
The Role of Monocytes in the Natural History of Idiopathic Pulmonary Fibrosis: A Systematic Literature Review

 , ,
, ,
Abstract
1. Introduction
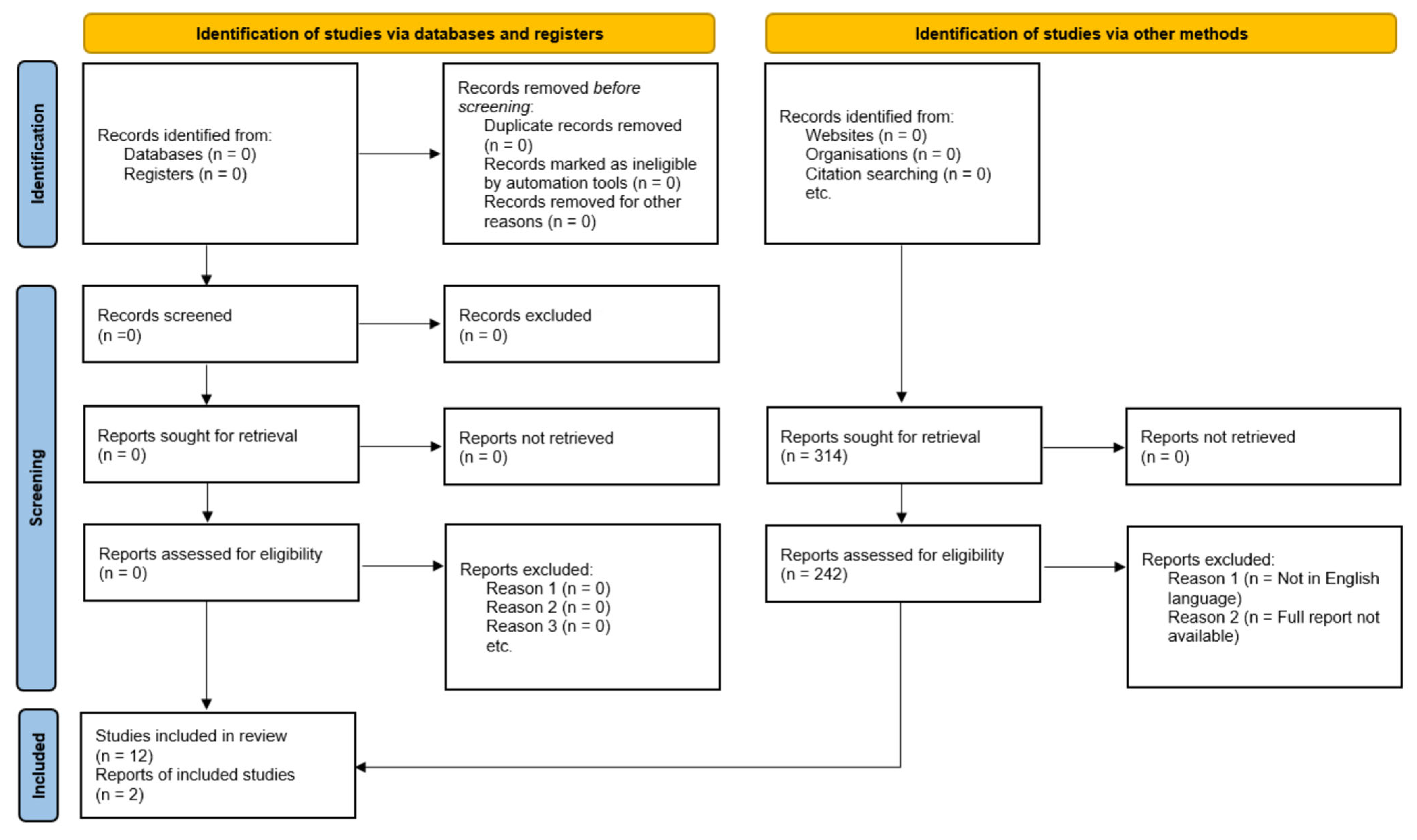
2. Methods
3. Results
Studies and Clinical Trials Associate Monocytes with ILD Diagnosis, Progression and Activity
4. Discussion
4.1. Immunophenotypes and Cytokines of IPF Monocytes
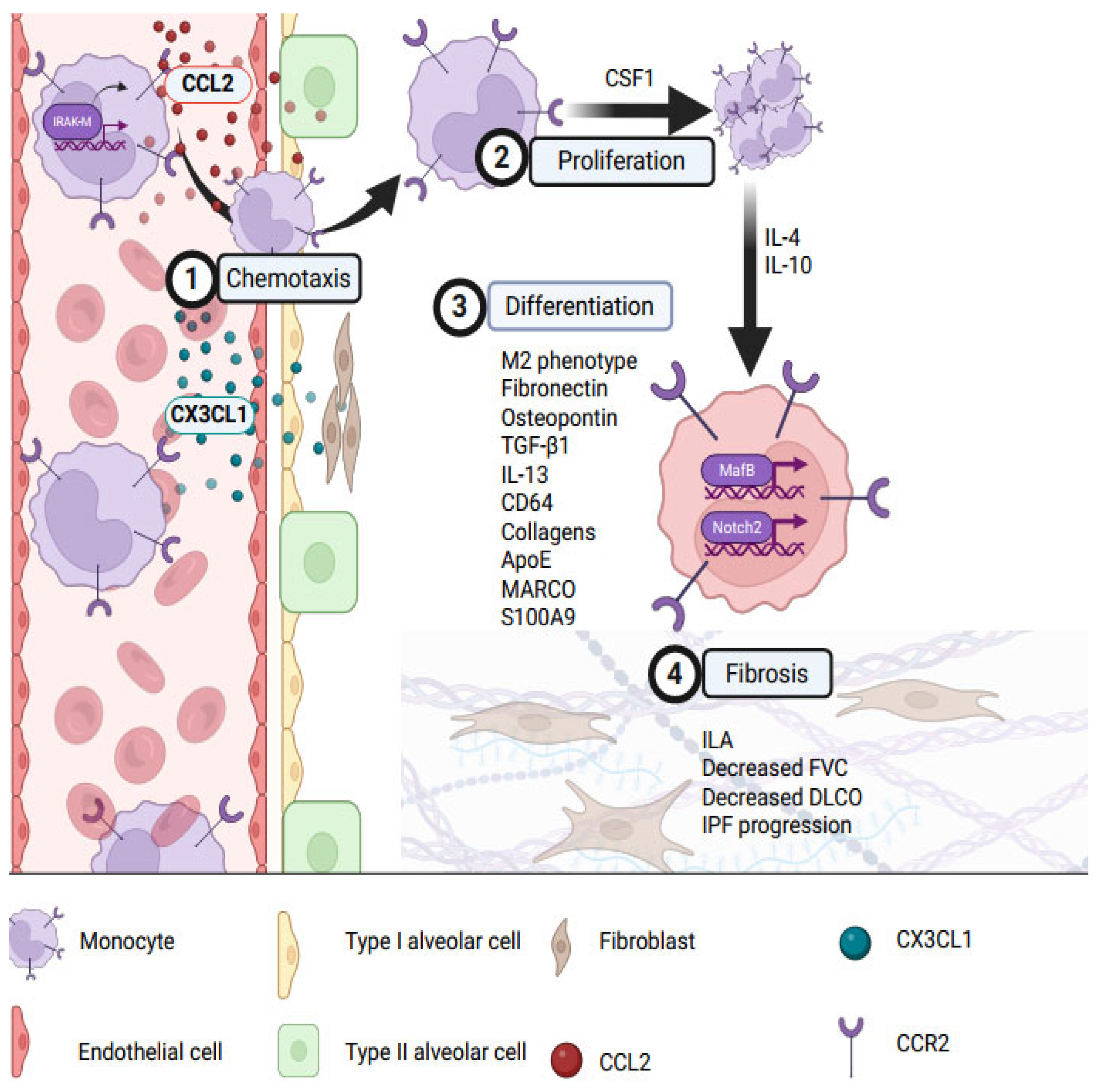
4.2. Monocyte Migration to IPF Lungs and Differentiation into Pro-Fibrotic Macrophages
4.3. Monocytes Induce Local Pulmonary Fibrosis
4.4. Progression from Pre-IPF to IPF: Role of the Monocyte
4.5. Potential Therapeutic Approaches
4.6. Role of Monocytes in Connective Tissue Disease ILD
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ApoE | apolipoprotein E |
| BLM-IPF | bleomycin-induced idiopathic pulmonary fibrosis |
| CSF1R | colony-stimulating factor 1 receptor |
| CT-ILD | connective tissue interstitial lung disease |
| DLCO | diffusing lung capacity for carbon monoxide |
| ERK | extracellular signal-regulated kinase |
| FVC | forced vital capacity |
| GM-CSF | granulocyte-monocyte colony stimulating factor |
| IIP | idiopathic interstitial pneumonia |
| ILD | interstitial lung disease |
| IPF | idiopathic pulmonary fibrosis |
| IRAK-M | interleukin-1 receptor-associated kinase |
| MDSC | monocyte-derived suppressor cell |
| Mo-AM | monocyte-derived alveolar macrophage |
| NSIP | non-specific interstitial pneumonia |
| RA-ILD | rheumatoid arthritis-related interstitial lung disease |
| ROS | reactive oxygen species |
| SP-D | surfactant protein D |
| TGF-β1 | transforming growth factor beta 1 |
| TIMP1 | tissue inhibitor of metalloproteinase 1 |
| TNF-α | tumor necrosis factor alpha |
| TR-AM | tissue resident alveolar macrophages |
| TREM | triggering receptors expressed on myeloid cells |
| UIP | usual interstitial pneumonia |
References
- Wakwaya, Y.; Brown, K.K. Idiopathic Pulmonary Fibrosis: Epidemiology, Diagnosis and Outcomes. Am. J. Med. Sci. 2019, 357, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Moss, B.J.; Ryter, S.W.; Rosas, I.O. Pathogenic Mechanisms Underlying Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. 2022, 17, 515–546. [Google Scholar] [CrossRef]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef]
- Abe, S.; Boyer, C.; Liu, X.; Wen, F.Q.; Kobayashi, T.; Fang, Q.; Wang, X.; Hashimoto, M.; Sharp, J.G.; Rennard, S.I. Cells derived from the circulation contribute to the repair of lung injury. Am. J. Respir. Crit. Care Med. 2004, 170, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.; Watanabe, S.; Verma, R.; Jablonski, R.P.; Chen, C.I.; Cheresh, P.; Markov, N.S.; Reyfman, P.A.; McQuattie-Pimentel, A.C.; Sichizya, L.; et al. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages. Eur. Respir. J. 2020, 55, 1900646. [Google Scholar] [CrossRef]
- Trzebanski, S.; Jung, S. Plasticity of monocyte development and monocyte fates. Immunol. Lett. 2020, 227, 66–78. [Google Scholar] [CrossRef]
- Guilliams, M.; Mildner, A.; Yona, S. Developmental and Functional Heterogeneity of Monocytes. Immunity 2018, 49, 595–613. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, P.B.; Marcovecchio, P.; Hamers, A.A.J.; Hedrick, C.C. Nonclassical Monocytes in Health and Disease. Annu. Rev. Immunol. 2019, 37, 439–456. [Google Scholar] [CrossRef]
- Ożańska, A.; Szymczak, D.; Rybka, J. Pattern of human monocyte subpopulations in health and disease. Scand. J. Immunol. 2020, 92, e12883. [Google Scholar] [CrossRef]
- Aegerter, H.; Lambrecht, B.N.; Jakubzick, C.V. Biology of lung macrophages in health and disease. Immunity 2022, 55, 1564–1580. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Evren, E.; Ringqvist, E.; Doisne, J.M.; Thaller, A.; Sleiers, N.; Flavell, R.A.; Di Santo, J.P.; Willinger, T. CD116+ fetal precursors migrate to the perinatal lung and give rise to human alveolar macrophages. J. Exp. Med. 2022, 219, e20210987. [Google Scholar] [CrossRef] [PubMed]
- Gibbings, S.L.; Thomas, S.M.; Atif, S.M.; McCubbrey, A.L.; Desch, A.N.; Danhorn, T.; Leach, S.M.; Bratton, D.L.; Henson, P.M.; Janssen, W.J.; et al. Three Unique Interstitial Macrophages in the Murine Lung at Steady State. Am. J. Respir. Cell Mol. Biol. 2017, 57, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.; Wang, Y.; Eubank, T.; Baran, C.; Nana-Sinkam, P.; Marsh, C. Survival of monocytes and macrophages and their role in health and disease. Front. Biosci. 2009, 14, 4079–4102. [Google Scholar] [CrossRef]
- McQuattie-Pimentel, A.C.; Budinger, G.R.S.; Ballinger, M.N. Monocyte-derived Alveolar Macrophages: The Dark Side of Lung Repair? Am. J. Respir. Cell Mol. Biol. 2018, 58, 5–6. [Google Scholar] [CrossRef]
