 3 | 7.89% | 2.88% |
| NRAS | 91 | 13 | 12.50% | |
| NRAS-12-13-STATUS | 96 | 8 | 61.54% | 7.69% |
| NRAS-61-STATUS | 99 | 5 | 38.46% | 4.80% |
| PIK3CA | 97 | 7 | 6.73% | |
| PIK3CA 542/545 | 99 | 6 | 85.71% | 5.77% |
| PIK3CA 1047 | 103 | 1 | 14.28% | 0.961% |
| BRAF | 97 | 7 | 6.73% | |
| AKT | 103 | 1 | 0.961% | |
| Combined gene mutations | 9 | 9.615% | ||
| KRAS + NRAS | 3 | 2.88% | ||
| KRAS + PIK3CA | 4 | 3.846% | ||
| KRAS + BRAF | 1 | 0.961% | ||
| KRAS + AKT1 | 1 | 0.961% | ||
| KRAS + NRAS + BRAF | 1 | 0.961% | ||
| Cytokine Subset | Top-Contributing Variables to Principal Component 1 (Based on Factor Loadings) | TIM3 | Gal-9 | ||
|---|---|---|---|---|---|
| p-Value | R-Value | p-Value | R-Value | ||
| Chemokine signaling | RANTES, SDF-1a, MCP1, CTACK, IL-8 IP-10 | *** | 0.5698 | *** | 0.5377 |
| Interleukin-10 signaling | IL-18, IL-8, RANTES, TNF-a, MCP1, MIP-1b, G-CSF, IP-10, MIP-1a, IL-10 | ** | 0.4809 | ** | 0.5036 |
| Interleukin-17 signaling pathway | TNF-a, IL-4, MCP3, Eotaxin, IL-8, GRO-a, MCP1, GM-CSF, IL-17, | ** | 0.4176 | ** | 0.4536 |
| NOD-Like receptor signaling pathway | IL-18, IL-8, IFN-a2, RANTES, TNF-a | ** | 0.4917 | ** | 0.5125 |
| Macrophage chemotaxis | RANTES, MCP1, IL-8, MIG, IP-10 | *** | 0.5278 | ** | 0.4353 |
| Positive regulation of lymphocyte migration | SDF-1a, RANTES, CTACK, MIP-1a, IP-10 | *** | 0.6044 | *** | 0.5516 |
| Protumor cytokines | IL-18, TNF-a, IL-8, RANTES, MCP3, MCP1 | ** | 0.4220 | ** | 0.4719 |
| Female | Male | All | |
|---|---|---|---|
| Age | 66.5 ± 9.64 | 65.0 ± 8.75 | 65 ± 9.22 |
| Tumor localization | |||
| Left-side tumor | 43 | 51 | 94 (71.76%) |
| Right-side tumor | 18 | 18 | 36 (27.48%) |
| T parameter | |||
| T1 | 2 | 5 | 7 (5.34%) |
| T2 | 12 | 9 | 21 (16.03%) |
| T3 | 41 | 46 | 87 (66.41%) |
| T4 | 8 | 8 | 16 (12.21%) |
| N parameter | |||
| N0 | 25 | 30 | 55 (41.98%) |
| N1 | 26 | 28 | 54 (41.22%) |
| N2 | 11 | 11 | 22 (16.79%) |
| M parameter | |||
| M0 | 55 | 56 | 111 (84.73%) |
| M1 | 7 | 13 | 20 (15.27%) |
| TNM Stage | |||
| I | 10 | 9 | 19 (14.50%) |
| II | 15 | 18 | 33 (25.19%) |
| III | 30 | 30 | 60 (45.80%) |
| IV | 7 | 12 | 19 (14.50%) |
| Histological Grading | |||
| High | 10 | 12 | 22 (16.79%) |
| Low | 52 | 38 | 109 (83.21%) |
| MSI Status (n = 79) | |||
| MSS tumors | 30 | 35 | 65 (82.28%) |
| MSI tumors | 9 | 5 | 14 (17.72%) |
| Gene | Exon | Amino Acid Change | Nucleotide Change | Cosmic ID |
|---|---|---|---|---|
