
In Silico Analysis of Post-COVID-19 Condition (PCC) Associated SNP rs9367106 Predicts the Molecular Basis of Abnormalities in the Lungs and Brain Functions

Abstract
1. Introduction
2. Results
2.1. The rs9367106 Resides Within the Long Noncoding RNAs, LINC01276 and AS-FOXP4 and Physically Interacts with the Coding FOXP4 Gene
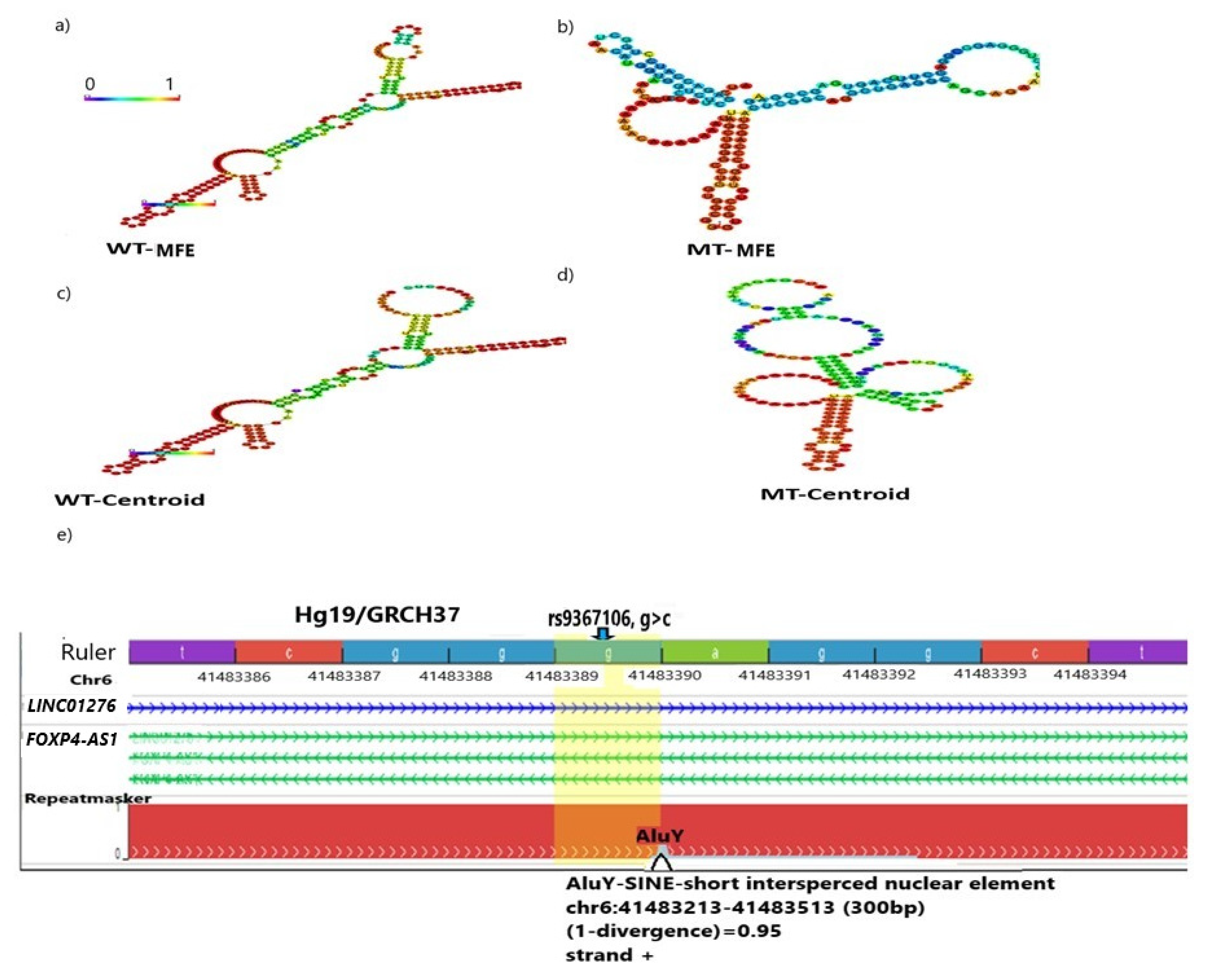
2.2. The G>C Transition of rs9367106 Extensively Alters the RNA Structure
2.3. The rs9367106 Disrupts an ALU/SINE Element
2.4. rs9367106 Carrying DNA Region Binds with Several Transcription Factors (TFs)
2.5. rs9367106 Affects the Expression of FOXP4 and MED20 Genes
2.6. The SNP-Carrying RNA Region Acts as a Distant Enhancer
2.7. The LINC01276 Targets the MED20 Gene That Extensively Expresses Tissues in the Brain and Modulates Sleep Disorder
2.8. FOXP4-AS1 Function
2.9. FOXP4 Could Alter the Functions of Lung Alveolar Cells and Brain Tissues Leading to PCC Phenotypes
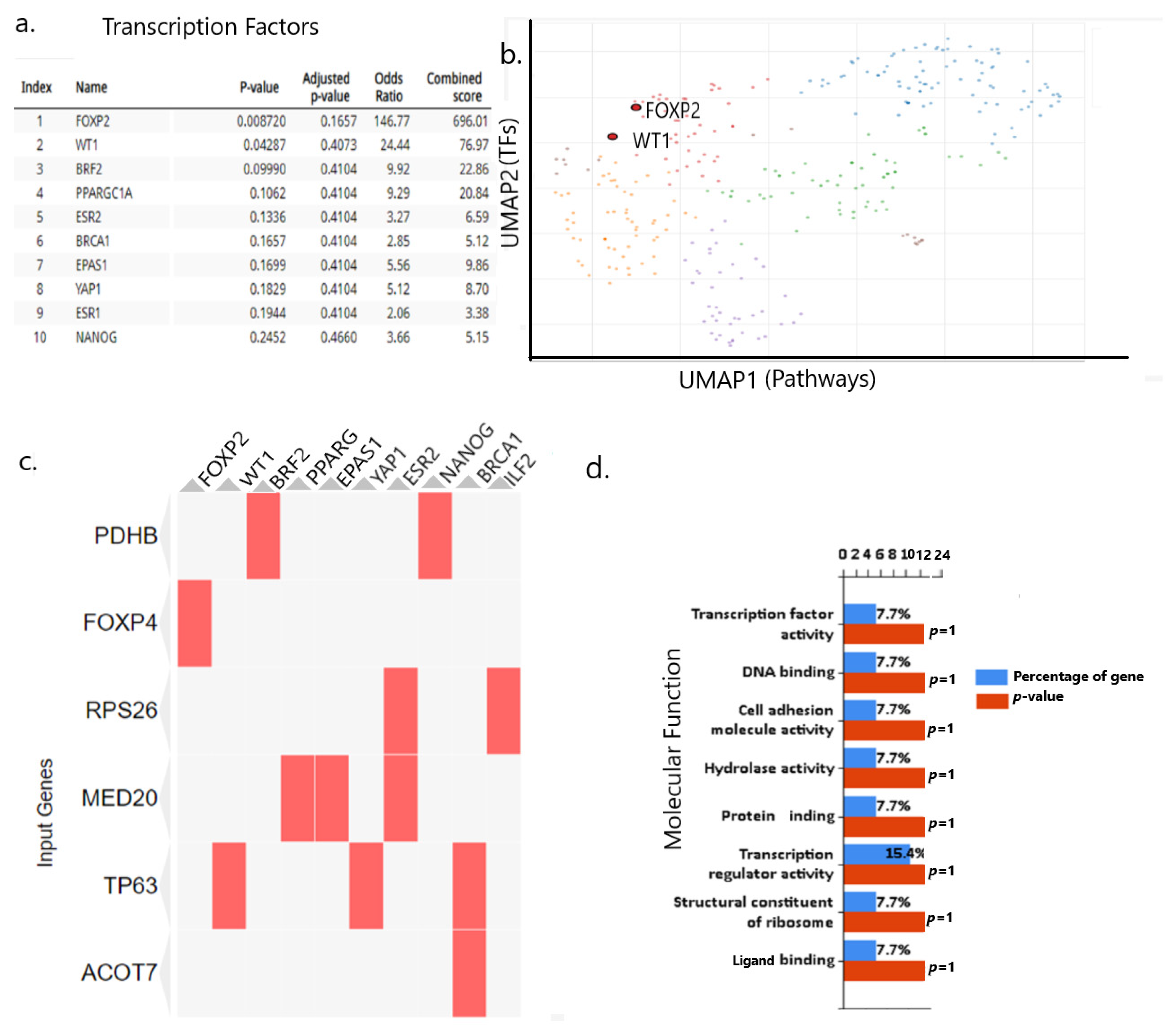
2.10. rs9367106 Co-Expressed Genes Induce FOXP4-FOXP2 and TP63 Enriched Pathway
3. Discussion
Limitation
4. Materials and Methods
4.1. Physical Interaction
4.2. eQTL Analysis
4.3. Expression
4.4. Enhancer Analysis
4.5. Transcription Factor Binding Sites
4.6. RNA Structure Modeling
4.7. ALU/SINE Sequence Identification
4.8. LINC01276 Target Identification
4.9. Co-Expression, Gene and Pathway Enrichment
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cabrera Martimbianco, A.L.; Pacheco, R.L.; Bagattini, Â.M.; Riera, R. Frequency, signs and symptoms, and criteria adopted for long COVID-19: A systematic review. Int. J. Clin. Pract. 2021, 75, e14357. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.D.; Lavelle, M.; Boursiquot, B.C.; Wan, E.Y. Long-term complications of COVID-19. Am. J. Physiol. Cell Physiol. 2022, 322, C1–C11. [Google Scholar] [CrossRef]
- Wong, A.C.; Devason, A.S.; Umana, I.C.; Cox, T.O.; Dohnalová, L.; Litichevskiy, L.; Perla, J.; Lundgren, P.; Etwebi, Z.; Izzo, L.T.; et al. Serotonin reduction in post-acute sequelae of viral infection. Cell 2023, 186, 4851–4867.e20. [Google Scholar] [CrossRef]
- Appelman, B.; Charlton, B.T.; Goulding, R.P.; Kerkhoff, T.J.; Breedveld, E.A.; Noort, W.; Offringa, C.; Bloemers, F.W.; van Weeghel, M.; Schomakers, B.V.; et al. Muscle abnormalities worsen after post-exertional malaise in long COVID. Nat. Commun. 2024, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Cervia-Hasler, C.; Brüningk, S.C.; Hoch, T.; Fan, B.; Muzio, G.; Thompson, R.C.; Ceglarek, L.; Meledin, R.; Westermann, P.; Emmenegger, M.; et al. Persistent complement dysregulation with signs of thromboinflammation in active Long Covid. Science 2024, 383, eadg7942. [Google Scholar] [CrossRef]
- Song, Y.; Myers, R.; Mehl, F.; Murphy, L.; Brooks, B.; Wilson, J.M.; Kadl, A.; Woodfolk, J.; Zeichner, S.L. ACE-2-like enzymatic activity is associated with immunoglobulin in COVID-19 patients. mBio 2024, 15, e0054124. [Google Scholar] [CrossRef]