- Hoogsteden, H.C.; van Dongen, J.J.; van Hal, P.T.; Delahaye, M.; Hop, W.; Hilvering, C. Phenotype of blood monocytes and alveolar macrophages in interstitial lung disease. Chest 1989, 95, 574–577. [Google Scholar] [CrossRef]
- Kim, J.S.; Axelsson, G.T.; Moll, M.; Anderson, M.R.; Bernstein, E.J.; Putman, R.K.; Hida, T.; Hatabu, H.; Hoffman, E.A.; Raghu, G.; et al. Associations of Monocyte Count and Other Immune Cell Types with Interstitial Lung Abnormalities. Am. J. Respir. Crit. Care Med. 2022, 205, 795–805. [Google Scholar] [CrossRef]
- Achaiah, A.; Rathnapala, A.; Pereira, A.; Bothwell, H.; Dwivedi, K.; Barker, R.; Benamore, R.; Hoyles, R.K.; Iotchkova, V.; Ho, L.P. Monocyte and neutrophil levels are potentially linked to progression to IPF for patients with indeterminate UIP CT pattern. BMJ Open Respir. Res. 2021, 8, e000899. [Google Scholar] [CrossRef]
- Scott, M.K.D.; Quinn, K.; Li, Q.; Carroll, R.; Warsinske, H.; Vallania, F.; Chen, S.; Carns, M.A.; Aren, K.; Sun, J.; et al. Increased monocyte count as a cellular biomarker for poor outcomes in fibrotic diseases: A retrospective, multicentre cohort study. Lancet Respir. Med. 2019, 7, 497–508. [Google Scholar] [CrossRef]
- Liu, Y.Z.; Saito, S.; Morris, G.F.; Miller, C.A., 3rd; Li, J.; Lefante, J.J. Proportions of resting memory T cells and monocytes in blood have prognostic significance in idiopathic pulmonary fibrosis. Genomics 2019, 111, 1343–1350. [Google Scholar] [CrossRef]
- Teoh, A.K.Y.; Jo, H.E.; Chambers, D.C.; Symons, K.; Walters, E.H.; Goh, N.S.; Glaspole, I.; Cooper, W.; Reynolds, P.; Moodley, Y.; et al. Blood monocyte counts as a potential prognostic marker for idiopathic pulmonary fibrosis: Analysis from the Australian IPF registry. Eur. Respir. J. 2020, 55, 1901855. [Google Scholar] [CrossRef]
- Kreuter, M.; Lee, J.S.; Tzouvelekis, A.; Oldham, J.M.; Molyneaux, P.L.; Weycker, D.; Atwood, M.; Kirchgaessler, K.U.; Maher, T.M. Monocyte Count as a Prognostic Biomarker in Patients with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 204, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Bernardinello, N.; Grisostomi, G.; Cocconcelli, E.; Castelli, G.; Petrarulo, S.; Biondini, D.; Saetta, M.; Spagnolo, P.; Balestro, E. The clinical relevance of lymphocyte to monocyte ratio in patients with Idiopathic Pulmonary Fibrosis (IPF). Respir. Med. 2022, 191, 106686. [Google Scholar] [CrossRef]
- Barratt, S.L.; Creamer, A.W.; Adamali, H.I.; Duckworth, A.; Fallon, J.; Fidan, S.; Nancarrow, T.; Wollerton, R.; Steward, M.; Gooptu, B.; et al. Use of peripheral neutrophil to lymphocyte ratio and peripheral monocyte levels to predict survival in fibrotic hypersensitivity pneumonitis (fHP): A multicentre retrospective cohort study. BMJ Open Respir. Res. 2021, 8, e001063. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Ichikado, K.; Anan, K.; Yasuda, Y.; Sekido, Y.; Suga, M.; Ichiyasu, H.; Sakagami, T. Monocyte count and the risk for acute exacerbation of fibrosing interstitial lung disease: A retrospective cohort study. Chronic Respir. Dis. 2020, 17, 1479973120909840. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Kass, D.J. Do Circulating Monocytes Promote and Predict Idiopathic Pulmonary Fibrosis Progression? Am. J. Respir. Crit. Care Med. 2021, 204, 9–11. [Google Scholar] [CrossRef]
- Yamashita, M.; Utsumi, Y.; Nagashima, H.; Nitanai, H.; Yamauchi, K. S100A9/CD163 expression profiles in classical monocytes as biomarkers to discriminate idiopathic pulmonary fibrosis from idiopathic nonspecific interstitial pneumonia. Sci. Rep. 2021, 11, 12135. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, P.; Morzadec, C.; de Latour, B.; Llamas-Gutierrez, F.; Luque-Paz, D.; Jouneau, S.; Vernhet, L. Soluble CD163 is produced by monocyte-derived and alveolar macrophages, and is not associated with the severity of idiopathic pulmonary fibrosis. Innate Immun. 2022, 28, 138–151. [Google Scholar] [CrossRef]
- Kapellos, T.S.; Bonaguro, L.; Gemünd, I.; Reusch, N.; Saglam, A.; Hinkley, E.R.; Schultze, J.L. Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases. Front. Immunol. 2019, 10, 2035. [Google Scholar] [CrossRef]
- Chen, S.; Saeed, A.F.U.H.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef]