| KRAS | 2 | G12A | c.35G>C | 522 |
| G12D | c.35G>A | 521 | ||
| G12R | c.34G>C | 518 | ||
| G12C | c.34G>T | 516 | ||
| G12S | c.34G>A | 517 | ||
| G12V | c.35G>T | 520 | ||
| G13D | c.38G>A | 532 | ||
| 3 | A59T | c.175G>A | 546 | |
| A59E | c.176C>A | 547 | ||
| A59G | c.176C>G | 28518 | ||
| Q61H | c.183A>C | 554 | ||
| Q61H | c.183A>T | 555 | ||
| Q61L | c.182A>T | 553 | ||
| Q61R | c.182A>G | 552 | ||
| 4 | K117N | c.351A>C | 19940 | |
| K117N | c.351A>T | 28519 | ||
| K117R | c.350A>G | 4696722 | ||
| K117E | c.349A>G | - | ||
| A146T | c.436G>A | 19404 | ||
| A146P | c.436G>C | 19905 | ||
| A146V | c.437C>T | 19900 | ||
| NRAS | 2 | G12D | c.35G>A | 564 |
| G12S | c.34G>A | 563 | ||
| G12C | c.34G>T | 562 | ||
| G13R | c.37G>C | 569 | ||
| G13V | c.38G>T | 574 | ||
| 3 | A59T | c.175G>A | 578 | |
| A59D | c.176C>A | 253327 | ||
| Q61K | c.181C>A | 580 | ||
| Q61L | c.182A>T | 583 | ||
| Q61R | c.182A>G | 584 | ||
| Q61H | c.183A>C | 586 | ||
| Q61H | c.183A>T | 585 | ||
| 4 | K117R | c.350A>G | - | |
| A146T | c.436G>A | 27174 | ||
| BRAF | 15 | V600E | c.1799T>A | 476 |
| V600E2 | c.1799-1800TG>AA | - | ||
| V600D | c.1799-1800TG>AT | 477 | ||
| V600K | c.1798-1799GT>AA | 473 | ||
| PIK3CA | 9 | E542K | c.1624G>A | 760 |
| E545K | c.1633G>A | 763 | ||
| E545Q | c.1633G>C | 27133 | ||
| 20 | H1047R | c.3140A>G | 775 | |
| H1047L | c.3140A>T | 776 | ||
| AKT1 | 4 | E17K | c.49G>A | 33765 |
| Process Name | Cytokines Involved | Origin |
|---|---|---|
| Positive regulation of immune system process | MIF, SCF, MCP1, SDF-1a, VEGFA, MCP3, MCSF, MIP-1a, IL-1a, IL-18, IL-6, RANTES, IL-5, TNF-b, LIF, IL-2, IL-1b, IL-7, IFN-g, IL-13, TNF-a, IL-10, IL-8, IL-4, IP-10, IL-15, IL-2Ra, IL-16, CTACK, IL-12p40, MIP-1b, IL-17 | GO |
| Chemokine signaling pathway | IL-8, MCP1, SDF-1a, GRO-a, IP-10, RANTES, MIP-1a, CTACK, Eotaxin, MCP3, MIP-1b | KEGG |
| Positive regulation of lymphocyte migration | IP-10, SDF-1a, MIP-1a, CTACK, MIP-1b, MCP3, RANTES | GO |
| Macrophage chemotaxis | MCP1, IL-8, Eotaxin, MIG, IP-10, GRO-a, MIP-1b, MCP3, RANTES, IL-1b | GO |
| PI3K-Akt signaling pathway | IL-2Ra, bNGF, IL-2, IL-3, IL-4, SCF, MCSF, IFN-a2, HGF, G-CSF, IL-7, PDGF-bb, BasicFGF, IL-6, VEGFA | KEGG |
| Leukocyte activation | IL-4, IL-15, IFN-g, SCF, IL-2Ra, IL-8, MCSF, IL-13, IL-18, MIP-1a, RANTES, IL-10, GM-CSF, IL-9, IL-7, IFN-a2, IL-2, IL-6, TNF-a | GO |
