
Positive AMPA and Kainate Receptor Modulators and Their Therapeutic Potential in CNS Diseases: A Comprehensive Review


Abstract
1. Introduction
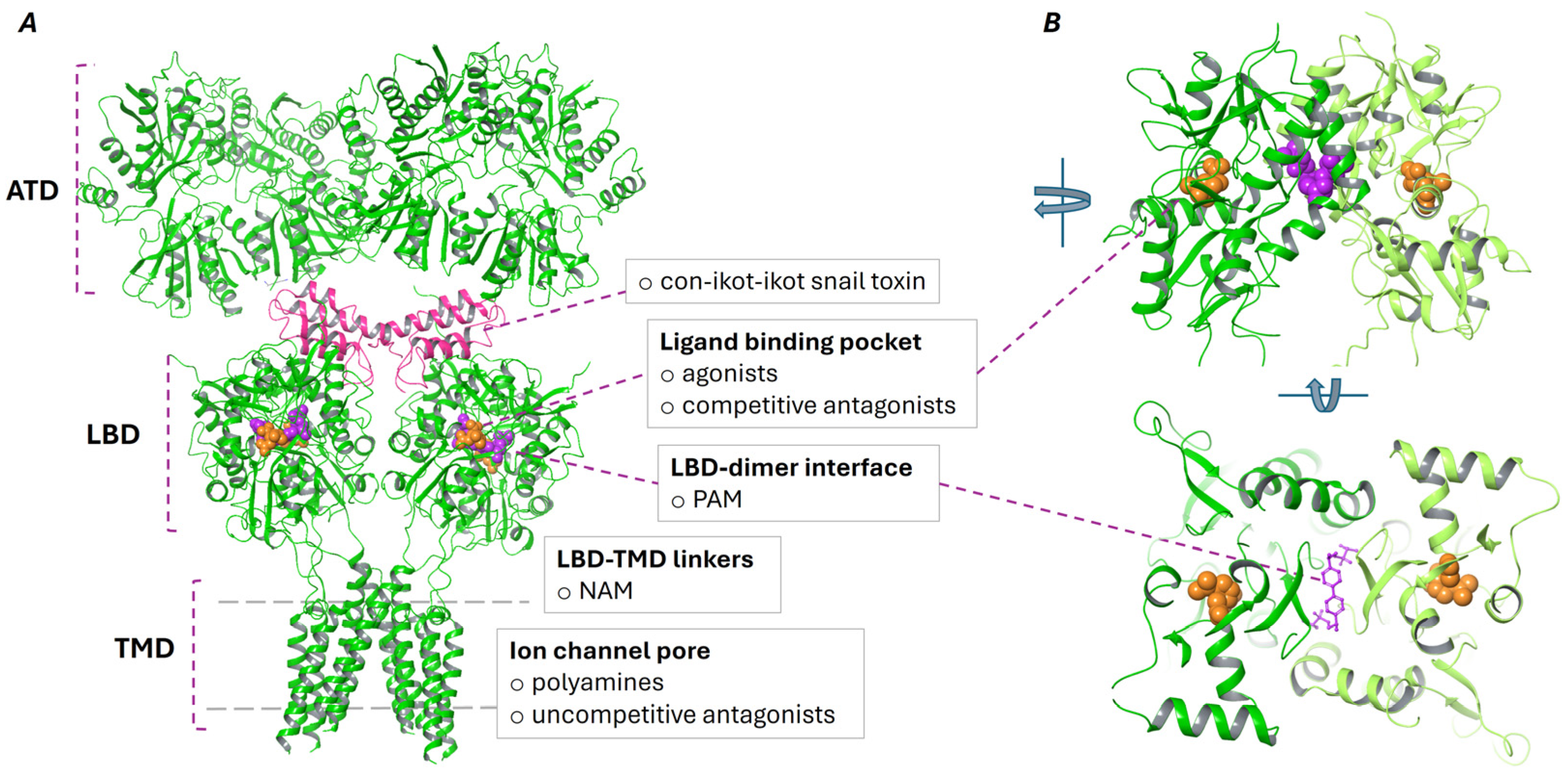
2. Structure and Pharmacological Modulation of AMPA and Kainate Receptors
3. Positive Allosteric Modulators of AMPA and Kainate Receptors
3.1. Benzamides and Related Structures
3.2. Benzothiadiazines and Ring-Fused Thiadiazines

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | EC50 [μM] | |||
|---|---|---|---|---|
| GluA2 | GluK1 | GluK2 | GluK3 | |
| BPAM121 (24) | 20.4 a | nd | no potentiation effect at 300 μM b | nd |
| BPAM344 (25) | 0.81 a (5-fold at 100 μM) b | 26 c (5-fold at 100 μM) b | 79 b 75 c (15-fold at 100 μM) b | 639 c (59-fold at 100 μM) b |
| BPAM521 (26) | 2.5 d | nd | 159 b (12-fold at 300 μM) b | nd |
| BPAM538 (27) | 0.002 a (46% at 100 μM) e | 58 e (130% at 100 μM) e | nd (32% at 100 μM) e | nd |
| 28 | 0.0134 f | nd | nd | nd |
| 29 | 0.0014 f | nd | nd | nd |
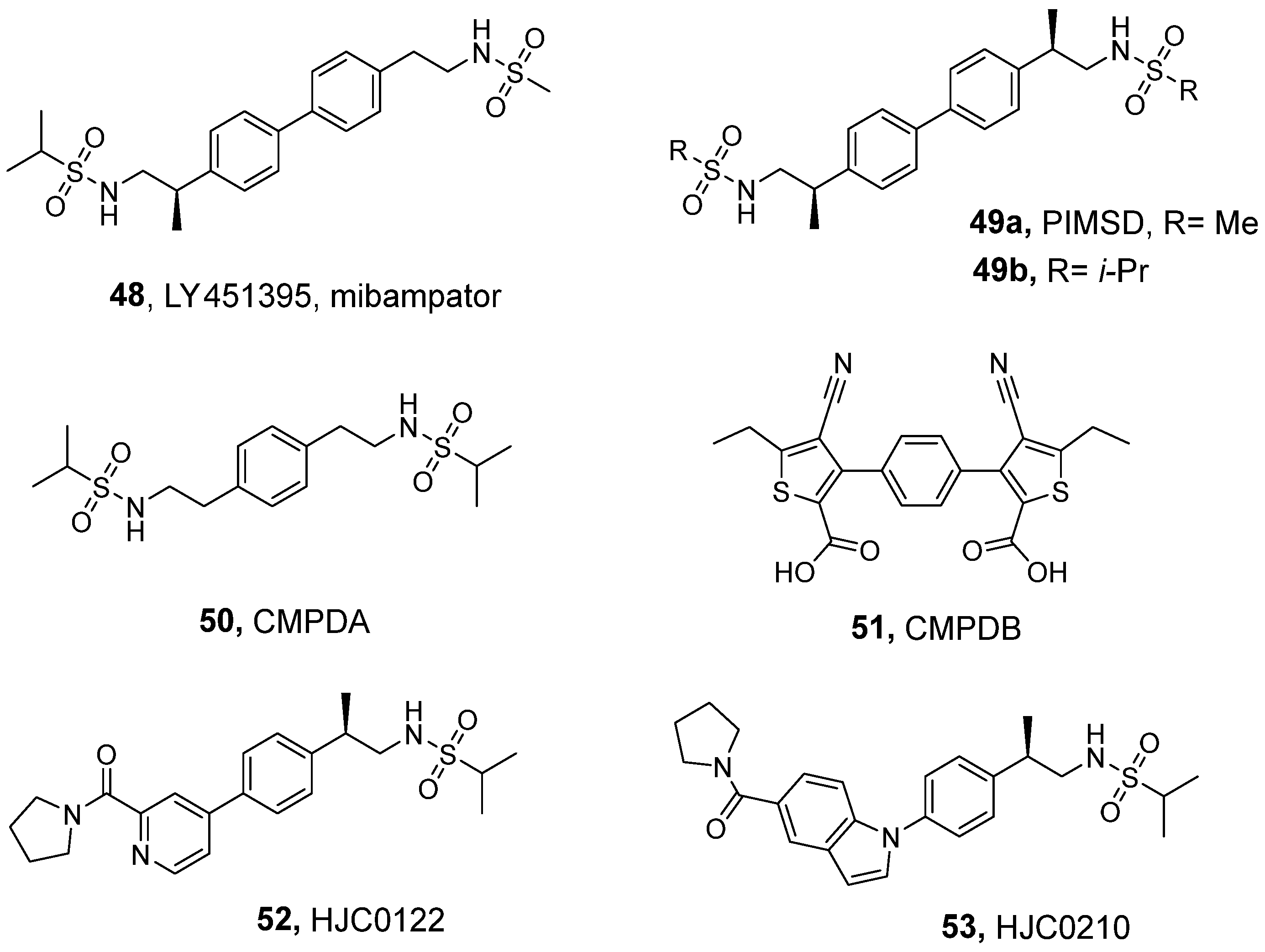
3.3. Sulfonamides and Related Dimeric Compounds
| Compound | EC50 [µM] (Emax [%]) | ||||||
|---|---|---|---|---|---|---|---|
| GluA1i | GluA1o | GluA2i | GluA2o | GluA3i | GluA4i | GluA4o | |
| LY404187 (40) | 5.65 a,b | nd | 0.15 a,b | 1.44 a,b | 1.66 a,b | 0.21 a,b 1.0 c (120) | nd |
| LY392098 (42) | 1.77 a,b | nd | 0.22 a,b | 0.55 a,b | 1.89 a,b | 0.20 a,b 1.2 c (120) | nd |
| PF-4778574 d,e (45) | nd | nd | 0.045 (111) | 0.090 (112) | nd | nd | nd |