- Xiang, G.A.; Zhang, Y.D.; Su, C.C.; Ma, Y.Q.; Li, Y.M.; Zhou, X.; Wei, L.Q.; Ji, W.J. Dynamic changes of mononuclear phagocytes in circulating, pulmonary alveolar and interstitial compartments in a mouse model of experimental silicosis. Inhal. Toxicol. 2016, 28, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Pechkovsky, D.V.; Prasse, A.; Kollert, F.; Engel, K.M.; Dentler, J.; Luttmann, W.; Friedrich, K.; Müller-Quernheim, J.; Zissel, G. Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin. Immunol. 2010, 137, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yang, K.; Bai, J.; Yi, J.; Gao, C.; Zhao, J.; Liang, S.; Wei, T.; Feng, L.; Song, L.; et al. Myeloid-specific blockade of Notch signaling alleviates murine pulmonary fibrosis through regulating monocyte-derived Ly6c(lo) MHCII(hi) alveolar macrophages recruitment and TGF-β secretion. FASEB J. 2020, 34, 11168–11184. [Google Scholar] [CrossRef]
- To, S.; Chavula, T.; Pedroza, M.; Smith, J.; Agarwal, S.K. Cadherin-11 Regulates Macrophage Development and Function. Front. Immunol. 2022, 13, 795337. [Google Scholar] [CrossRef]
- Fraser, E.; Denney, L.; Antanaviciute, A.; Blirando, K.; Vuppusetty, C.; Zheng, Y.; Repapi, E.; Iotchkova, V.; Taylor, S.; Ashley, N.; et al. Multi-Modal Characterization of Monocytes in Idiopathic Pulmonary Fibrosis Reveals a Primed Type I Interferon Immune Phenotype. Front. Immunol. 2021, 12, 623430. [Google Scholar] [CrossRef]
- Moos, P.J.; Cheminant, J.R.; Cowman, S.; Noll, J.; Wang, Q.; Musci, T.; Venosa, A. Spatial and phenotypic heterogeneity of resident and monocyte-derived macrophages during inflammatory exacerbations leading to pulmonary fibrosis. Front. Immunol. 2024, 15, 1425466. [Google Scholar] [CrossRef]
- Cui, H.; Banerjee, S.; Xie, N.; Hussain, M.; Jaiswal, A.; Liu, H.; Kulkarni, T.; Antony, V.B.; Liu, R.-M.; Colonna, M.; et al. TREM2 promotes lung fibrosis via controlling alveolar macrophage survival and pro-fibrotic activity. Nat. Commun. 2025, 16, 1761. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rigau, A.R.; Xie, W.; Huang, L.; Shao, X.; Li, Y.-N.; Matei, A.-E.; Ye, W.; Zou, H.; Pinello, L.; et al. Mapping spatially-resolved transcriptomes in systemic sclerosis. bioRxiv 2025, 84, 9–10. [Google Scholar] [CrossRef]
- Reader, B.F.; Sethuraman, S.; Hay, B.R.; Thomas Becket, R.V.; Karpurapu, M.; Chung, S.; Lee, Y.G.; Christman, J.W.; Ballinger, M.N. IRAK-M Regulates Monocyte Trafficking to the Lungs in Response to Bleomycin Challenge. J. Immunol. 2020, 204, 2661–2670. [Google Scholar] [CrossRef]
- Bailey, J.I.; Puritz, C.H.; Senkow, K.J.; Markov, N.S.; Diaz, E.; Jonasson, E.; Yu, Z.; Swaminathan, S.; Lu, Z.; Fenske, S.; et al. Profibrotic monocyte-derived alveolar macrophages are expanded in patients with persistent respiratory symptoms and radiographic abnormalities after COVID-19. Nat. Immunol. 2024, 25, 2097–2109. [Google Scholar] [CrossRef]
- Vanneste, D.; Bai, Q.; Hasan, S.; Peng, W.; Pirottin, D.; Schyns, J.; Maréchal, P.; Ruscitti, C.; Meunier, M.; Liu, Z.; et al. MafB-restricted local monocyte proliferation precedes lung interstitial macrophage differentiation. Nat. Immunol. 2023, 24, 827–840. [Google Scholar] [CrossRef]
- Cruz Tleugabulova, M.; Melo, S.P.; Wong, A.; Arlantico, A.; Liu, M.; Webster, J.D.; Lau, J.; Lechner, A.; Corak, B.; Hodgins, J.J.; et al. Induction of a distinct macrophage population and protection from lung injury and fibrosis by Notch2 blockade. Nat. Commun. 2024, 15, 9575. [Google Scholar] [CrossRef]
- Osterholzer, J.J.; Olszewski, M.A.; Murdock, B.J.; Chen, G.H.; Erb-Downward, J.R.; Subbotina, N.; Browning, K.; Lin, Y.; Morey, R.E.; Dayrit, J.K.; et al. Implicating exudate macrophages and Ly-6C(high) monocytes in CCR2-dependent lung fibrosis following gene-targeted alveolar injury. J. Immunol. 2013, 190, 3447–3457. [Google Scholar] [CrossRef] [PubMed]
- Puukila, S.; Lawrence, M.D.; De Pasquale, C.G.; Bersten, A.D.; Bihari, S.; McEvoy-May, J.; Nemec-Bakk, A.; Dixon, D.L. Monocyte chemotactic protein (MCP)-1 (CCL2) and its receptor (CCR2) are elevated in chronic heart failure facilitating lung monocyte infiltration and differentiation which may contribute to lung fibrosis. Cytokine 2023, 161, 156060. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, A.; Lo Re, S.; Chantry, M.; Izquierdo Carerra, X.; Uwambayinema, F.; Ricci, D.; Devosse, R.; Ibouraadaten, S.; Brombin, L.; Palmai-Pallag, M.; et al. CCR2(+) monocytic myeloid-derived suppressor cells (M-MDSCs) inhibit collagen degradation and promote lung fibrosis by producing transforming growth factor-β1. J. Pathol. 2017, 243, 320–330. [Google Scholar] [CrossRef]