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Perelman, C.; Sepulveda, R.; Rebolledo, P.A.; Cuapio, A.; Villapol, S. More than 50 long-term effects of COVID-19: A systematic review and meta-analysis. Sci. Rep. 2021, 11, 6144. [Google Scholar] [CrossRef]
- COVID-19 Host Genetics Initiative. Mapping the human genetic architecture of COVID-19. Nature 2021, 600, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernández, J.; Prati, D.; Baselli, G.; Asselta, R.; et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar]
- Wang, F.; Huang, S.; Gao, R.; Zhou, Y.; Lai, C.; Li, Z.; Xian, W.; Qian, X.; Li, Z.; Huang, Y.; et al. Initial Whole Genome Sequencing and Analysis of the HostGenetic Contribution to COVID-19 Severity and Susceptibility. Cell Discovery 2020, 6, 83. [Google Scholar] [CrossRef]
- Maiti, A.K. The African-American population with a low allele frequency of SNP rs1990760 (T allele) in IFIH1 predicts less IFN-beta expression and potential vulnerability to COVID-19 infection. Immunogenetics 2020, 72, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Langton, D.J.; Bourke, S.C.; Lie, B.A.; Reiff, G.; Natu, S.; Darlay, R.; Burn, J.; Echevarria, C. The influence of HLA genotype on susceptibility to, and severity of, COVID-19 infection. medRxiv 2021. [Google Scholar] [CrossRef]
- Ledford, H. Gene linked to long COVID found in analysis of thousands of patients. Nature 2023, 619, 445. [Google Scholar] [CrossRef]
- Lammi, V.; Nakanishi, T.; Jones, S.E.; Andrews, S.J.; Karjalainen, J.; Cortés, B.; O’Brien, H.E.; Ochoa-Guzman, A.; Fulton-Howard, B.E.; Broberg, M.; et al. Genome-wide association study of Long COVID-19. Nat. Genet. 2025, 57, 1402–1417. [Google Scholar] [CrossRef] [PubMed]
- Tak, Y.G.; Farnham, P.J. Making sense of GWAS: Using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome. Epigenetics Chromatin. 2015, 8, 57. [Google Scholar] [CrossRef]
- Molineros, J.E.; Maiti, A.K.; Sun, C.; Looger, L.L.; Han, S.; Kim-Howard, X.; Glenn, S.; Adler, A.; Kelly, J.A.; Niewold, T.B.; et al. Admixture mapping in lupus identifies multiple functional variants within IFIH1 associated with apoptosis, inflammation, and autoantibody production. PLoS Genet. 2013, 9, e1003222. [Google Scholar] [CrossRef] [PubMed]
- Krietenstein, N.; Abraham, S.; Venev, S.V.; Abdennur, N.; Gibcus, J.; Hsieh, T.H.S.; Parsi, K.M.; Yang, L.; Maehr, R.; Mirny, L.A.; et al. Ultrastructural Details of Mammalian Chromosome Architecture. Mol. Cell. 2020, 78, 554–565.e7. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Sanyal, A.; Lajoie, B.R.; Jain, G.; Dekker, J. The long-range interaction landscape of gene promoters. Nature 2012, 489, 109–113. [Google Scholar] [CrossRef]
- Tang, Z.; Luo, O.J.; Li, X.; Zheng, M.; Zhu, J.J.; Szalaj, P.; Trzaskoma, P.; Magalska, A.; Wlodarczyk, J.; Ruszczycki, B.; et al. CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 2015, 163, 1611–1627. [Google Scholar] [CrossRef]
- Jin, F.; Li, Y.; Dixon, J.R.; Selvaraj, S.; Ye, Z.; Lee, A.Y.; Yen, C.A.; Schmitt, A.D.; Espinoza, C.A.; Ren, B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 2013, 503, 290–294. [Google Scholar] [CrossRef]