| BIIB104 (46) | 0.3 f | 22f | 0.024 d,g (124) | 0.880 d,g (108) | nd | 0.30 f | 9.0 f |
| UoS12258 a,h (47) | 10.0 | nd | 2.69 (101) | nd | 6.76 | 2.24 | nd |
| Compound | EC50 [nM] | Emax |
|---|---|---|
| LY404187 (40) | 1300 a | 45.3-fold a 83% at 100 nM b |
| LY392098 (42) | 1700 c | 31.0-fold c 180% at 1µM b |
| PF-4701475 (43) d,e | 123 | 147% |
| 44 d,e | <10 | nd |
| PF-4778574 (45) d,f | 919 | 162% |
| BIIB104 (46) d,g | 310 | 110% |
| UoS12258 (47) h | 7.94 | nd |
3.4. Trifluoromethylpyrazoles
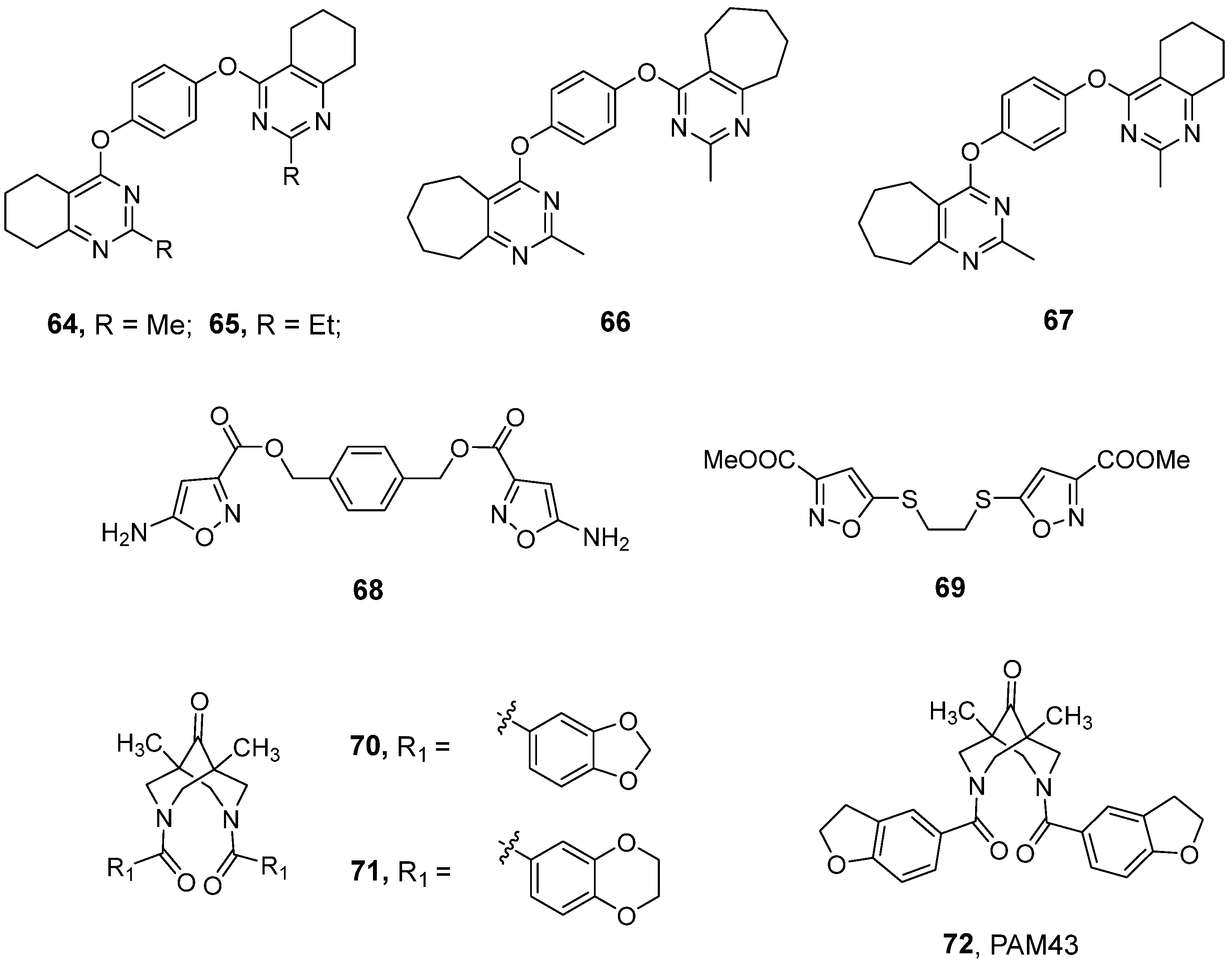
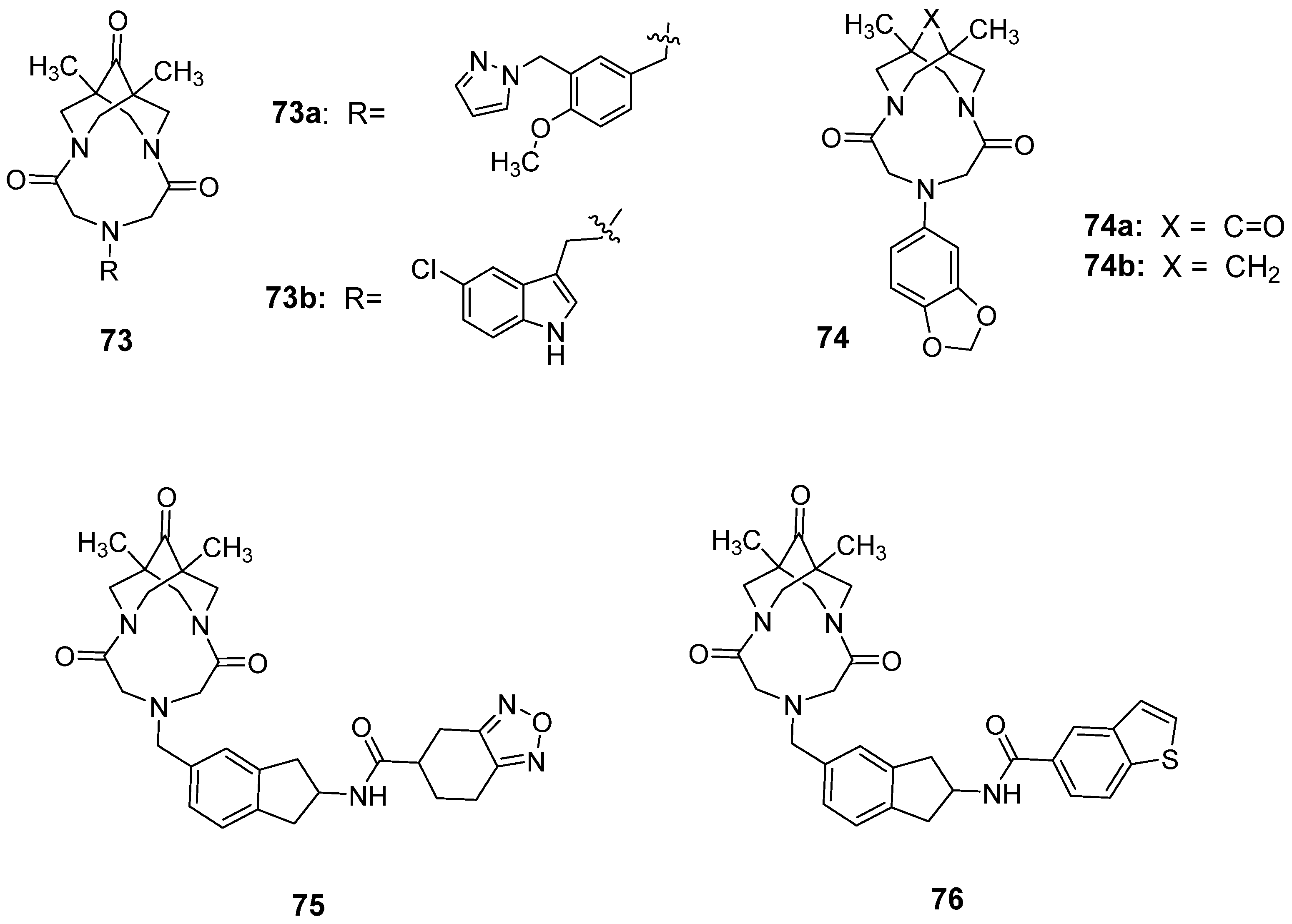
3.5. Recently Developed Bivalent Ligands
| Compound | Number of Neurons | Potentiation Effects [%] Observed at a Specific Modulator Concentration (Control 100%) | Ref. | |||||
|---|---|---|---|---|---|---|---|---|
| 10−12 | 10−11 | 10−10 | 10−9 | 10−8 | 10−7 | |||
| 64 | 7 | 108 | 132 | 143 | 170 | 123 | 85 | [150] |
| 67 | 5 | 105 | 138 | 149 | 177 | 163 | 155 | [150] |
| 68 | 4 | 141 | 172 | 152 | 144 | 129 | 113 | [153] |
| 69 | 3 | 100 | 139 | 177 | 163 | 146 | 141 | [151] |
| 72 | 13 | 100 | 122 | 152 | 162 | 145 | 136 | [154] |
| 73a | 3-6 | 143 | 129 | 117 | 122 | 109 | 110 | [155] |
| 73b | 6 | - | 118 | 124 | 132 | 125 | 119 | [156] |
| 74a | 7 | 111 | 141 | 152 | 162 | 148 | 135 | [157] |
| 74b | 7 | 89 | 87 | 84 | 75 | 68 | 61 | [157] |
3.6. Miscellaneous Chemotypes
3.7. Con-Ikot-Ikot and ConA
4. Clinical Relevance and Therapeutic Applications
| Intervention | Condition | Stage of Development Status | Reference |