- Greiffo, F.R.; Viteri-Alvarez, V.; Frankenberger, M.; Dietel, D.; Ortega-Gomez, A.; Lee, J.S.; Hilgendorff, A.; Behr, J.; Soehnlein, O.; Eickelberg, O.; et al. CX3CR1–fractalkine axis drives kinetic changes of monocytes in fibrotic interstitial lung diseases. Eur. Respir. J. 2020, 55, 1900460. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.; Suchankova, M.; Ganovska, M.; Leksa, V.; Sandor, F.; Tedlova, E.; Konig, B.; Bucova, M. The Role of CX3CL1 and ADAM17 in Pathogenesis of Diffuse Parenchymal Lung Diseases. Diagnostics 2021, 11, 1074. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, J.; Hu, H.; Zhou, Y.; Lin, Z.; Jing, H.; Sun, B. Multiomic analysis of monocyte-derived alveolar macrophages in idiopathic pulmonary fibrosis. J. Transl. Med. 2024, 22, 598. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A.; Kaminski, N. Idiopathic pulmonary fibrosis: Aberrant recapitulation of developmental programs? PLoS Med. 2008, 5, e62. [Google Scholar] [CrossRef]
- Perrot, C.Y.; Karampitsakos, T.; Herazo-Maya, J.D. Monocytes and macrophages: Emerging mechanisms and novel therapeutic targets in pulmonary fibrosis. Am. J. Physiol. Cell Physiol. 2023, 325, C1046–C1057. [Google Scholar] [CrossRef]
- Gibbons, M.A.; MacKinnon, A.C.; Ramachandran, P.; Dhaliwal, K.; Duffin, R.; Phythian-Adams, A.T.; van Rooijen, N.; Haslett, C.; Howie, S.E.; Simpson, A.J.; et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef]
- Guilliams, M.; De Kleer, I.; Henri, S.; Post, S.; Vanhoutte, L.; De Prijck, S.; Deswarte, K.; Malissen, B.; Hammad, H.; Lambrecht, B.N. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 2013, 210, 1977–1992. [Google Scholar] [CrossRef] [PubMed]
- Herazo-Maya, J.D.; Sun, J.; Molyneaux, P.L.; Li, Q.; Villalba, J.A.; Tzouvelekis, A.; Lynn, H.; Juan-Guardela, B.M.; Risquez, C.; Osorio, J.C.; et al. Validation of a 52-gene risk profile for outcome prediction in patients with idiopathic pulmonary fibrosis: An international, multicentre, cohort study. Lancet Respir. Med. 2017, 5, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.B.; Paine, R., 3rd; Christensen, P.J.; Moore, T.A.; Sitterding, S.; Ngan, R.; Wilke, C.A.; Kuziel, W.A.; Toews, G.B. Protection from pulmonary fibrosis in the absence of CCR2 signaling. J. Immunol. 2001, 167, 4368–4377. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K. Abandoning M1/M2 for a Network Model of Macrophage Function. Circ. Res. 2016, 119, 414–417. [Google Scholar] [CrossRef]
- Vancheri, C.; Sortino, M.A.; Tomaselli, V.; Mastruzzo, C.; Condorelli, F.; Bellistrí, G.; Pistorio, M.P.; Canonico, P.L.; Crimi, N. Different expression of TNF-alpha receptors and prostaglandin E2Production in normal and fibrotic lung fibroblasts: Potential implications for the evolution of the inflammatory process. Am. J. Respir. Cell Mol. Biol. 2000, 22, 628–634. [Google Scholar] [CrossRef]
- Vancheri, C.; Mastruzzo, C.; Tomaselli, V.; Sortino, M.A.; D’Amico, L.; Bellistrí, G.; Pistorio, M.P.; Salinaro, E.T.; Palermo, F.; Mistretta, A.; et al. Normal human lung fibroblasts differently modulate interleukin-10 and interleukin-12 production by monocytes: Implications for an altered immune response in pulmonary chronic inflammation. Am. J. Respir. Cell Mol. Biol. 2001, 25, 592–599. [Google Scholar] [CrossRef]
- Moore, B.B.; Coffey, M.J.; Christensen, P.; Sitterding, S.; Ngan, R.; Wilke, C.A.; McDonald, R.; Phare, S.M.; Peters-Golden, M.; Paine, R., 3rd; et al. GM-CSF regulates bleomycin-induced pulmonary fibrosis via a prostaglandin-dependent mechanism. J. Immunol. 2000, 165, 4032–4039. [Google Scholar] [CrossRef]
- Mayr, C.H.; Santacruz, D.; Jarosch, S.; Bleck, M.; Dalton, J.; McNabola, A.; Lempp, C.; Neubert, L.; Rath, B.; Kamp, J.C.; et al. Spatial transcriptomic characterization of pathologic niches in IPF. Sci. Adv. 2024, 10, eadl5473. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; Wang, J.; Yang, F.; Luo, W.; Huang, J.; Chen, M.; Wang, S.; Li, C.; Zhang, W.; et al. ZC3H4 regulates infiltrating monocytes, attenuating pulmonary fibrosis through IL-10. Respir. Res. 2022, 23, 204. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, N.; Willis, G.R.; Fernandez-Gonzalez, A.; Reis, M.; Nassiri, S.; Mitsialis, S.A.; Kourembanas, S. Mesenchymal stromal cell exosomes prevent and revert experimental pulmonary fibrosis through modulation of monocyte phenotypes. JCI Insight 2019, 4, e128060. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Jiang, D.; Banerjee, S.; Xie, N.; Kulkarni, T.; Liu, R.-M.; Duncan, S.R.; Liu, G. Monocyte-derived alveolar macrophage apolipoprotein E participates in pulmonary fibrosis resolution. JCI Insight 2020, 5, e134539. [Google Scholar] [CrossRef]
- Franzén, L.; Olsson Lindvall, M.; Hühn, M.; Ptasinski, V.; Setyo, L.; Keith, B.P.; Collin, A.; Oag, S.; Volckaert, T.; Borde, A.; et al. Mapping spatially resolved transcriptomes in human and mouse pulmonary fibrosis. Nat. Genet. 2024, 56, 1725–1736. [Google Scholar] [CrossRef]