- Li, Y.; Huang, W.; Niu, L.; Umbach, D.M.; Covo, S.; Li, L. Characterization of constitutive CTCF/cohesin loci: A possible role in establishing topological domains in mammalian genomes. BMC Genom. 2013, 14, 553. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Witt, H.; Katsumura, K.; Ye, Z.; Wang, Q.; Bresnick, E.H.; Farnham, P.J.; Jin, V.X. Integration of Hi-C and ChIP-seq data reveals distinct types of chromatin linkages. Nucleic Acids Res. 2012, 40, 7690–7704. [Google Scholar] [CrossRef] [PubMed]
- Fullwood, M.J.; Liu, M.H.; Pan, Y.F.; Liu, J.; Xu, H.; Mohamed, Y.B.; Orlov, Y.L.; Velkov, S.; Ho, A.; Mei, P.H.; et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 2009, 462, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Salameh, T.J.; Wang, X.; Song, F.; Zhang, B.; Wright, S.M.; Khunsriraksakul, C.; Ruan, Y.; Yue, F. A supervised learning framework for chromatin loop detection in genome-wide contact maps. Nat. Commun. 2020, 11, 3428. [Google Scholar] [CrossRef]
- Liddicoat, B.J.; Piskol, R.; Chalk, A.M.; Ramaswami, G.; Higuchi, M.; Hartner, J.C.; Li, J.B.; Seeburg, P.H.; Walkley, C.R. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 2015, 349, 1115–1120. [Google Scholar] [CrossRef]
- Liu, C.; Wang, Q.; Wang, Y.; Wang, G.; Wang, L.; Chen, H.; Jiao, T.; Hu, C.; Lei, X.; Guo, L.; et al. Augmentation of anti-MDA5 antibody implies severe disease in COVID-19 patients. medRxiv 2020. [Google Scholar] [CrossRef]
- Sampaio, N.G.; Chauveau, L.; Hertzog, J.; Bridgeman, A.; Fowler, G.; Moonen, J.P.; Dupont, M.; Russell, R.A.; Noerenberg, M.; Rehwinkel, J. The RNA sensor MDA5 detects SARS-CoV-2 infection. Sci. Rep. 2021, 11, 13638. [Google Scholar] [CrossRef]
- Maiti, A.K. MDA5 Is a Major Determinant of Developing Symptoms in Critically Ill COVID-19 Patients. Clin. Rev. Allergy Immunol. 2024, 67, 58–72. [Google Scholar] [CrossRef]
- Ryan, F.J.; Hope, C.M.; Masavuli, M.G.; Lynn, M.A.; Mekonnen, Z.A.; Yeow, A.E.L.; Garcia-Valtanen, P.; Al-Delfi, Z.; Gummow, J.; Ferguson, C.; et al. Long-term perturbation of the peripheral immune system months after SARS-CoV-2 infection. BMC Med. 2022, 20, 26. [Google Scholar] [CrossRef]
- Jiang, Y.; Qian, F.; Bai, X.; Liu, Y.; Wang, Q.; Ai, B.; Han, X.; Shi, S.; Zhang, J.; Li, X.; et al. SEdb: A comprehensive human super-enhancer database. Nucleic Acids Res. 2019, 47, D235–D243. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Wood, J.; Jaycox, J.R.; Dhodapkar, R.M.; Lu, P.; Gehlhausen, J.R.; Tabachnikova, A.; Greene, K.; Tabacof, L.; Malik, A.A.; et al. Distinguishing features of long COVID identified through immune profiling. Nature 2023, 623, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Delorey, T.M.; Ziegler, C.G.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, Å.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017, bax028. [Google Scholar] [CrossRef]