|---|---|---|---|
| CX516, ampalex (5) | Schizophrenia | Phase II/Phase III Completed (2007) | [176], NCT00235352 |
| Fragile X syndrome/autism | Phase II Completed (2005) | [174], NCT00054730 | |
| Mild cognitive impairment | Phase II Completed (2004) | [173], NCT00040443 | |
| Alzheimer’s disease/dementia | Phase II Completed (2005) | NCT00001662 | |
| CX691, farampator (7) | Major depressive disorder | Phase II Terminated (2007) | NCT00113022 |
| Cognitive deficits in schizophrenia | Phase II Withdrawn (2009) | [175], NCT00425815 | |
| CX717 (8) | Attention deficit hyperactivity disorder | Phase II Completed (2006) | NCT03375021 |
| CX1739 (9) | Opiate-induced respiratory depression | Phase II Uknown status (2016) | NCT02735629 |
| ORG-26576 (10) | Major depressive disorder | Phase II Completed (2008) | [177], NCT00610649 |
| Attention deficit hyperactivity disorder | Phase II Completed (2009) | [178], NCT00610441 | |
| S47445, CX1632, turlampator (18) | Alzheimer’s disease | Phase II Completed (2017) | [179], NCT02626572 |
| Major depressive disorder | Phase II Completed (2017) | NCT02805439 | |
| S18986 (22) | Mild cognitive impairment | Phase II Terminated (2006) | [180], NCT00202540 |
| TAK-653, osavampator (37) | Major depressive disorder | Phase II Completed (2024) | [181], NCT05203341 |
| Major depressive disorder | Phase III Recruiting | NCT06786624 | |
| BIIB104 (46) | Hearing loss | Phase I Completed (2013) | [182], NCT01518920 |