- Hunninghake, G.M.; Quesada-Arias, L.D.; Carmichael, N.E.; Martinez Manzano, J.M.; Poli De Frías, S.; Baumgartner, M.A.; DiGianni, L.; Gampala-Sagar, S.N.; Leone, D.A.; Gulati, S.; et al. Interstitial Lung Disease in Relatives of Patients with Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.A.; Planchart Ferretto, M.A.; Maeda, A.H.; Perez Garcia, M.F.; Carmichael, N.E.; Gulati, S.; Rice, M.B.; Goldberg, H.J.; Putman, R.K.; Hatabu, H.; et al. Progressive Interstitial Lung Disease in Relatives of Patients with Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2023, 207, 211–214. [Google Scholar] [CrossRef]
- Rose, J.A.; Steele, M.P.; Kosak Lopez, E.J.; Axelsson, G.T.; Galecio Chao, A.G.; Waich, A.; Regan, K.; Gulati, S.; Maeda, A.H.; Sultana, S.; et al. Protein biomarkers of interstitial lung abnormalities in relatives of patients with pulmonary fibrosis. Eur. Respir. J. 2025, 65, 2401349. [Google Scholar] [CrossRef]
- Takenouchi, Y.; Kitakaze, K.; Tsuboi, K.; Okamoto, Y. Growth differentiation factor 15 facilitates lung fibrosis by activating macrophages and fibroblasts. Exp. Cell Res. 2020, 391, 112010. [Google Scholar] [CrossRef]
- Wang, S.; Li, J.; Wu, C.; Lei, Z.; Wang, T.; Huang, X.; Zhang, S.; Liu, Y.; Bi, X.; Zheng, F.; et al. Single-Cell RNA Sequencing Reveals Monocyte-Derived Interstitial Macrophages with a Pro-Fibrotic Phenotype in Bleomycin-Induced Pulmonary Fibrosis. Int. J. Mol. Sci. 2024, 25, 11669. [Google Scholar] [CrossRef]
- Silva-Bermudez, L.S.; Klüter, H.; Kzhyshkowska, J.G. Macrophages as a Source and Target of GDF-15. Int. J. Mol. Sci. 2024, 25, 7313. [Google Scholar] [CrossRef]
- Hunninghake, G.M. Interstitial lung abnormalities: Erecting fences in the path towards advanced pulmonary fibrosis. Thorax 2019, 74, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Unterman, A.; Zhao, A.Y.; Neumark, N.; Schupp, J.C.; Ahangari, F.; Cosme, C., Jr.; Sharma, P.; Flint, J.; Stein, Y.; Ryu, C.; et al. Single-Cell Profiling Reveals Immune Aberrations in Progressive Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2024, 210, 484–496. [Google Scholar] [CrossRef]
- Ryerson, C.J.; Vittinghoff, E.; Ley, B.; Lee, J.S.; Mooney, J.J.; Jones, K.D.; Elicker, B.M.; Wolters, P.J.; Koth, L.L.; King, T.E., Jr.; et al. Predicting survival across chronic interstitial lung disease: The ILD-GAP model. Chest 2014, 145, 723–728. [Google Scholar] [CrossRef]
- Hirata, M.; Hara, Y.; Fujii, H.; Murohashi, K.; Saigusa, Y.; Zhao, S.; Kobayashi, M.; Nagasawa, R.; Tagami, Y.; Izawa, A.; et al. ILD-GAP combined with the monocyte ratio could be a better prognostic prediction model than ILD-GAP in patients with interstitial lung diseases. BMC Pulm. Med. 2024, 24, 16. [Google Scholar] [CrossRef]
- Karampitsakos, T.; Milas, T.; Tsiri, P.; Katsaras, M.; Koletsis, E.; Vachlas, K.; Malakounidou, E.; Zarkadi, E.; Tsirikos, G.; Georgiopoulou, V.; et al. Elevated peripheral blood monocyte count is associated with prolonged postoperative hospitalization and functional decline in patients with interstitial lung diseases undergoing surgical lung biopsy. Eur. Rev. Med. Pharmacol. Sci. 2024, 28, 3683–3696. [Google Scholar] [CrossRef] [PubMed]
- Tsuneyoshi, S.; Zaizen, Y.; Tominaga, M.; Matama, G.; Umemoto, S.; Ohno, S.; Takaki, R.; Yano, R.; Murotani, K.; Okamoto, M.; et al. Clinical significance of high monocyte counts for the continuous treatment with nintedanib. BMC Pulm. Med. 2023, 23, 242. [Google Scholar] [CrossRef]
- Shao, G.; Thöne, P.; Kaiser, B.; Lamprecht, B.; Lang, D. Functional Improvement at One Year in Fibrotic Interstitial Lung Diseases—Prognostic Value of Baseline Biomarkers and Anti-Inflammatory Therapies. Diagnostics 2024, 14, 1544. [Google Scholar] [CrossRef] [PubMed]
- Brody, S.L.; Gunsten, S.P.; Luehmann, H.P.; Sultan, D.H.; Hoelscher, M.; Heo, G.S.; Pan, J.; Koenitzer, J.R.; Lee, E.C.; Huang, T.; et al. Chemokine Receptor 2-targeted Molecular Imaging in Pulmonary Fibrosis. A Clinical Trial. Am. J. Respir. Crit. Care Med. 2021, 203, 78–89. [Google Scholar] [CrossRef]
- Farooq, H.; Luehmann, H.P.; Koenitzer, J.R.; Heo, G.S.; Sultan, D.H.; Kulkarni, D.H.; Gunsten, S.P.; Sashti, R.M.; Huang, T.; Keller, A.R.; et al. Molecular imaging in experimental pulmonary fibrosis reveals that nintedanib unexpectedly modulates CCR2 immune cell infiltration. eBioMedicine 2024, 110, 105431. [Google Scholar] [CrossRef]
- Huang, J.; Maier, C.; Zhang, Y.; Soare, A.; Dees, C.; Beyer, C.; Harre, U.; Chen, C.W.; Distler, O.; Schett, G.; et al. Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 1941–1948. [Google Scholar] [CrossRef]
