

PCSK9 Inhibitor Inclisiran Attenuates Cardiotoxicity Induced by Sequential Anthracycline and Trastuzumab Exposure via NLRP3 and MyD88 Pathway Inhibition


 ,
,  ,
,  , ,
, ,  ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
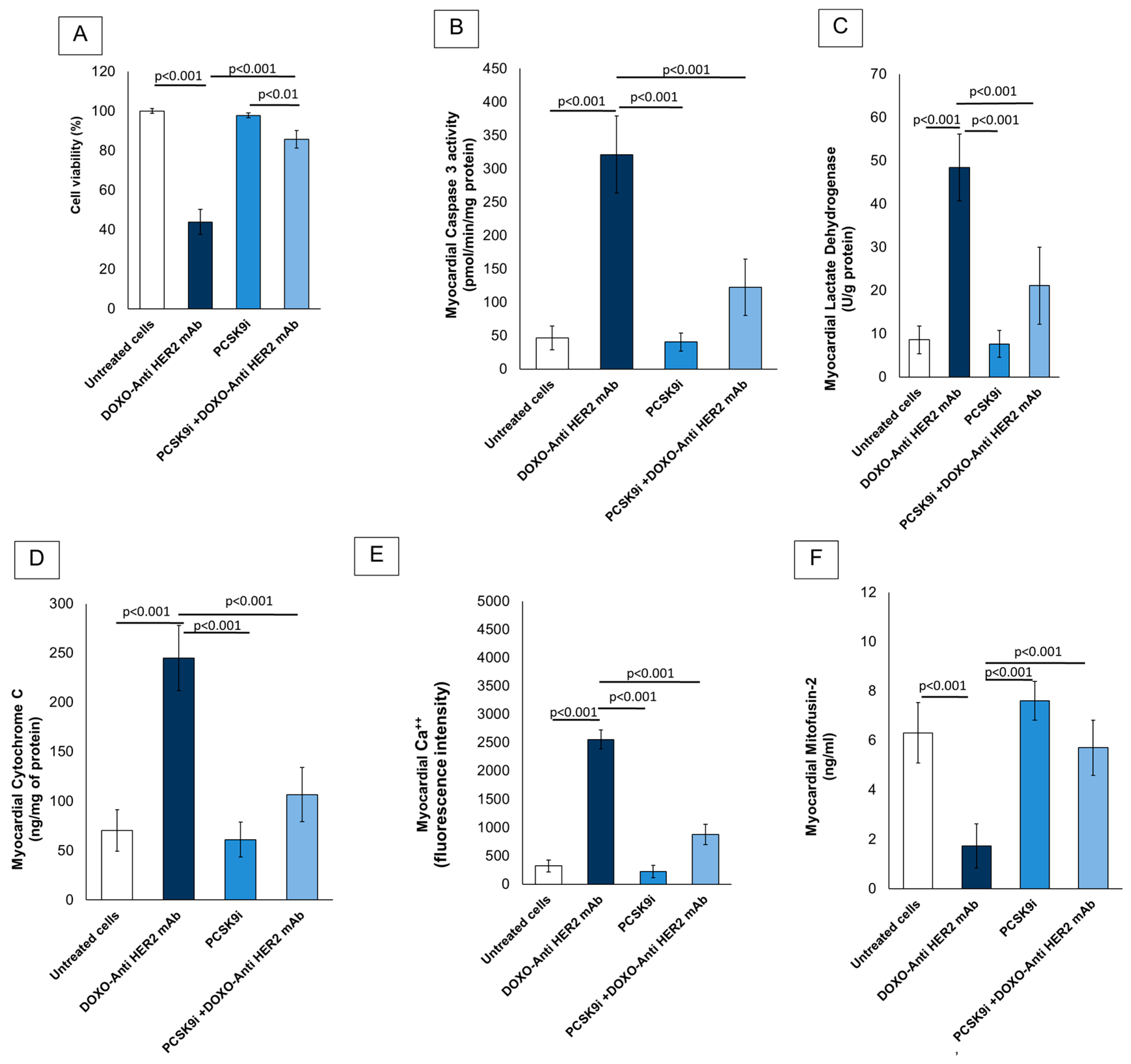
2.1. Cardioprotective Properties of Inclisiran During Anthracyclines and Trastuzumab Therapy in Sequential Regimen