- Vodopiutz, J.; Schmook, M.T.; Konstantopoulou, V.; Plecko, B.; Greber-Platzer, S.; Creus, M.; Seidl, R.; Janecke, A.R. MED20 mutation associated with infantile basal ganglia degeneration and brain atrophy. Eur. J. Pediatr. 2015, 174, 113–118. [Google Scholar] [CrossRef]
- Yao, Y.; Jia, Y.; Wen, Y.; Cheng, B.; Cheng, S.; Liu, L.; Yang, X.; Meng, P.; Chen, Y.; Li, C.E.; et al. Genome-Wide Association Study and Genetic Correlation Scan Provide Insights into Its Genetic Architecture of Sleep Health Score in the UK Biobank Cohort. Nat. Sci. Sleep. 2022, 14, 1–12. [Google Scholar] [CrossRef]
- Li, J.; Lian, Y.; Yan, C.; Cai, Z.; Ding, J.; Ma, Z.; Peng, P.; Wang, K. Long non-coding RNA FOXP4-AS1 is an unfavourable prognostic factor and regulates proliferation and apoptosis in colorectal cancer. Cell Prolif. 2017, 50, e12312. [Google Scholar] [CrossRef]
- Liang, J.; Wang, D.; Qiu, G.; Zhu, X.; Liu, J.; Li, H.; Guo, P. Long Noncoding RNA FOXP4-AS1 Predicts Unfavourable Prognosis and Regulates Proliferation and Invasion in Hepatocellular Carcinoma. Biomed. Res. Int. 2021, 2021, 8850656. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ge, D.; Chen, X.I.; Qiu, J.; Yin, Z.; Zheng, S.; Jiang, C. FOXP4-AS1 participates in the development and progression of osteosarcoma by downregulating LATS1 via binding to LSD1 and EZH2. Biochem. Biophys. Res. Commun. 2018, 502, 493–500. [Google Scholar] [CrossRef]
- Wu, X.; Xiao, Y.; Zhou, Y.; Zhou, Z.; Yan, W. LncRNA FOXP4-AS1 is activated by PAX5 and promotes the growth of prostate cancer by sequestering miR-3184-5p to upregulate FOXP4. Cell Death Dis. 2019, 10, 472. [Google Scholar] [CrossRef]
- Yang, Y.; Qiu, R.; Weng, Q.; Xu, Z.; Song, J.; Zhao, S.; Meng, M.; Zhang, D.; Kong, C.; Wang, H.; et al. MLL4 Regulates the Progression of Non-Small-Cell Lung Cancer by Regulating the PI3K/AKT/SOX2 Axis. Cancer Res. Treat. 2023, 55, 778–803. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, K.; Okada, Y.; Takahashi, A.; Kamatani, Y.; Momozawa, Y.; Ashikawa, K.; Kunitoh, H.; Matsumoto, S.; Takano, A.; Shimizu, K.; et al. Association of variations in HLA class II and other loci with susceptibility to EGFR-mutated lung adenocarcinoma. Nat. Commun. 2016, 7, 12451. [Google Scholar] [CrossRef]
- Luo, Y.S.; Zhang, K.; Cheng, Z.S. Absence of Association between a Long COVID and Severe COVID-19 Risk Variant of. Front. Genet. 2023, 14, 1258829. [Google Scholar] [CrossRef] [PubMed]
- Del Viso, F.; Zhou, D.; Thiffault, I.; Lawson, C.; Cross, L.; Jenkins, J.; Rush, E.; Saunders, C. Recurrent FOXP4 nonsense variant in two unrelated patients: Association with neurodevelopmental disease and congenital diaphragmatic hernia. Am. J. Med. Genet. A 2023, 191, 259–264. [Google Scholar] [CrossRef]
- Snijders Blok, L.; Vino, A.; Den Hoed, J.; Underhill, H.R.; Monteil, D.; Li, H.; Reynoso Santos, F.J.; Chung, W.K.; Amaral, M.D.; Schnur, R.E.; et al. Heterozygous variants that disturb the transcriptional repressor activity of FOXP4 cause a developmental disorder with speech/language delays and multiple congenital abnormalities. Genet. Med. 2021, 23, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Co, M.; Anderson, A.G.; Konopka, G. FOXP transcription factors in vertebrate brain development, function, and disorders. Wiley Interdiscip. Rev. Dev. Biol. 2020, 9, e375. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; COVID-19 Genomics UK Consortium; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef]