| Ketamine-induced cognitive impairment | Phase I Completed (2014) | [183], NCT01749098 | |
| Cognitive impairment in schizophrenia | Phase II Completed (2022) | [184], NCT03745820 | |
| UoS12258, GSK729327 (47) | Schizophrenia | Phase I Completed (2009) | NCT00448890 |
| LY451395, mibampator (48) | Alzheimer’s disease | Phase II Completed (2003) | NCT00051909 |
| Aggression and agitation associated with AD | Phase II Completed (2011) | [185], NCT00843518 |
5. Illicit Use and Potential Abuse as ‘Smart Drugs’
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hansen, K.B.; Wollmuth, L.P.; Bowie, D.; Furukawa, H.; Menniti, F.S.; Sobolevsky, A.I.; Swanson, G.T.; Swanger, S.A.; Greger, I.H.; Nakagawa, T.; et al. Structure, function, and pharmacology of glutamate receptor ion channels. Pharmacol. Rev. 2021, 73, 1469–1658. [Google Scholar] [CrossRef]
- Valbuena, S.; Lerma, J. Non-canonical signaling, the hidden life of ligand-gated ion channels. Neuron 2016, 92, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Chałupnik, P.; Szymańska, E. Kainate receptor antagonists: Recent advances and therapeutic perspective. Int. J. Mol. Sci. 2023, 24, 1908. [Google Scholar] [CrossRef]
- Guo, C.; Ma, Y.-Y. Calcium permeable-AMPA receptors and excitotoxicity in neurological disorders. Front. Neural Circuits 2021, 15, 711564. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, E.; von Engelhardt, J. AMPA receptor complex constituents: Control of receptor assembly, membrane trafficking and subcellular localization. Mol. Cell. Neurosci. 