| Inflammatory response | IL-9, CTACK, Eotaxin, MCP1, IFN-a2, IL-1Ra, IL-2Ra, IFN-g, IL-15, IL-1a, IL-6, IL-17, IL-4, MCP3, MIP-1a, IL-18, CTACK, MIF, TNF-a, RANTES, MCSF, MIG, IL-1b, IL-5, IL-10, IL-8, IL-13, IP-10, MIP-1b | GO |
| Interleukin-10 signaling | MCP1, MCSF, IL-8, IL-18, IL-6, GM-CSF, LIF, IL-10, IL-1Ra, IL-1a, IP-10, GRO-a, MIP-1a, IL-1b, MIP-1b, G-CSF, RANTES, TNF-a | KEGG |
| NOD-like receptor signaling pathway | IL-8, TNF-a, MCP1, IFN-a2, GRO-a, IL-6, IL-1b, RANTES, IL-18 | KEGG |
| Protumor cytokines | IL-1a, IL-1b, IL-6, IL-8, IL-17, IL-18, TNF-a, RANTES, MCP1, MCP3, MIF, G-CSF, GM-CSF | Own literature review |
| IL-17 signaling | MCP1, IL-8, IL-1b, IL-4, IL-5, IL-6, IL-13, IL-17, GM-CSF, IFN-g, G-CSF, TNF-a, MCP3, Eotaxin, GRO-a, IP-10 | KEGG |
| Positive regulation of cytokine production | IL-9, IL-12p70, GM-CSF, IL-10, HGF, IL-2, IL-15, IL-1b, IL-18, IFN-g, IL-7, IL-4, TNF-a, IL-17, MIF, TNF-b, IL-16, IL-13, MIP-1a, IL-1a, IL-6 | GO |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ochman, B.; Kot, A.; Mielcarska, S.; Kula, A.; Dawidowicz, M.; Hudy, D.; Szrot, M.; Piecuch, J.; Waniczek, D.; Czuba, Z.; et al. TIM-3/Galectin-9 Immune Axis in Colorectal Cancer in Relation to KRAS, NRAS, BRAF, PIK3CA, AKT1 Mutations, MSI Status, and the Cytokine Milieu. Int. J. Mol. Sci. 2025, 26, 6735. https://doi.org/10.3390/ijms26146735
Ochman B, Kot A, Mielcarska S, Kula A, Dawidowicz M, Hudy D, Szrot M, Piecuch J, Waniczek D, Czuba Z, et al. TIM-3/Galectin-9 Immune Axis in Colorectal Cancer in Relation to KRAS, NRAS, BRAF, PIK3CA, AKT1 Mutations, MSI Status, and the Cytokine Milieu. International Journal of Molecular Sciences. 2025; 26(14):6735. https://doi.org/10.3390/ijms26146735
Chicago/Turabian StyleOchman, Błażej, Anna Kot, Sylwia Mielcarska, Agnieszka Kula, Miriam Dawidowicz, Dorota Hudy, Monika Szrot, Jerzy Piecuch, Dariusz Waniczek, Zenon Czuba, and et al. 2025. "TIM-3/Galectin-9 Immune Axis in Colorectal Cancer in Relation to KRAS, NRAS, BRAF, PIK3CA, AKT1 Mutations, MSI Status, and the Cytokine Milieu" International Journal of Molecular Sciences 26, no. 14: 6735. https://doi.org/10.3390/ijms26146735
APA StyleOchman, B., Kot, A., Mielcarska, S., Kula, A., Dawidowicz, M., Hudy, D., Szrot, M., Piecuch, J., Waniczek, D., Czuba, Z., & Świętochowska, E. (2025). TIM-3/Galectin-9 Immune Axis in Colorectal Cancer in Relation to KRAS, NRAS, BRAF, PIK3CA, AKT1 Mutations, MSI Status, and the Cytokine Milieu. International Journal of Molecular Sciences, 26(14), 6735. https://doi.org/10.3390/ijms26146735