- Bange, E.M.; Han, N.A.; Wileyto, P.; Kim, J.Y.; Gouma, S.; Robinson, J.; Greenplate, A.R.; Hwee, M.A.; Porterfield, F.; Owoyemi, O.; et al. CD8+ T cells contribute to survival in patients with COVID-19 and hematologic cancer. Nat. Med. 2021, 27, 1280–1289. [Google Scholar] [CrossRef]
- Agerer, B.; Koblischke, M.; Gudipati, V.; Montaño-Gutierrez, L.F.; Smyth, M.; Popa, A.; Genger, J.W.; Endler, L.; Florian, D.M.; Mühlgrabner, V.; et al. SARS-CoV-2 mutations in MHC-I-restricted epitopes evade CD8. Sci. Immunol. 2021, 6, eabg6461. [Google Scholar] [CrossRef]
- Lamers, M.M.; van den Hoogen, B.G.; Haagmans, B.L. ADAR1: “Editor-in-Chief” of Cytoplasmic Innate Immunity. Front. Immunol. 2019, 10, 1763. [Google Scholar] [CrossRef]
- Jung, C.; Kmiec, D.; Koepke, L.; Zech, F.; Jacob, T.; Sparrer, K.M.; Kirchhoff, F. Omicron: What Makes the Latest SARS-CoV-2 Variant of Concern So Concerning? J. Virol. 2022, 96, e0207721. [Google Scholar] [CrossRef]
- Mourier, T.; Sadykov, M.; Carr, M.J.; Gonzalez, G.; Hall, W.W.; Pain, A. Host-directed editing of the SARS-CoV-2 genome. Biochem. Biophys. Res. Commun. 2021, 538, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Amado-Rodríguez, L.; Del Riego, E.S.; de Ona, J.G.; Alonso, I.L.; Gil-Pena, H.; López-Martínez, C.; Martín-Vicente, P.; Lopez-Vazquez, A.; Lopez, A.G.; Cuesta-Llavona, E.; et al. Effects of IFIH1 rs1990760 variants on systemic inflammation and outcome in critically ill COVID-19 patients in an observational translational study. Elife 2022, 11, e73012. [Google Scholar] [CrossRef] [PubMed]
- Muñiz-Banciella, M.G.; Albaiceta, G.M.; Amado-Rodríguez, L.; Del Riego, E.S.; Alonso, I.L.; López-Martínez, C.; Martín-Vicente, P.; García-Clemente, M.; Hermida-Valverde, T.; Enríquez-Rodriguez, A.I.; et al. Age-dependent effect of the IFIH1/MDA5 gene variantson the risk of critical COVID-19. Immunogenetics 2022, 75, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Mehdipour, P. ADAR1: From basic mechanisms to inhibitors. Trends Cell Biol. 2024, 35, 59–73. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, X. TRABID targets DDB2 for deubiquitination to promote proliferation of hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2022, 625, 23–30. [Google Scholar] [CrossRef]
- Nakagawa, H.; Akamatsu, S.; Takata, R.; Takahashi, A.; Kubo, M.; Nakamura, Y. Prostate cancer genomics, biology, and risk assessment through genome-wide association studies. Cancer Sci. 2012, 103, 607–613. [Google Scholar] [CrossRef]
- Robinson, J.T.; Turner, D.; Durand, N.C.; Thorvaldsdóttir, H.; Mesirov, J.P.; Aiden, E.L. Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cell Syst. 2018, 6, 256–258.e1. [Google Scholar] [CrossRef]