2018, 91, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Kamalova, A.; Nakagawa, T. AMPA receptor structure and auxiliary subunits. J. Physiol. 2021, 599, 453–469. [Google Scholar] [CrossRef]
- Ishii, T.; Stolz, J.R.; Swanson, G.T. Auxiliary proteins are the predominant determinants of differential efficacy of clinical candidates acting as AMPA receptor positive allosteric modulators. Mol. Pharmacol. 2020, 97, 336–350. [Google Scholar] [CrossRef]
- Fernandes, H.B.; Catches, J.S.; Petralia, R.S.; Copits, B.A.; Xu, J.; Russell, T.A.; Swanson, G.T.; Contractor, A. High-affinity kainate receptor subunits are necessary for ionotropic but not metabotropic signaling. Neuron 2009, 63, 818–829. [Google Scholar] [CrossRef]
- Pøhlsgaard, J.; Frydenvang, K.; Madsen, U.; Kastrup, J.S. Lessons from more than 80 structures of the GluA2 ligand-binding domain in complex with agonists, antagonists and allosteric modulators. Neuropharmacology 2011, 60, 135–150. [Google Scholar] [CrossRef]
- Gangwar, S.P.; Yen, L.Y.; Yelshanskaya, M.V.; Korman, A.; Jones, D.R.; Sobolevsky, A.I. Modulation of GluA2–γ5 synaptic complex desensitization, polyamine block and antiepileptic perampanel inhibition by auxiliary subunit cornichon-2. Nat. Struct. Mol. Biol. 2023, 30, 1481–1494. [Google Scholar] [CrossRef]