- Soldano, S.; Smith, V.; Montagna, P.; Gotelli, E.; Campitiello, R.; Pizzorni, C.; Paolino, S.; Sulli, A.; Cere, A.; Cutolo, M. Nintedanib downregulates the profibrotic M2 phenotype in cultured monocyte-derived macrophages obtained from systemic sclerosis patients affected by interstitial lung disease. Arthritis Res. Ther. 2024, 26, 74. [Google Scholar] [CrossRef] [PubMed]
- Toda, M.; Mizuguchi, S.; Minamiyama, Y.; Yamamoto-Oka, H.; Aota, T.; Kubo, S.; Nishiyama, N.; Shibata, T.; Takemura, S. Pirfenidone suppresses polarization to M2 phenotype macrophages and the fibrogenic activity of rat lung fibroblasts. J. Clin. Biochem. Nutr. 2018, 63, 58–65. [Google Scholar] [CrossRef]
- Ishii, D.; Kawasaki, T.; Sato, H.; Tatsumi, K.; Imamoto, T.; Yoshioka, K.; Abe, M.; Hasegawa, Y.; Ohara, O.; Suzuki, T. Effects of Anti-Fibrotic Drugs on Transcriptome of Peripheral Blood Mononuclear Cells in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2024, 25, 3750. [Google Scholar] [CrossRef]
- Ying, H.; Fang, M.; Hang, Q.Q.; Chen, Y.; Qian, X.; Chen, M. Pirfenidone modulates macrophage polarization and ameliorates radiation-induced lung fibrosis by inhibiting the TGF-β1/Smad3 pathway. J. Cell. Mol. Med. 2021, 25, 8662–8675. [Google Scholar] [CrossRef]
- Morris, E.A.; Parvizi, R.; Orzechowski, N.M.; Whitfield, M.L.; Pioli, P.A. Mycophenolate mofetil directly modulates myeloid viability and pro-fibrotic activation of human macrophages. Rheumatology 2024, 64, 3125–3133. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Brown, K.K.; Du Bois, R.M.; Frankel, S.K.; Cosgrove, G.P.; Fernandez-Perez, E.R.; Huie, T.J.; Krishnamoorthy, M.; Meehan, R.T.; Olson, A.L.; et al. Mycophenolate mofetil improves lung function in connective tissue disease-associated interstitial lung disease. J. Rheumatol. 2013, 40, 640–646. [Google Scholar] [CrossRef]
- Suissa, S.; Suissa, K. Antifibrotics and Reduced Mortality in Idiopathic Pulmonary Fibrosis: Immortal Time Bias. Am. J. Respir. Crit. Care Med. 2023, 207, 105–109. [Google Scholar] [CrossRef]
- Xiang, J.; Cheng, S.; Feng, T.; Wu, Y.; Xie, W.; Zhang, M.; Xu, X.; Zhang, C. Neotuberostemonine attenuates bleomycin-induced pulmonary fibrosis by suppressing the recruitment and activation of macrophages. Int. Immunopharmacol. 2016, 36, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Ouyang, B.; Tang, W.; Wang, N.; Yang, F.; Shi, H.; Zhang, Z.; Yu, H.; Chen, M.; Wei, Y.; et al. Icariside II modulates pulmonary fibrosis via PI3K/Akt/β-catenin pathway inhibition of M2 macrophage program. Phytomedicine 2024, 130, 155687. [Google Scholar] [CrossRef]
- Okabe, Y.; Toda, E.; Urushiyama, H.; Terashima, Y.; Kunugi, S.; Kajimoto, Y.; Terasaki, M.; Matsushima, K.; Saito, A.; Yamauchi, Y.; et al. Antifibrotic effect of disulfiram on bleomycin-induced lung fibrosis in mice and its impact on macrophage infiltration. Sci. Rep. 2024, 14, 23653. [Google Scholar] [CrossRef]
- Guo, Z.; Li, S.; Zhang, N.; Kang, Q.; Zhai, H. Schisandra Inhibit Bleomycin-Induced Idiopathic Pulmonary Fibrosis in Rats via Suppressing M2 Macrophage Polarization. BioMed Res. Int. 2020, 2020, 5137349. [Google Scholar] [CrossRef]
- Hong, S.-Y.; Lu, Y.-T.; Chen, S.-Y.; Hsu, C.-F.; Lu, Y.-C.; Wang, C.-Y.; Huang, K.-L. Targeting pathogenic macrophages by the application of SHP-1 agonists reduces inflammation and alleviates pulmonary fibrosis. Cell Death Dis. 2023, 14, 352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ohno, S.; Steer, B.; Klee, S.; Staab-Weijnitz, C.A.; Wagner, D.; Lehmann, M.; Stoeger, T.; Königshoff, M.; Adler, H. S100a4 Is Secreted by Alternatively Activated Alveolar Macrophages and Promotes Activation of Lung Fibroblasts in Pulmonary Fibrosis. Front. Immunol. 2018, 9, 1216. [Google Scholar] [CrossRef] [PubMed]
- Sennello, J.A.; Misharin, A.V.; Flozak, A.S.; Berdnikovs, S.; Cheresh, P.; Varga, J.; Kamp, D.W.; Budinger, G.R.; Gottardi, C.J.; Lam, A.P. Lrp5/β-Catenin Signaling Controls Lung Macrophage Differentiation and Inhibits Resolution of Fibrosis. Am. J. Respir. Cell Mol. Biol. 2017, 56, 191–201. [Google Scholar] [CrossRef]
- Okazaki, H.; Sato, S.; Koyama, K.; Morizumi, S.; Abe, S.; Azuma, M.; Chen, Y.; Goto, H.; Aono, Y.; Ogawa, H.; et al. The novel inhibitor PRI-724 for Wnt/β-catenin/CBP signaling ameliorates bleomycin-induced pulmonary fibrosis in mice. Exp. Lung Res. 2019, 45, 188–199. [Google Scholar] [CrossRef] [PubMed]
- El-Demerdash, E. Anti-inflammatory and antifibrotic effects of methyl palmitate. Toxicol. Appl. Pharmacol. 2011, 254, 238–244. [Google Scholar] [CrossRef]