- Zheng, Z.; Huang, D.; Wang, J.; Zhao, K.; Zhou, Y.; Guo, Z.; Zhai, S.; Xu, H.; Cui, H.; Yao, H.; et al. QTLbase: An integrative resource for quantitative trait loci across multiple human molecular phenotypes. Nucleic Acids Res. 2020, 48, D983–D991. [Google Scholar] [CrossRef]
- Messeguer, X.; Escudero, R.; Farré, D.; Núñez, O.; Martínez, J.; Albà, M.M. PROMO: Detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002, 18, 333–334. [Google Scholar] [CrossRef]
- Farré, D.; Roset, R.; Huerta, M.; Adsuara, J.E.; Roselló, L.; Albà, M.M.; Messeguer, X. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 2003, 31, 3651–3653. [Google Scholar] [CrossRef]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neuböck, R.; Hofacker, I.L. The Vienna RNA websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Höner zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Pathan, M.; Keerthikumar, S.; Ang, C.S.; Gangoda, L.; Quek, C.Y.; Williamson, N.A.; Mouradov, D.; Sieber, O.M.; Simpson, R.J.; Salim, A.; et al. FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics 2015, 15, 2597–2601. [Google Scholar] [CrossRef]
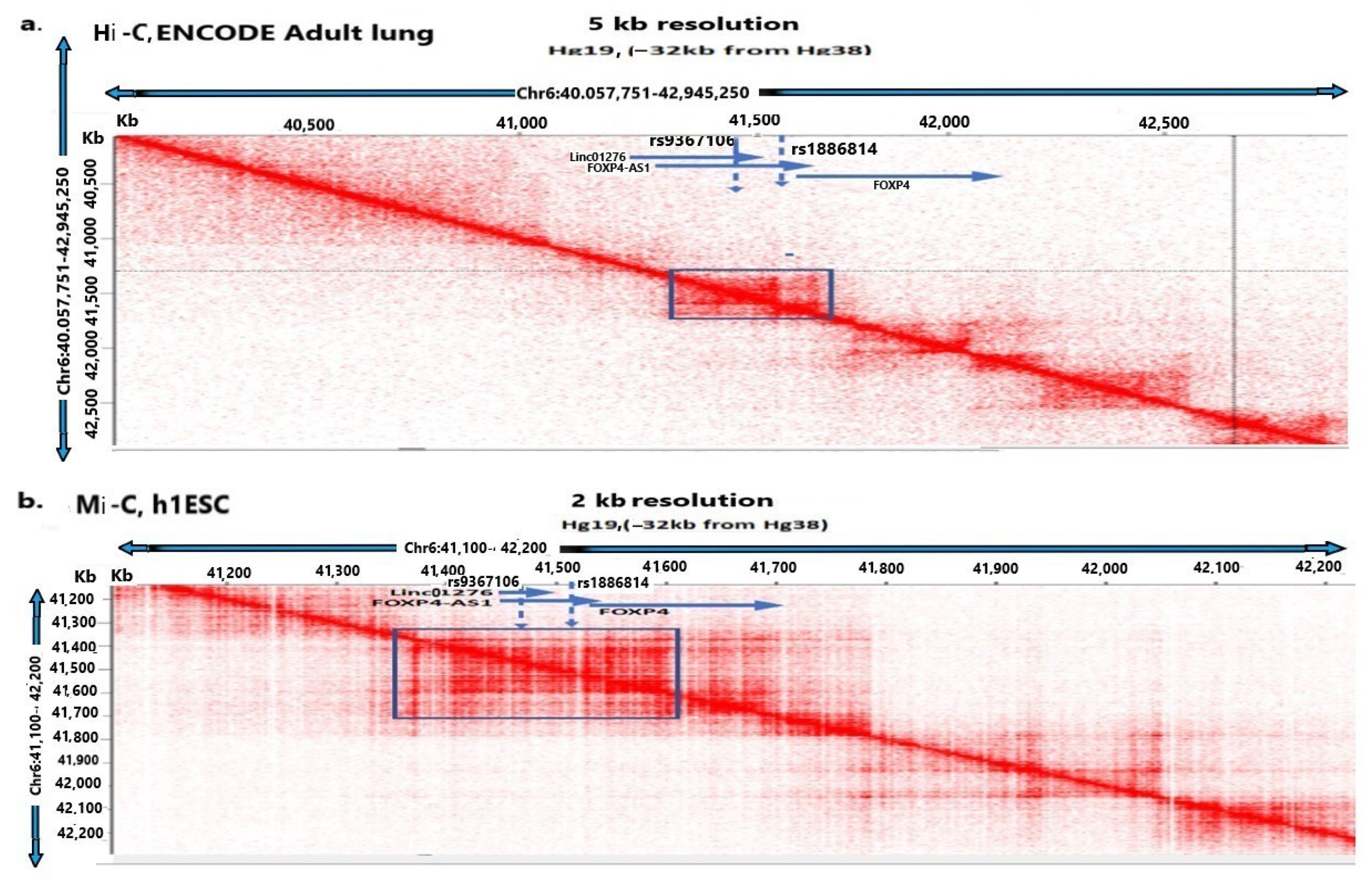


 and interacts with FOXP4
and interacts with FOXP4  . The purple color
. The purple color  is the antisense FOXP4-AS1. (c) Mechanism of altered regulatory function by G>C transition of the SNP rs9367106. LINC01276 and FOXP4-AS1 RNA interact with FOXP4, and LINC01276 also interact with MED20 to develop PCC phenotypes in neuronal and lung tissues.
and interacts with FOXP4 . The purple color is the antisense FOXP4-AS1. (c) Mechanism of altered regulatory function by G>C transition of the SNP rs9367106. LINC01276 and FOXP4-AS1 RNA interact with FOXP4, and LINC01276 also interact with MED20 to develop PCC phenotypes in neuronal and lung tissues.
is the antisense FOXP4-AS1. (c) Mechanism of altered regulatory function by G>C transition of the SNP rs9367106. LINC01276 and FOXP4-AS1 RNA interact with FOXP4, and LINC01276 also interact with MED20 to develop PCC phenotypes in neuronal and lung tissues.
and interacts with FOXP4 . The purple color is the antisense FOXP4-AS1. (c) Mechanism of altered regulatory function by G>C transition of the SNP rs9367106. LINC01276 and FOXP4-AS1 RNA interact with FOXP4, and LINC01276 also interact with MED20 to develop PCC phenotypes in neuronal and lung tissues.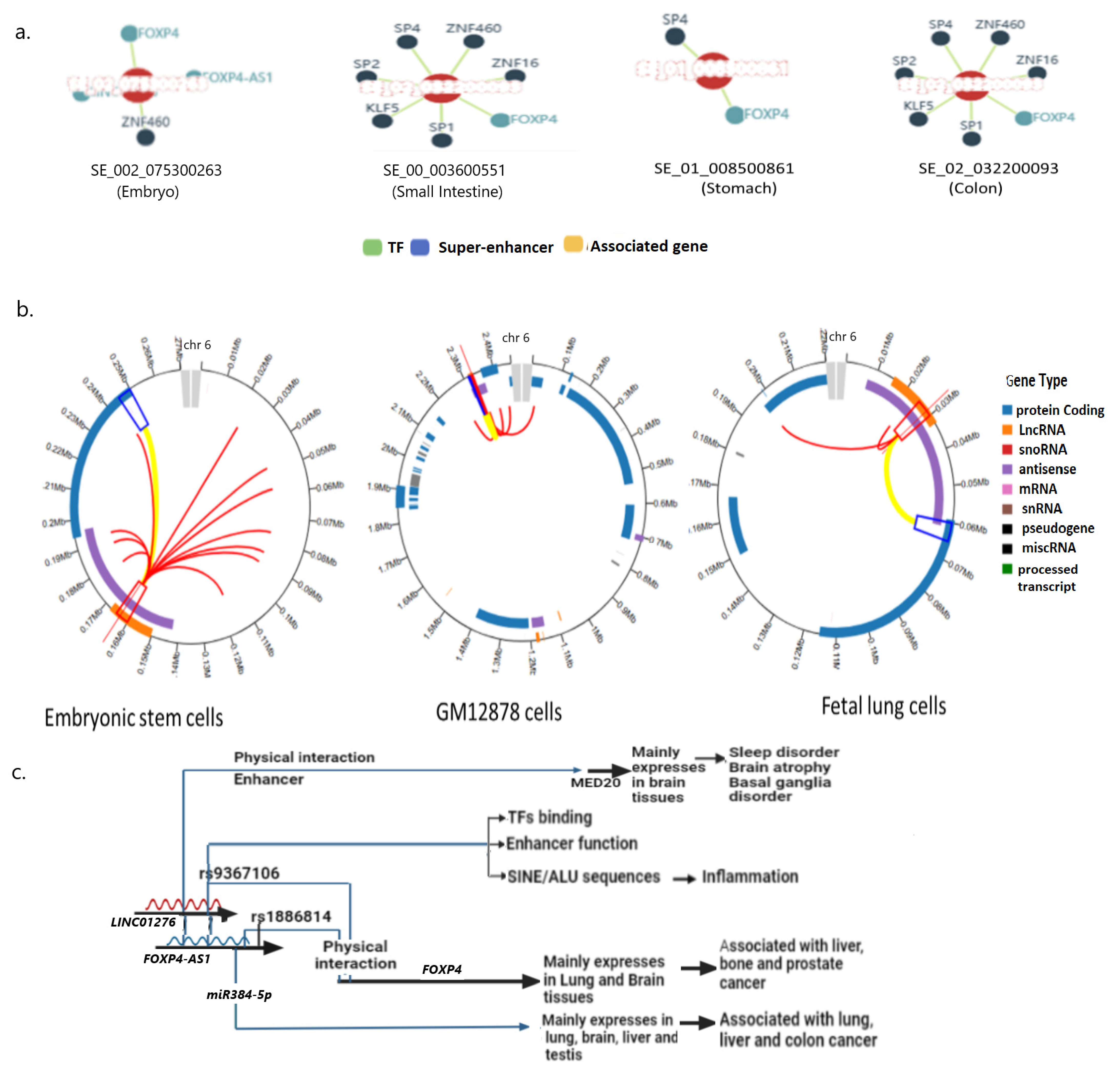

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contact Domain (TADs) | ||||||||||
| Cells | Methods | Built | Chr | 1st Chr | 1st Chr | Chr | 2nd Chr | 2nd Chr | Experiments/ Database | |
| h1ESC | Mi-C | Hg19 | 6 | 41,335,000 | 41,565,000 | 6 | 41,335,000 | 41,565,000 | [17] | |
| hff6 | Mi-C | Hg19 | 6 | 41,330,000 | 41,565,000 | 6 | 41,330,000 | 41,565,000 | [17] | |
| GM12987 | Hi-C | HG19 | 6 | 41,390,000 | 41,560,000 | 6 | 41,390,000 | 41,560,000 | [18] | |
| K662 | Hi-C | Hg19 | 6 | 41,430,000 | 41,440,000 | 6 | 41,540,000 | 41,550,000 | [18] | |
| Loop | ||||||||||
| h1ESC | Mi-C | Hg19 | 6 | 41,335,000 | 41,340,000 | 6 | 41,650,000 | 41,655,000 | [17] | |