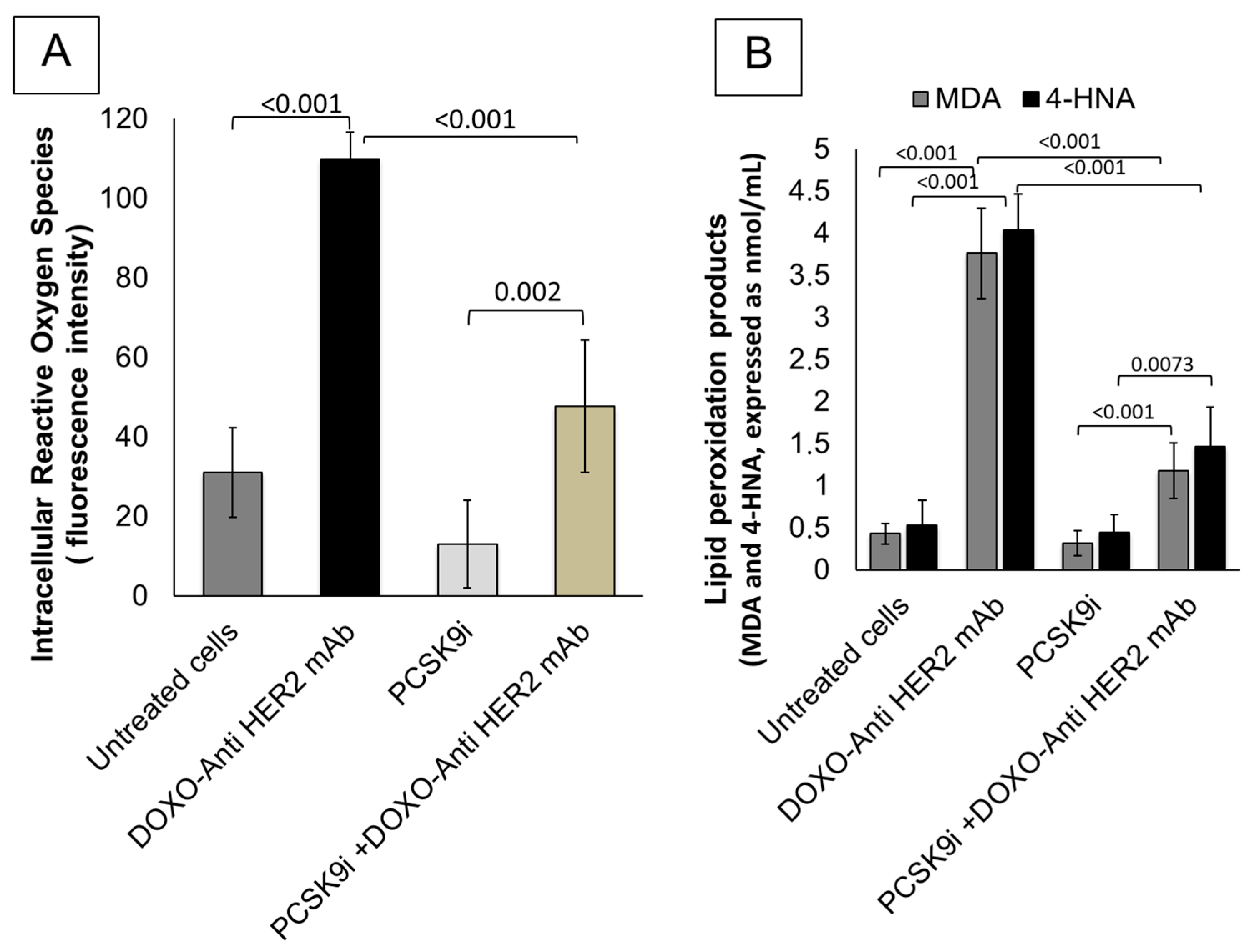
2.2. Effects of Inclisiran on Intracellular Reactive Oxygen Species and Lipid Peroxidation Levels During Exposure to Anthracyclines and Trastuzumab
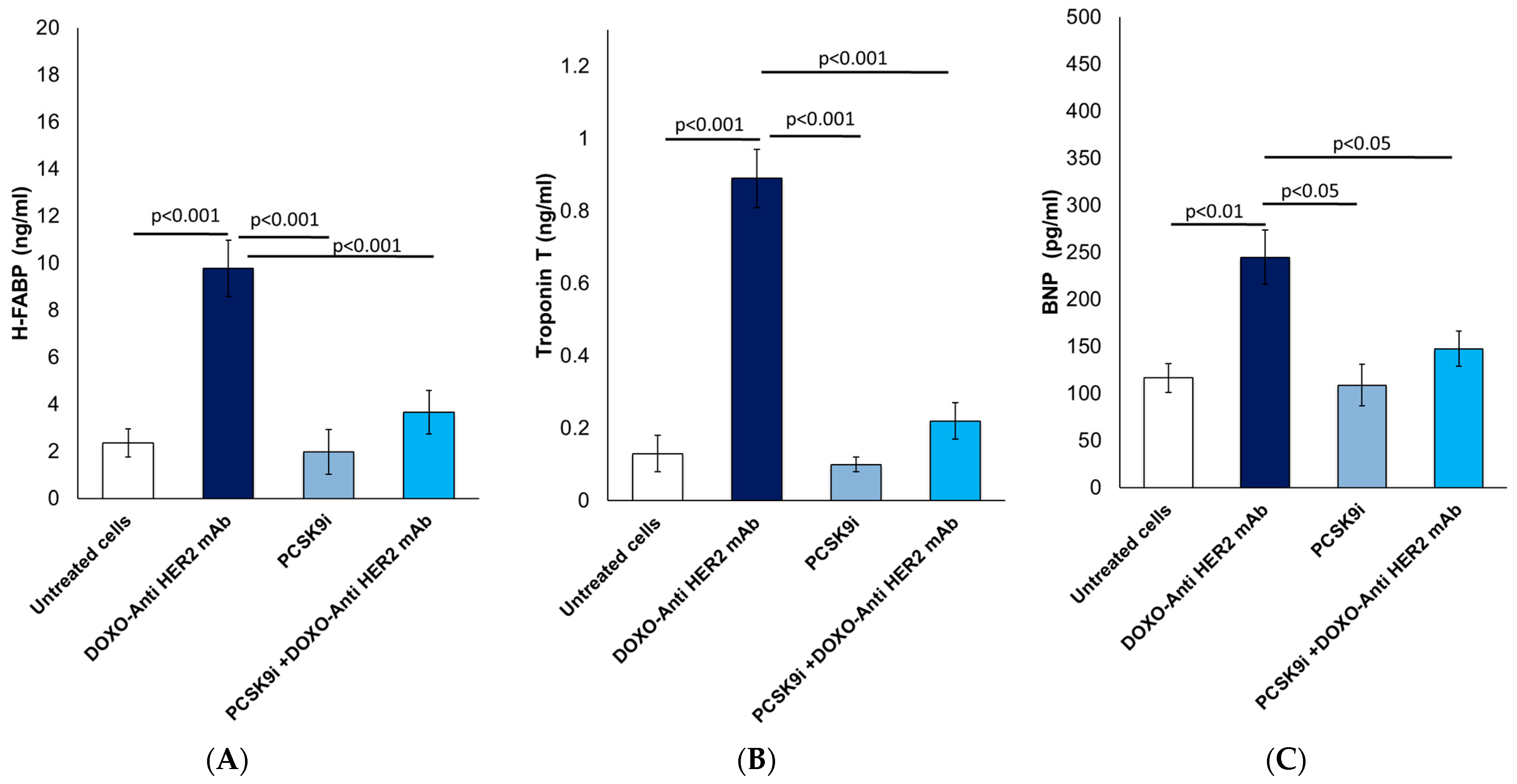
2.3. Biomarkers of Heart Failure: Analysis of H-FABP, Troponin-T and BNP
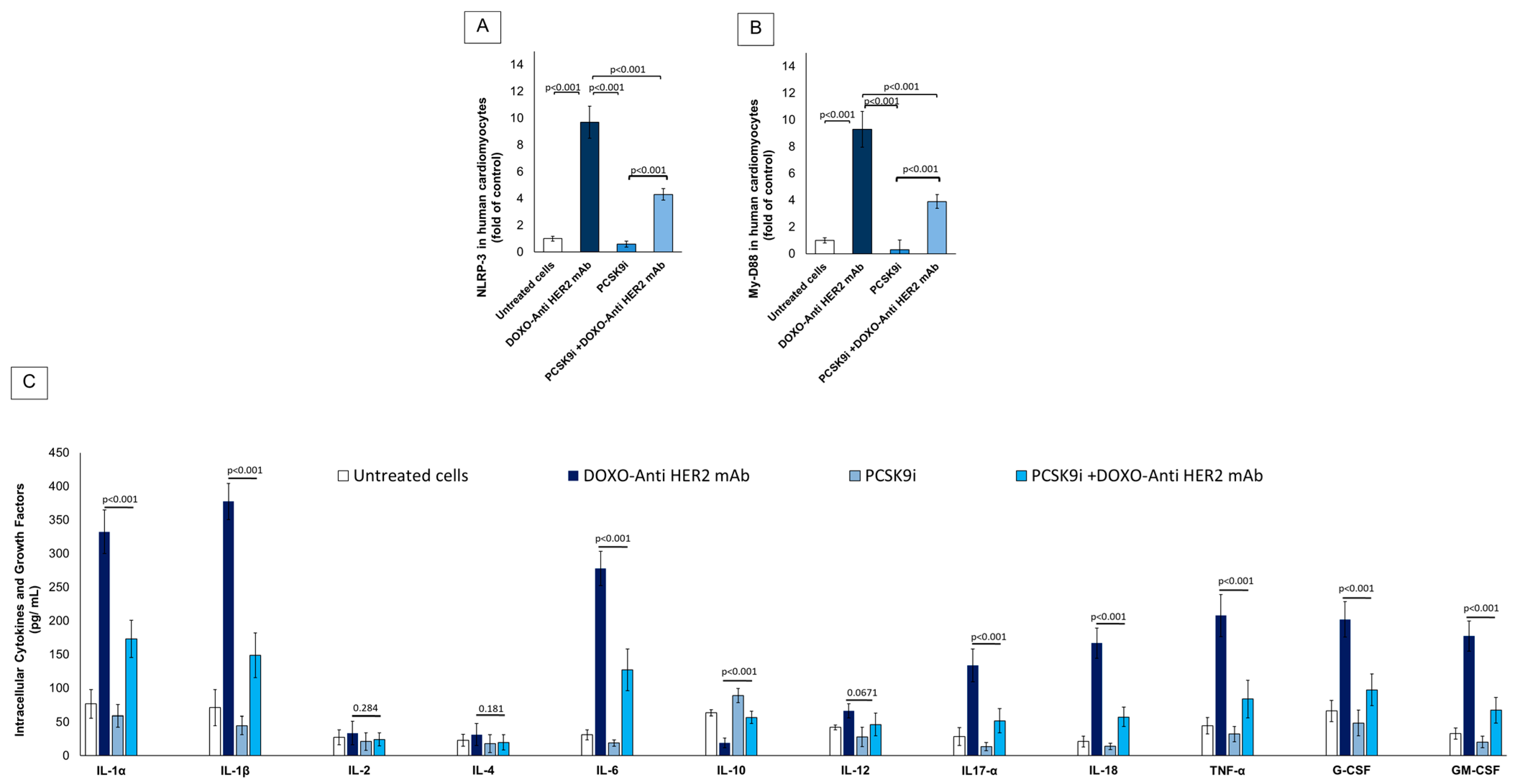