- Pirotte, B.; Francotte, P.; Goffin, E.; De Tullio, P. AMPA receptor positive allosteric modulators: A patent review. Expert. Opin. Ther. Pat. 2013, 23, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Partin, K.M. AMPA receptor potentiators: From drug design to cognitive enhancement. Curr. Opin. Pharmacol. 2015, 20, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Frydenvang, K.; Pickering, D.S.; Kastrup, J.S. Structural basis for positive allosteric modulation of AMPA and kainate receptors. J. Physiol. 2022, 600, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Sobolevsky, A.I.; Rosconi, M.P.; Gouaux, E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 2009, 462, 745–756. [Google Scholar] [CrossRef]
- Jin, R.; Clark, S.; Weeks, A.M.; Dudman, J.T.; Gouaux, E.; Partin, K.M. Mechanism of positive allosteric modulators acting on AMPA receptors. J. Neurosci. 2005, 25, 9027–9036. [Google Scholar] [CrossRef]
- Ptak, C.P.; Ahmed, A.H.; Oswald, R.E. Probing the allosteric modulator binding site of GluR2 with thiazide derivatives. Biochemistry 2009, 48, 8594–8602. [Google Scholar] [CrossRef]
- Ahmed, H.; Ptak, C.P.; Oswald, R.E. Molecular mechanism of flop selectivity and subsite recognition for an AMPA receptor allosteric modulator: Structures of GluA2 and GluA3 in complexes with PEPA. Biochemistry 2010, 49, 2843–2850. [Google Scholar] [CrossRef]
- Golubeva, E.A.; Lavrov, M.I.; Radchenko, E.V.; Palyulin, V.A. Diversity of AMPA receptor ligands: Chemotypes, binding modes, mechanisms of action, and therapeutic effects. Biomolecules 2022, 13, 56. [Google Scholar] [CrossRef]