| K562 | Hi-C | Hg19 | 6 | 41,330,000 | 41,340,000 | 6 | 41,560,000 | 41,570,000 | [18] | |
| K562 | Hi-C | Hg19 | 6 | 41,430,000 | 41,440,000 | 6 | 41,540,000 | 41,550,000 | [18] | |
| GM12978 | Hi-C | Hg19 | 6 | 41,330,000 | 41,565,000 | 6 | 41,330,000 | 41,565,000 | [20] | |
| GM12978 | Hi-C | Hg19 | 6 | 41,430,934 | 41,440,274 | 6 | 41,532,231 | 41,533,943 | [19] | |
| GM12978 | Hi-C | Hg19 | 6 | 41,430,934 | 41,440,274 | 6 | 41,549,065 | 41,559,381 | [19] | |
| IMR90 | Hi-C | Hg19 | 6 | 41,332,022 | 41,692,022 | 6 | 41,332,022 | 41,692,022 | [21] | |
| K562 | CTCF/Chia-pet | Hg19 | 6 | 41,335,405 | 41,335,912 | 6 | 41,430,559 | 41,431,171 | [22] | |
| K562 | CTCF/Chia-pet | Hg19 | 6 | 41,431,132 | 41,431,741 | 6 | 41,651,489 | 41,652,021 | [22] | |
| K562 | Hi-C | Hg19 | 6 | 41,437,792 | 41,441,855 | 6 | 41,442,012 | 41,447,536 | [23] | |
| K562 | Hi-C | Hg19 | 6 | 41,472,346 | 41,533,996 | 6 | 41,480,511 | 41,550,119 | [23] | |
| Peak | ||||||||||
| h1ESC | Mi-C | Hg19 | 6 | 41,465,000 | 41,470,000 | 6 | 41,515,000 | 41,520,000 | [17] | |
| hFF6 | Mi-C | Hg19 | 6 | 41,390,000 | 41,395,000 | 6 | 41,555,000 | 41,560,000 | [17] | |
| Hg19 | |||
|---|---|---|---|
| START Region | END Region | Gene | Gene Start/End |
| chr6:41,480,772–41,483,123 | chr6:41,559,468–41,562,360 | FOXP4/FOXP4-AS1 | chr6:41,514,164–41,570,122/chr6:41,462,591–41,516,359 |
| chr6:41,480,772–41,483,123 | chr6:41,559,468–41,562,360 | FOXP4/FOXP4-AS1 | chr6:41,514,164–41,570,122/chr6:41,462,591–41,516,359 |
| chr6:41,482,545–41,484,504 | chr6:41,491,670–41,493,377 | FOXP4/FOXP4-AS1 | chr6:41,514,164–41,570,122/chr6:41,462,591–41,516,359 |
| chr6:41,482,796–41,484,928 | chr6:41,485,202–41,488,839 | FOXP4/FOXP4-AS1 | chr6:41,514,164–41,570,122/chr6:41,462,591–41,516,359 |
| chr6:41,487,810–41,489,374 | chr6:41,507,169–41,508,917 | FOXP4/FOXP4-AS1 | chr6:41,514,164–41,570,122/chr6:41,462,591–41,516,359 |
| chr6:41,489,724–41,492,551 | chr6:41,509,027–41,511,261 | FOXP4/FOXP4-AS1 | chr6:41,514,164–41,570,122/chr6:41,462,591–41,516,359 |
| chr6:41,482,545–41,484,504 | chr6:41,491,670–41,493,377 | FOXP4/FOXP4-AS1 | chr6:41,514,164–41,570,122/chr6:41,462,591–41,516,359 |
| chr6:41,488,184–41,490,839 | chr6:41,494,174–41,495,933 | FOXP4/FOXP4-AS1 | chr6:41,514,164–41,570,122/chr6:41,462,591–41,516,359 |
| chr6:41,482,796–41,484,928 | chr6:41,485,202–41,488,839 | FOXP4/FOXP4-AS1 | chr6:41,514,164–41,570,122/chr6:41,462,591–41,516,359 |
| chr6:41,482,796–41,484,928 | chr6:41,485,202–41,488,839 | FOXP4/FOXP4-AS1 | chr6:41,514,164–41,570,122/chr6:41,462,591–41,516,359 |
| chr6:41,482,545–41,484,504 | chr6:41,491,670–41,493,377 | FOXP4/FOXP4-AS1 | chr6:41,514,164–41,570,122/chr6:41,462,591–41,516,359 |
| chr6:41,488,184–41,490,839 | chr6:41,494,174–41,495,933 | FOXP4/FOXP4-AS1 | chr6:41,514,164–41,570,122/chr6:41,462,591–41,516,359 |
| chr6:41,478,170–41,480,141 | chr6:41,497,521–41,499,884 | FOXP4/FOXP4-AS1 | chr6:41,514,164–41,570,122/chr6:41,462,591–41,516,359 |