2.4. Effect of Inclisiran on NLRP-3, MyD-88 and Cytokines During Exposure to Anthracyclines and Trastuzumab Therapy in Sequential Regimen
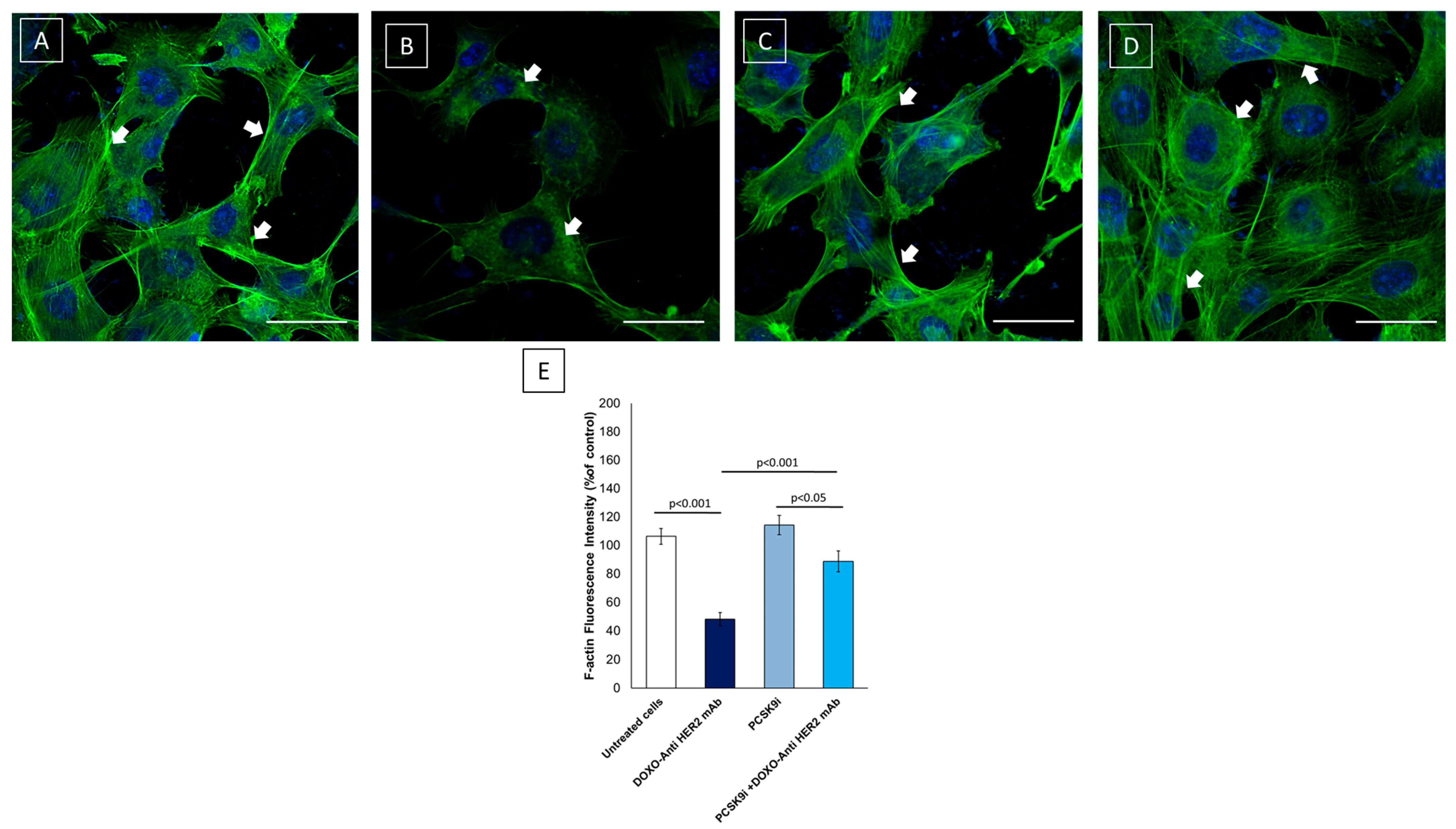
2.5. Confocal Laser Scanning Microscope (CLSM) Analysis of F-Actin Expression and Morphology in Cardiomyocytes
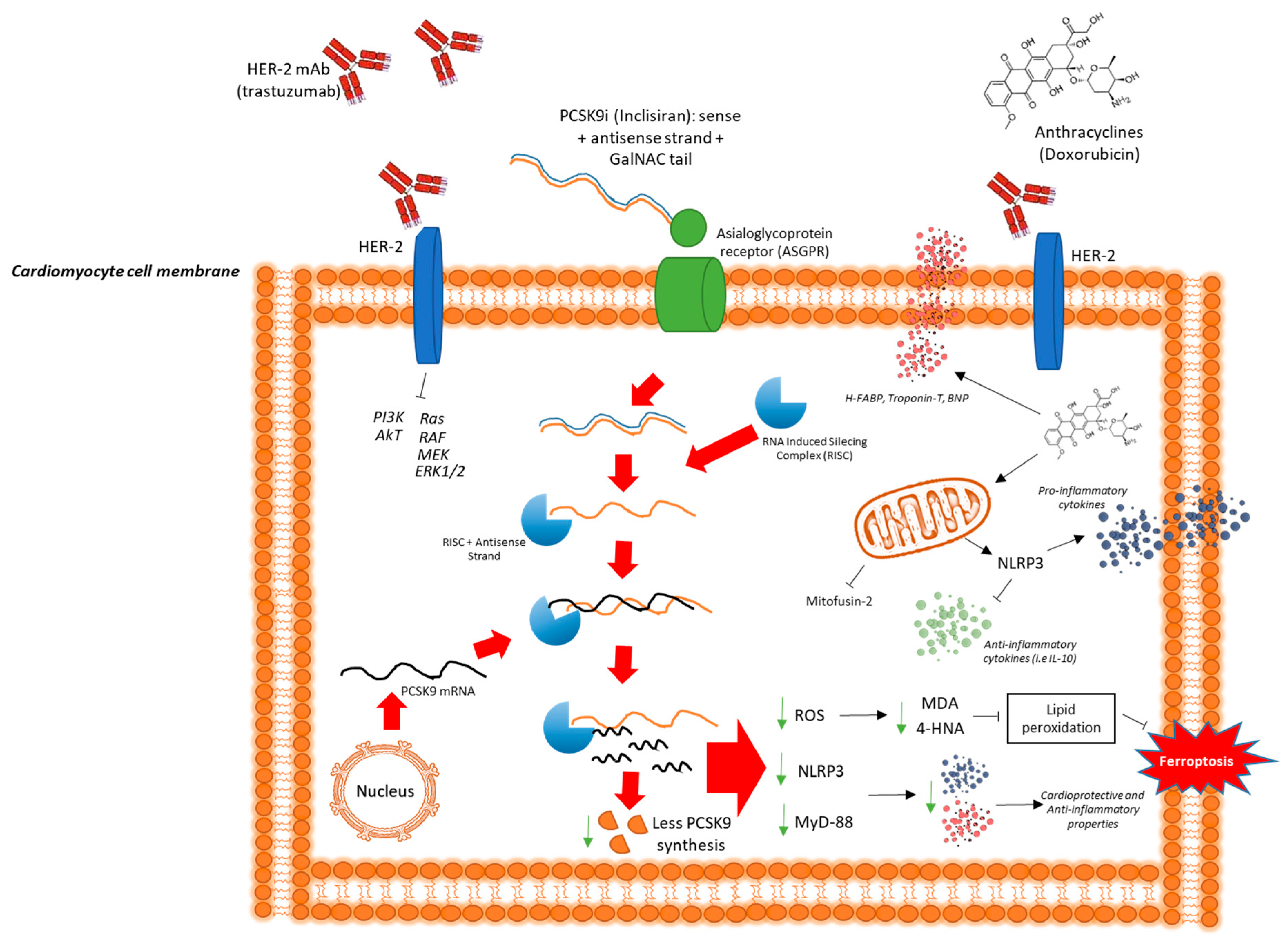
3. Discussion
4. Materials and Methods
- ○
- Control group (untreated cells): cells were kept in DMEM/F12 10% FBS for 24 h;
- ○
- Inclisiran group (PCSK9i): cardiomyocytes were treated for 24 h with inclisiran (200 nM);
- ○
- Doxorubicin + Trastuzumab group (Doxo-Tra): Doxorubicin and trastuzumab sequentially treated cells were treated with doxorubicin (1 µM) for 4 h, and then the medium was replaced with a fresh one, and trastuzumab (200 nM) was added for 20 h.