- Nakagome, K.; Dohi, M.; Okunishi, K.; Tanaka, R.; Miyazaki, J.; Yamamoto, K. In vivo IL-10 gene delivery attenuates bleomycin induced pulmonary fibrosis by inhibiting the production and activation of TGF-beta in the lung. Thorax 2006, 61, 886–894. [Google Scholar] [CrossRef]
- Solomon, J.J.; Danoff, S.K.; Woodhead, F.A.; Hurwitz, S.; Maurer, R.; Glaspole, I.; Dellaripa, P.F.; Gooptu, B.; Vassallo, R.; Cox, P.G.; et al. Safety, tolerability, and efficacy of pirfenidone in patients with rheumatoid arthritis-associated interstitial lung disease: A randomised, double-blind, placebo-controlled, phase 2 study. Lancet Respir. Med. 2023, 11, 87–96. [Google Scholar] [CrossRef]
- Kulshrestha, R.; Pandey, A.; Jaggi, A.; Bansal, S. Beneficial effects of N-acetylcysteine on protease-antiprotease balance in attenuating bleomycin-induced pulmonary fibrosis in rats. Iran. J. Basic Med. Sci. 2020, 23, 396–405. [Google Scholar] [CrossRef]
- Bohdziewicz, A.; Pawlik, K.K.; Maciejewska, M.; Sikora, M.; Alda-Malicka, R.; Czuwara, J.; Rudnicka, L. Future Treatment Options in Systemic Sclerosis—Potential Targets and Ongoing Clinical Trials. J. Clin. Med. 2022, 11, 1310. [Google Scholar] [CrossRef]
- Alapati, D.; Zacharias, W.J.; Hartman, H.A.; Rossidis, A.C.; Stratigis, J.D.; Ahn, N.J.; Coons, B.; Zhou, S.; Li, H.; Singh, K.; et al. In utero gene editing for monogenic lung disease. Sci. Transl. Med. 2019, 11, eaav8375. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Zhu, L.; Zhou, Z.; Chen, P.; Liu, S.; Locy, M.L.; Thannickal, V.J.; Zhou, Y. Reversing Mechanoinductive DSP Expression by CRISPR/dCas9-mediated Epigenome Editing. Am. J. Respir. Crit. Care Med. 2018, 198, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Bonella, F.; Spagnolo, P.; Ryerson, C. Current and Future Treatment Landscape for Idiopathic Pulmonary Fibrosis. Drugs 2023, 83, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, J.; Sgalla, G.; Richeldi, L. An up-to-date review of approved and emerging antibody therapies for idiopathic pulmonary fibrosis. Expert Opin. Biol. Ther. 2023, 23, 1239–1244. [Google Scholar] [CrossRef]
- Chang, X.; Xing, L.; Wang, Y.; Yang, C.-X.; He, Y.-J.; Zhou, T.-J.; Gao, X.-D.; Li, L.; Hao, H.-P.; Jiang, H.-L. Monocyte-derived multipotent cell delivered programmed therapeutics to reverse idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba3167. [Google Scholar] [CrossRef]
- Bernstein, E.J.; Denton, C.P.; Huang, S.; Khanna, D. Baseline absolute monocyte count predicts lung function decline among patients with systemic sclerosis-associated interstitial lung disease: A post hoc analysis from the focuSSced trial. Semin. Arthritis Rheum. 2024, 65, 152376. [Google Scholar] [CrossRef]
- Mathai, S.K.; Gulati, M.; Peng, X.; Russell, T.R.; Shaw, A.C.; Rubinowitz, A.N.; Murray, L.A.; Siner, J.M.; Antin-Ozerkis, D.E.; Montgomery, R.R.; et al. Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab. Investig. 2010, 90, 812–823. [Google Scholar] [CrossRef]
- Poole, J.A.; Cole, K.E.; Thiele, G.M.; Talmadge, J.E.; England, B.R.; Nelson, A.J.; Gleason, A.; Schwab, A.; Gaurav, R.; Duryee, M.J.; et al. Expansion of distinct peripheral blood myeloid cell subpopulations in patients with rheumatoid arthritis-associated interstitial lung disease. Int. Immunopharmacol. 2024, 127, 111330. [Google Scholar] [CrossRef] [PubMed]
- Poole, J.A.; Schwab, A.; Thiele, G.M.; England, B.R.; Nelson, A.J.; Gleason, A.; Duryee, M.J.; Bailey, K.L.; Romberger, D.J.; Hershberger, D.; et al. Unique transcriptomic profile of peripheral blood monocytes in rheumatoid arthritis-associated interstitial lung disease. Rheumatology 2024, keae572. [Google Scholar] [CrossRef]
- Hayashi, M.; Aoki, A.; Asakawa, K.; Sakagami, T.; Kikuchi, T.; Takada, T. Cytokine profiles of amyopathic dermatomyositis with interstitial lung diseases treated with mycophenolate. Respirol. Case Rep. 2017, 5, e00235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chan, C.C.Y.; Cheung, K.F.; Chau, M.K.M.; Yap, D.Y.H.; Ma, M.K.M.; Chan, K.W.; Yung, S.; Chan, T.M. Effect of mycophenolate and rapamycin on renal fibrosis in lupus nephritis. Clin. Sci. 2019, 133, 1721–1744. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, E.R. Combining rituximab with mycophenolate for the treatment of interstitial lung disease. Eur. Respir. J. 2023, 61, 2300614. [Google Scholar] [CrossRef]