| chr6:41,482,545–41,484,504 | chr6:41,491,670–41,493,377 | FOXP4/FOXP4-AS1 | chr6:41,514,164–41,570,122/chr6:41,462,591–41,516,359 |
| Hg19 | ||||||
|---|---|---|---|---|---|---|
| START Region | Gene | END Region | Gene | Method | Cells | Attached Antibody |
| chr6:41,474,351–41,477,033 | rs9367106/LINC01276 (chr6:41,470,182–41,487,590) | chr:41,484,872–41,500,890 | MED20 (chr6:41,873,092–41,888,877) | Hi-C | IMR90 | NA |
| chr6:41,464,534–41,475,625 | rs9367106/LINC01276 (chr6:41,470,182–41,487,590) | chr:41,484,872–41,488,927 | MED20 (chr6:41,873,092–41,888,877) | Hi-C | IMR90 | NA |
| chr6:41,482,503–41,486,148 | rs9367106/LINC01276 (chr6:41,470,182–41,487,590) | chr6:41,512,979–4,151,495 | MED20 (chr6:41,873,092–41,888,877) | Chia-PET | K562 | POL2RA |
| chr6:41,437,907–41,440,085 | rs9367106/LINC01276 (chr6:41,470,182–41,487,590) | chr6:41,484,931–41,487,417 | MED20 (chr6:41,873,092–41,888,877) | Chia-PET | MCF7 | POL2RA |
| chr6:41,482,503–41,486,148 | rs9367106/LINC01276 (chr6:41,470,182–41,487,590) | chr6:41,512,979–41,514,951 | MED20 (chr6:41,873,092–41,888,877) | Chia-PET | MCF7 | POL2RA |
| chr6:41,472,346–41,533,996 | rs9367106/LINC01276 (chr6:41,470,182–41,487,590) | chr6:41,480,511–41,550,119 | MED20 (chr6:41,873,092–41,888,877) | Chia-PET | K562 | POL2RA |
| Region | Australia | |||
| Ethnicity | No ethnicity reported | |||
| Co-morbidities | Not reported | |||
| Age | Matched (~equivalent) | |||
| Disease severity | N | Male | Female | Clinical symptoms |
| Healthy subjects | 14 | 7 | 7 | no known significant systemic diseases, and negative anti-Spike and anti-RBD serology |
| Mild | 50 | 26 | 24 | COVID-19 disease severity was scored as per CDC descriptors (https://www.cdc.gov/covid/hcp/clinical-care/management-and-treatment.html (accessed on 6 July 2025)) |
| Moderate | 6 | 2 | 4 | “ |
| Severe | 7 | 5 | 2 | “ |
| Critical | 6 | 3 | 3 | “ |
| Total | 83 | 43 | 40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiti, A.K. In Silico Analysis of Post-COVID-19 Condition (PCC) Associated SNP rs9367106 Predicts the Molecular Basis of Abnormalities in the Lungs and Brain Functions. Int. J. Mol. Sci. 2025, 26, 6680. https://doi.org/10.3390/ijms26146680
Maiti AK. In Silico Analysis of Post-COVID-19 Condition (PCC) Associated SNP rs9367106 Predicts the Molecular Basis of Abnormalities in the Lungs and Brain Functions. International Journal of Molecular Sciences. 2025; 26(14):6680. https://doi.org/10.3390/ijms26146680
Chicago/Turabian StyleMaiti, Amit K. 2025. "In Silico Analysis of Post-COVID-19 Condition (PCC) Associated SNP rs9367106 Predicts the Molecular Basis of Abnormalities in the Lungs and Brain Functions" International Journal of Molecular Sciences 26, no. 14: 6680. https://doi.org/10.3390/ijms26146680
APA StyleMaiti, A. K. (2025). In Silico Analysis of Post-COVID-19 Condition (PCC) Associated SNP rs9367106 Predicts the Molecular Basis of Abnormalities in the Lungs and Brain Functions. International Journal of Molecular Sciences, 26(14), 6680. https://doi.org/10.3390/ijms26146680