- ○
- Doxorubicin + Trastuzumab + Inclisiran: cardiomyocytes were treated as specified in Doxo-Tra group but cells were always co-exposed to inclisiran 200 nM).
4.1. Assessment of Cell Survival, Lactate Dehydrogenase, Cytochrome C Release and Caspase-3 Activity During Exposure to Inclisiran Alone or Combined to Anthracyclines and Trastuzumab
4.2. Intracellular Calcium Content and Lipid Peroxidation
4.3. Mitofusin-2 Intracellular Expression
4.4. Biomarkers of Cardiotoxicity: Troponin-T, H-FABP and BNP Quantification
4.5. F-Actin Expression Assessed Using Confocal Laser Scanning Microscope
4.6. Quantification of NLRP-3, MyD-88 Cellular Expression
4.7. Quantitative Profiling of Intracellular Cytokines in Human Cardiomyocytes
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PCSK9i | Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitor |
| HF | Heart Failure |
| BNP | B-type Natriuretic Peptide |
| H-FABP | Heart-type Fatty Acid Binding Protein |
| DOXO | Doxorubicin |
| TRA | Trastuzumab |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| CTRCD | Cancer Therapy-Related Cardiac Dysfunction |
| NLRP3 | NOD-like Receptor Family Pyrin Domain Containing 3 |
| MyD88 | Myeloid Differentiation Primary Response 88 |
| IL | Interleukin |
| TNF-α | Tumor Necrosis Factor Alpha |
| IFN-γ | Interferon Gamma |
| G-CSF | Granulocyte Colony-Stimulating Factor |
| GM-CSF | Granulocyte-Macrophage Colony-Stimulating Factor |
| CCL2 | Chemokine (C-C motif) Ligand 2 (a.k.a. MCP-1) |
| LDH | Lactate Dehydrogenase |
| ATP | Adenosine Triphosphate |
| ROS | Reactive Oxygen Species |
| MDA | Malondialdehyde |
| 4-HNA | 4-Hydroxynonenal |
| OXPHOS | Oxidative Phosphorylation |
| NDUFB8 | NADH:Ubiquinone Oxidoreductase Subunit B8 (Complex I) |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| DAPI | 4′,6-Diamidino-2-Phenylindole (nuclear stain) |
| F-actin | Filamentous Actin |
| CLSM | Confocal Laser Scanning Microscope |
| PI3K | Phosphoinositide 3-Kinase |
| Akt | Protein Kinase B |
| ERK | Extracellular Signal-Regulated Kinase |
| eNOS | Endothelial Nitric Oxide Synthase |
| NO | Nitric Oxide |
| DMEM | Dulbecco’s Modified Eagle Medium |
| FBS | Fetal Bovine Serum |
| PBS | Phosphate Buffered Saline |
| BSA | Bovine Serum Albumin |
| SGLT2i | Sodium–Glucose Cotransporter-2 Inhibitor |
References
- Camilli, M.; Cipolla, C.M.; Dent, S.; Minotti, G.; Cardinale, D.M. Anthracycline Cardiotoxicity in Adult Cancer Patients: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol. 2024, 6, 655–677. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Henriksen, P.A.; Rankin, S.; Lang, N.N. Cardioprotection in Patients at High Risk of Anthracycline-Induced Cardiotoxicity: JACC: CardioOncology Primer. JACC CardioOncol. 2023, 5, 292–297. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Quagliariello, V.; Canale, M.L.; Bisceglia, I.; Iovine, M.; Paccone, A.; Maurea, C.; Scherillo, M.; Merola, A.; Giordano, V.; Palma, G.; et al. Sodium-glucose cotransporter 2 inhibitor dapagliflozin prevents ejection fraction reduction, reduces myocardial and renal NF-κB expression and systemic pro-inflammatory biomarkers in models of short-term doxorubicin cardiotoxicity. Front. Cardiovasc. Med. 2024, 11, 1289663. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dempsey, N.; Rosenthal, A.; Dabas, N.; Kropotova, Y.; Lippman, M.; Bishopric, N.H. Trastuzumab-induced cardiotoxicity: A review of clinical risk factors, pharmacologic prevention, and cardiotoxicity of other HER2-directed therapies. Breast Cancer Res. Treat. 2021, 188, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Keefe, D.L. Trastuzumab-associated cardiotoxicity. Cancer 2002, 95, 1592–1600, Erratum in Cancer 2003, 97, 1136. [Google Scholar] [CrossRef] [PubMed]
- Hamirani, Y.; Fanous, I.; Kramer, C.M.; Wong, A.; Salerno, M.; Dillon, P. Anthracycline- and trastuzumab-induced cardiotoxicity: A retrospective study. Med. Oncol. 2016, 33, 82. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nicolazzi, M.A.; Carnicelli, A.; Fuorlo, M.; Scaldaferri, A.; Masetti, R.; Landolfi, R.; Favuzzi, A.M.R. Anthracycline and trastuzumab-induced cardiotoxicity in breast cancer. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 2175–2185. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E. Inclisiran: A Review in Hypercholesterolemia. Am. J. Cardiovasc. Drugs 2023, 23, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Koren, M.J.; Rodriguez, F.; East, C.; Toth, P.P.; Watwe, V.; Abbas, C.A.; Sarwat, S.; Kleeman, K.; Kumar, B.; Ali, Y.; et al. An “Inclisiran First” Strategy vs. Usual Care in Patients With Atherosclerotic Cardiovascular Disease. J. Am. Coll. Cardiol. 2024, 83, 1939–1952. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.S.; Koenig, W.; Landmesser, U.; Leiter, L.A.; Raal, F.J.; Schwartz, G.G.; Lesogor, A.; Maheux, P.; Stratz, C.; Zang, X.; et al. Safety and Tolerability of Inclisiran for Treatment of Hypercholesterolemia in 7 Clinical Trials. J. Am. Coll. Cardiol. 2023, 82, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Quagliariello, V.; Bisceglia, I.; Berretta, M.; Iovine, M.; Canale, M.L.; Maurea, C.; Giordano, V.; Paccone, A.; Inno, A.; Maurea, N. PCSK9 Inhibitors in Cancer Patients Treated with Immune-Checkpoint Inhibitors to Reduce Cardiovascular Events: New Frontiers in Cardioncology. Cancers 2023, 15, 1397. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Repp, M.L.; Edwards, M.D.; Burch, C.S.; Rao, A.; Chinyere, I.R. PCSK9 Inhibitors and Anthracyclines: The Future of Cardioprotection in Cardio-Oncology. Hearts 2024, 5, 375–388. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ray, K.K.; Troquay, R.P.T.; Visseren, F.L.J.; Leiter, L.A.; Scott Wright, R.; Vikarunnessa, S.; Talloczy, Z.; Zang, X.; Maheux, P.; Lesogor, A.; et al. Long-term efficacy and safety of inclisiran in patients with high cardiovascular risk and elevated LDL cholesterol (ORION-3): Results from the 4-year open-label extension of the ORION-1 trial. Lancet Diabetes Endocrinol. 2023, 11, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yan, B.; Tai, S.; Zhou, S.; Zheng, X.L. PCSK9: Associated with cardiac diseases and their risk factors? Arch. Biochem. Biophys. 2021, 704, 108717. [Google Scholar] [CrossRef] [PubMed]
- Jong, J.; Pinney, J.R.; Packard, R.R.S. Anthracycline-induced cardiotoxicity: From pathobiology to identification of molecular targets for nuclear imaging. Front. Cardiovasc. Med. 2022, 9, 919719. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, S.Y.; Kim, S.J.; Kim, B.J.; Rah, S.Y.; Chung, S.M.; Im, M.J.; Kim, U.H. Doxorubicin-induced reactive oxygen species generation and intracellular Ca2+ increase are reciprocally modulated in rat cardiomyocytes. Exp. Mol. Med. 2006, 38, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Shinlapawittayatorn, K.; Chattipakorn, S.C.; Chattipakorn, N. The effects of doxorubicin on cardiac calcium homeostasis and contractile function. J. Cardiol. 2022, 80, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Chen, J.; Liu, Y.; Fletcher, M.; Jensen, B.C.; Cheng, Z. Doxorubicin induces cardiomyocyte apoptosis and atrophy through cyclin-dependent kinase 2-mediated activation of forkhead box O1. J. Biol. Chem. 2020, 295, 4265–4276. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wei, S.; Ma, W.; Li, X.; Jiang, C.; Sun, T.; Li, Y.; Zhang, B.; Li, W. Involvement of ROS/NLRP3 Inflammasome Signaling Pathway in Doxorubicin-Induced Cardiotoxicity. Cardiovasc. Toxicol. 2020, 20, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Ma, W.; Yang, Y.; Sun, T.; Jiang, C.; Liu, J.; Zhang, B.; Li, W. Trastuzumab potentiates doxorubicin-induced cardiotoxicity via activating the NLRP3 inflammasome in vivo and in vitro. Biochem. Pharmacol. 2023, 214, 115662. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A.G.; Mezzaroma, E.; Toldo, S.; Melendez, G.C.; Franco, R.L.; Lesnefsky, E.J.; Abbate, A.; Hundley, W.G.; Salloum, F.N. NLRP3-mediated inflammation in cardio-oncology: Sterile yet harmful. Transl. Res. 2023, 252, 9–20. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lin, M.; Xiong, W.; Wang, S.; Li, Y.; Hou, C.; Li, C.; Li, G. The Research Progress of Trastuzumab-Induced Cardiotoxicity in HER-2-Positive Breast Cancer Treatment. Front. Cardiovasc. Med. 2022, 8, 821663. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wei, S.; Ma, W.; Xie, S.; Liu, S.; Xie, N.; Li, W.; Zhang, B.; Liu, J. Hyperoside Protects Trastuzumab-Induced Cardiotoxicity via Activating the PI3K/Akt Signaling Pathway. Cardiovasc. Drugs Ther. 2025, 39, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Belger, C.; Abrahams, C.; Imamdin, A.; Lecour, S. Doxorubicin-induced cardiotoxicity and risk factors. Int. J. Cardiol. Heart Vasc. 2023, 50, 101332. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Moilanen, T.; Jokimäki, A.; Tenhunen, O.; Koivunen, J.P. Trastuzumab-induced cardiotoxicity and its risk factors in real-world setting of breast cancer patients. J. Cancer Res. Clin. Oncol. 2018, 144, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Gabani, M.; Castañeda, D.; Nguyen, Q.M.; Choi, S.K.; Chen, C.; Mapara, A.; Kassan, A.; Gonzalez, A.A.; Khataei, T.; Ait-Aissa, K.; et al. Association of Cardiotoxicity With Doxorubicin and Trastuzumab: A Double-Edged Sword in Chemotherapy. Cureus 2021, 13, e18194. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, Y.; Chen, H.; Hong, L.; Wang, H.; Li, B.; Zhang, M.; Li, J.; Yang, L.; Liu, F. Inclisiran: A new generation of lipid-lowering siRNA therapeutic. Front. Pharmacol. 2023, 14, 1260921. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huang, F.; Dai, Q.; Zhou, Y.; Guan, J.; Wu, J.; Dong, Y.; Lv, J. Inclisiran in Cardiovascular Health: A Review of Mechanisms, Efficacy, and Future Prospects. Med. Sci. Monit. 2025, 31, e946439. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Barkas, F.; Ray, K. An update on inclisiran for the treatment of elevated LDL cholesterol. Expert Opin. Pharmacother. 2024, 25, 349–358. [Google Scholar] [CrossRef] [PubMed]