- Khanna, D.; Albera, C.; Fischer, A.; Khalidi, N.; Raghu, G.; Chung, L.; Chen, D.; Schiopu, E.; Tagliaferri, M.; Seibold, J.R.; et al. An Open-label, Phase II Study of the Safety and Tolerability of Pirfenidone in Patients with Scleroderma-associated Interstitial Lung Disease: The LOTUSS Trial. J. Rheumatol. 2016, 43, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Spino, C.; Bernstein, E.; Goldin, J.; Tashkin, D.; Roth, M. Combination Therapy of Mycophenolate Mofetil and Pirfenidone vs. Mycophenolate Alone: Results from the Scleroderma Lung Study III. In Proceedings of the ACR Convergence 2022, Philadelphia, PA, USA, 10–14 November 2022. [Google Scholar]
- Goldman, N.R.; Nihtyanova, S.I.; Beesley, C.F.; Wells, A.U.; Denton, C.P.; Renzoni, E.A.; Mageed, R.; Ong, V.H. Tocilizumab and rituximab for systemic sclerosis interstitial lung disease: A real-world cohort analysis. Rheumatology 2025, keaf006. [Google Scholar] [CrossRef]
- Khanna, D.; Lin, C.J.F.; Furst, D.E.; Wagner, B.; Zucchetto, M.; Raghu, G.; Martinez, F.J.; Goldin, J.; Siegel, J.; Denton, C.P. Long-Term Safety and Efficacy of Tocilizumab in Early Systemic Sclerosis-Interstitial Lung Disease: Open-Label Extension of a Phase 3 Randomized Controlled Trial. Am. J. Respir. Crit. Care Med. 2022, 205, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Pu, J.; Wang, Y.; Wu, Z.; Liang, Y.; Song, J.; Pan, S.; Han, F.; Yang, L.; Xu, X.; et al. Tofacitinib in the treatment of primary Sjögren’s syndrome-associated interstitial lung disease: Study protocol for a prospective, randomized, controlled and open-label trial. BMC Pulm. Med. 2023, 23, 473. [Google Scholar] [CrossRef]
- Johnson, S.R.; Bernstein, E.J.; Bolster, M.B.; Chung, J.H.; Danoff, S.K.; George, M.D.; Khanna, D.; Guyatt, G.; Mirza, R.D.; Aggarwal, R.; et al. 2023 American College of Rheumatology (ACR)/American College of Chest Physicians (CHEST) Guideline for the Treatment of Interstitial Lung Disease in People with Systemic Autoimmune Rheumatic Diseases. Arthritis Rheumatol. 2024, 76, 1182–1200. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Authors | Main Findings | n | Reference |
|---|---|---|---|
| Kim et al. | Elevated monocyte counts are associated with ILA development, 5-year ILA progression and reduced FVC. ILA patients have a higher activated monocyte percentage. | 7396 | [17] |
| Achaia et al. | Elevated monocyte (and neutrophil) counts predict progression from indeterminate UIP on chest CT scans to definitive IPF. | 48 | [18] |
| Scott et al. | Elevated CD14+ monocyte counts are associated with shorter transplant-free survival and mortality. | 7459 | [19] |
| Liu et al. | High monocyte proportion predicts shorter survival time in IPF patients. | 94 | [20] |
| Teoh et al. | Monocyte counts predict reduced DLCO and poorer survival in IPF patients. | 231 | [21] |
| Kreuter et al. | IPF patients with higher monocyte counts are at higher-risk of IPF progression, all-cause hospitalization and 1-year mortality. | 2067 | [22] |
| Bernardinello et al. | Lymphocyte-to-monocyte ratio below 4.18 predicts worse survival in newly diagnosed IPF patients. | 77 | [23] |
| Kawamura et al. | Elevated monocyte counts at initiation of antifibrotic therapy for fibrosing ILDs develop earlier and more frequent exacerbations. | 122 | [25] |
| Yamashita et al. | S100A9+CD163− are reduced, and S100A9−CD163+ re elevated, in NSIP compared to IPF. In IPF patients, S100A9+CD163− monocytes are associated with serum surfactant protein D (a marker of alveolar damage). | 70 | [27] |
| Chauvin et al. | Soluble CD163 does not correlate with IPF prognosis. | 155 | [28] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lema, D.; Kosak Lopez, E.; Lam, J.; Tskhakaia, I.; Gonzalez Moret, Y.; Abdollahi, S. The Role of Monocytes in the Natural History of Idiopathic Pulmonary Fibrosis: A Systematic Literature Review. Int. J. Mol. Sci. 2025, 26, 6538. https://doi.org/10.3390/ijms26136538
Lema D, Kosak Lopez E, Lam J, Tskhakaia I, Gonzalez Moret Y, Abdollahi S. The Role of Monocytes in the Natural History of Idiopathic Pulmonary Fibrosis: A Systematic Literature Review. International Journal of Molecular Sciences. 2025; 26(13):6538. https://doi.org/10.3390/ijms26136538
Chicago/Turabian StyleLema, Diego, Esteban Kosak Lopez, Justin Lam, Irakli Tskhakaia, Yurilu Gonzalez Moret, and Shahrzad Abdollahi. 2025. "The Role of Monocytes in the Natural History of Idiopathic Pulmonary Fibrosis: A Systematic Literature Review" International Journal of Molecular Sciences 26, no. 13: 6538. https://doi.org/10.3390/ijms26136538
APA StyleLema, D., Kosak Lopez, E., Lam, J., Tskhakaia, I., Gonzalez Moret, Y., & Abdollahi, S. (2025). The Role of Monocytes in the Natural History of Idiopathic Pulmonary Fibrosis: A Systematic Literature Review. International Journal of Molecular Sciences, 26(13), 6538. https://doi.org/10.3390/ijms26136538