- German, C.A.; Shapiro, M.D. Small Interfering RNA Therapeutic Inclisiran: A New Approach to Targeting PCSK9. BioDrugs 2020, 34, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dec, A.; Niemiec, A.; Wojciechowska, E.; Maligłówka, M.; Bułdak, Ł.; Bołdys, A.; Okopień, B. Inclisiran-A Revolutionary Addition to a Cholesterol-Lowering Therapy. Int. J. Mol. Sci. 2023, 24, 6858. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rogula, S.; Błażejowska, E.; Gąsecka, A.; Szarpak, Ł.; Jaguszewski, M.J.; Mazurek, T.; Filipiak, K.J. Inclisiran-Silencing the Cholesterol, Speaking up the Prognosis. J. Clin. Med. 2021, 10, 2467. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mohamed, A.A.; Ray, K.K. Inclisiran and cardiovascular events: A comprehensive review of efficacy, safety, and future perspectives. Curr. Opin. Cardiol. 2023, 38, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Raal, F.J.; Kallend, D.G.; Jaros, M.J.; Koenig, W.; Leiter, L.A.; Landmesser, U.; Schwartz, G.G.; Lawrence, D.; Friedman, A.; et al. Inclisiran and cardiovascular events: A patient-level analysis of phase III trials. Eur. Heart J. 2023, 44, 129–138. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, Z.; Zhang, S.; Xiao, Y.; Zhang, W.; Wu, S.; Qin, T.; Yue, Y.; Qian, W.; Li, L. NLRP3 Inflammasome and Inflammatory Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 4063562. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bracey, N.A.; Beck, P.L.; Muruve, D.A.; Hirota, S.A.; Guo, J.; Jabagi, H.; Wright JRJr Macdonald, J.A.; Lees-Miller, J.P.; Roach, D.; Semeniuk, L.M.; et al. The Nlrp3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1β. Exp. Physiol. 2013, 98, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Khair, M.; Khair, M.; Vangaveti, V.N.; Malabu, U.H. The role of the NLRP3 inflammasome in atherosclerotic disease: Systematic review and meta-analysis. J. Cardiol. 2024, 84, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, X.; Lei, W.; Hou, Y.; Zhang, Y.; Tang, R.; Yang, Z.; Tian, Y.; Zhu, Y.; Wang, C.; et al. The NLRP3 inflammasome in fibrosis and aging: The known unknowns. Ageing Res. Rev. 2022, 79, 101638. [Google Scholar] [CrossRef] [PubMed]
- Artlett, C.M. The Mechanism and Regulation of the NLRP3 Inflammasome during Fibrosis. Biomolecules 2022, 12, 634. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bayer, A.L.; Smolgovsky, S.; Ngwenyama, N.; Hernández-Martínez, A.; Kaur, K.; Sulka, K.; Amrute, J.; Aronovitz, M.; Lavine, K.; Sharma, S.; et al. T-Cell MyD88 Is a Novel Regulator of Cardiac Fibrosis Through Modulation of T-Cell Activation. Circ. Res. 2023, 133, 412–429. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xie, Y.; Yu, H.; Ye, Y.; Wang, J.; Yang, Z.; Zhou, E. Activation of Ferroptosis and NF-κB/NLRP3/MAPK Pathways in Methylmercury-Induced Hepatotoxicity. Toxicol. Ind. Health 2025, 41, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, B.; Chen, W.; Li, W.; Yang, H.; Pan, D. Effects of Inclisiran, Alirocumab, Evolocumab, and Evinacumab on Lipids: A Network Meta-Analysis. Rev. Cardiovasc. Med. 2025, 26, 25248. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Quagliariello, V.; Bonelli, A.; Caronna, A.; Lombari, M.C.; Conforti, G.; Libutti, M.; Iaffaioli, R.V.; Berretta, M.; Botti, G.; Maurea, N. SARS-CoV-2 infection: NLRP3 inflammasome as plausible target to prevent cardiopulmonary complications? Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9169–9171. [Google Scholar] [CrossRef] [PubMed]
- Quagliariello, V.; De Laurentiis, M.; Cocco, S.; Rea, G.; Bonelli, A.; Caronna, A.; Lombari, M.C.; Conforti, G.; Berretta, M.; Botti, G.; et al. NLRP3 as Putative Marker of Ipilimumab-Induced Cardiotoxicity in the Presence of Hyperglycemia in Estrogen-Responsive and Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 7802. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Steffens, D.; Bramlage, P.; Scheeff, C.; Kasner, M.; Hassanein, A.; Friebel, J.; Rauch-Kröhnert, U. PCSK9 inhibitors and cardiovascular outcomes. Expert Opin. Biol. Ther. 2020, 20, 35–47. [Google Scholar] [CrossRef] [PubMed]
- McClintick, D.J.; Giugliano, R.P. Ups and downs in PCSK9 inhibition in the cardiovascular arena: A review. Curr. Opin. Lipidol. 2023, 34, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Chen, Y.; Huang, Y.; Sun, X.; Shi, X. The role of cardiac microenvironment in cardiovascular diseases: Implications for therapy. Hum. Cell 2024, 37, 607–624. [Google Scholar] [CrossRef] [PubMed]
- Vo, J.B.; Ramin, C.; Veiga, L.H.S.; Brandt, C.; Curtis, R.E.; Bodelon, C.; Barac, A.; Roger, V.L.; Feigelson, H.S.; Buist, D.S.M.; et al. Long-term cardiovascular disease risk after anthracycline and trastuzumab treatments in US breast cancer survivors. J. Natl. Cancer Inst. 2024, 116, 1384–1394. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Blanter, J.B.; Frishman, W.H. The Preventive Role of Angiotensin Converting Enzyme Inhibitors/Angiotensin-II Receptor Blockers and β-Adrenergic Blockers in Anthracycline- and Trastuzumab-Induced Cardiotoxicity. Cardiol. Rev. 2019, 27, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Avula, V.; Sharma, G.; Kosiborod, M.N.; Vaduganathan, M.; Neilan, T.G.; Lopez, T.; Dent, S.; Baldassarre, L.; Scherrer-Crosbie, M.; Barac, A.; et al. SGLT2 Inhibitor Use and Risk of Clinical Events in Patients With Cancer Therapy-Related Cardiac Dysfunction. JACC Heart Fail. 2024, 12, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, M.; Marzocco, S.; Franceschelli, S.; Popolo, A. Trastuzumab and Doxorubicin Sequential Administration Increases Oxidative Stress and Phosphorylation of Connexin 43 on Ser368. Int. J. Mol. Sci. 2022, 23, 6375. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Andreata, F.; Bonizzi, A.; Sevieri, M.; Truffi, M.; Monieri, M.; Sitia, L.; Silva, F.; Sorrentino, L.; Allevi, R.; Zerbi, P.; et al. Co-administration of H-ferritin-doxorubicin and Trastuzumab in neoadjuvant setting improves efficacy and prevents cardiotoxicity in HER2 + murine breast cancer model. Sci. Rep. 2020, 10, 11425. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Qiu, Q.; Shen, T.; Wang, Q.; Yu, X.; Jia, N.; He, Q. Cardiac shock wave therapy protects cardiomyocytes from hypoxia-induced injury by modulating miR-210. Mol. Med. Rep. 2020, 21, 631–640. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liao, B.; Tian, X. CTRP12 alleviates cardiomyocyte ischemia-reperfusion injury via regulation of KLF15. Mol. Med. Rep. 2022, 26, 247. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tan, X.; Wang, D.B.; Lu, X.; Wei, H.; Zhu, R.; Zhu, S.S.; Jiang, H.; Yang, Z.J. Doxorubicin induces apoptosis in H9c2 cardiomyocytes: Role of overexpressed eukaryotic translation initiation factor 5A. Biol. Pharm. Bull. 2010, 33, 1666–1672. [Google Scholar] [CrossRef] [PubMed]
- Quagliariello, V.; Berretta, M.; Bisceglia, I.; Giacobbe, I.; Iovine, M.; Giordano, V.; Arianna, R.; Barbato, M.; Izzo, F.; Maurea, C.; et al. The sGCa Vericiguat Exhibit Cardioprotective and Anti-Sarcopenic Effects through NLRP-3 Pathways: Potential Benefits for Anthracycline-Treated Cancer Patients. Cancers 2024, 16, 1487. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, Y.; Chen, Y.; Zhang, M.; Tang, Y.; Xie, Y.; Huang, X.; Li, Y. Doxorubicin induces sarcoplasmic reticulum calcium regulation dysfunction via the decrease of SERCA2 and phospholamban expressions in rats. Cell Biochem. Biophys. 2014, 70, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Vitale, R.; Marzocco, S.; Popolo, A. Role of Oxidative Stress and Inflammation in Doxorubicin-Induced Cardiotoxicity: A Brief Account. Int. J. Mol. Sci. 2024, 25, 7477. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tang, H.; Tao, A.; Song, J.; Liu, Q.; Wang, H.; Rui, T. Doxorubicin-induced cardiomyocyte apoptosis: Role of mitofusin 2. Int. J. Biochem. Cell Biol. 2017, 88, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Parry, T.L.; Brown, D.I.; Mota, R.I.; Huang, W.; Beak, J.Y.; Sola, M.; Zhou, C.; Hicks, S.T.; Caughey, M.C.; et al. Doxorubicin Exposure Causes Subacute Cardiac Atrophy Dependent on the Striated Muscle-Specific Ubiquitin Ligase MuRF1. Circ. Heart Fail. 2019, 12, e005234. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shi, S.; Qin, Z.; Liu, C.; Zhao, Y.; Bai, X.; Sun, C.; Li, X.; Cong, W.; Yuan, X.; Sun, L.; et al. The function of PCSK9 in doxorubicin-induced cardiotoxicity and its underlying mechanism. Sci. Rep. 2025, 15, 22067. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Godinho, R.; Noto, A.; Fenwick, C.; Stravodimou, A.; Hugelshofer, S.; Peters, S.; Hullin, R.; Obeid, M. Cytokine storm complicated by cardiogenic shock induced by anti-HER2 therapies. J. Immunother. Cancer 2023, 11, e006942. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cortés-Ríos, J.; Zárate, A.M.; Figueroa, J.D.; Medina, J.; Fuentes-Lemus, E.; Rodríguez-Fernández, M.; Aliaga, M.; López-Alarcón, C. Protein quantification by bicinchoninic acid (BCA) assay follows complex kinetics and can be performed at short incubation times. Anal. Biochem. 2020, 608, 113904. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quagliariello, V.; Berretta, M.; Bisceglia, I.; Iovine, M.; Barbato, M.; Arianna, R.; Canale, M.L.; Paccone, A.; Inno, A.; Scherillo, M.; et al. PCSK9 Inhibitor Inclisiran Attenuates Cardiotoxicity Induced by Sequential Anthracycline and Trastuzumab Exposure via NLRP3 and MyD88 Pathway Inhibition. Int. J. Mol. Sci. 2025, 26, 6617. https://doi.org/10.3390/ijms26146617
Quagliariello V, Berretta M, Bisceglia I, Iovine M, Barbato M, Arianna R, Canale ML, Paccone A, Inno A, Scherillo M, et al. PCSK9 Inhibitor Inclisiran Attenuates Cardiotoxicity Induced by Sequential Anthracycline and Trastuzumab Exposure via NLRP3 and MyD88 Pathway Inhibition. International Journal of Molecular Sciences. 2025; 26(14):6617. https://doi.org/10.3390/ijms26146617
Chicago/Turabian StyleQuagliariello, Vincenzo, Massimiliano Berretta, Irma Bisceglia, Martina Iovine, Matteo Barbato, Raffaele Arianna, Maria Laura Canale, Andrea Paccone, Alessandro Inno, Marino Scherillo, and et al. 2025. "PCSK9 Inhibitor Inclisiran Attenuates Cardiotoxicity Induced by Sequential Anthracycline and Trastuzumab Exposure via NLRP3 and MyD88 Pathway Inhibition" International Journal of Molecular Sciences 26, no. 14: 6617. https://doi.org/10.3390/ijms26146617
APA StyleQuagliariello, V., Berretta, M., Bisceglia, I., Iovine, M., Barbato, M., Arianna, R., Canale, M. L., Paccone, A., Inno, A., Scherillo, M., Oliva, S., Cadeddu Dessalvi, C., Mauriello, A., Maurea, C., Fonderico, C., Maratea, A. C., Gabrielli, D., & Maurea, N. (2025). PCSK9 Inhibitor Inclisiran Attenuates Cardiotoxicity Induced by Sequential Anthracycline and Trastuzumab Exposure via NLRP3 and MyD88 Pathway Inhibition. International Journal of Molecular Sciences, 26(14), 6617. https://doi.org/10.3390/ijms26146617