- Imbesi, M.; Uz, T.; Manev, R.; Sharma, R.P.; Manev, H. Minocycline increases phosphorylation and membrane insertion of neuronal GluR1 receptors. Neurosci. Lett. 2008, 447, 134–137. [Google Scholar] [CrossRef]
- Chen, L.; Dürr, K.L.; Gouaux, E. X-ray structures of AMPA receptor–cone snail toxin complexes illuminate activation mechanism. Science 2014, 345, 1021–1026. [Google Scholar] [CrossRef]
- Winblad, B. Piracetam: A review of pharmacological properties and clinical uses. CNS Drug Rev. 2005, 11, 169–182. [Google Scholar] [CrossRef]
- Malykh, A.G.; Sadaie, M.R. Piracetam and piracetam-like drugs: From basic science to novel clinical applications to CNS disorders. Drugs 2010, 70, 287–312. [Google Scholar] [CrossRef] [PubMed]
- Black, M.D. Therapeutic potential of positive AMPA modulators and their relationship to AMPA receptor subunits. A review of preclinical data. Psychopharmacology 2005, 179, 154–163. [Google Scholar] [CrossRef]
- Koliaki, C.C.; Messini, C.; Tsolaki, M. Clinical efficacy of aniracetam, either as monotherapy or combined with cholinesterase inhibitors, in patients with cognitive impairment: A comparative open study. CNS Neurosci. Ther. 2012, 18, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.; Reeta, K.H.; Sharma, U.; Suri, V.; Singh, S. AMPA receptor modulation through sequential treatment with perampanel and aniracetam mitigates post-stroke damage in experimental model of ischemic stroke. Naunyn Schmiedebergs Arch. Pharmacol. 2023, 396, 3529–3545. [Google Scholar] [CrossRef]
- Love, R.W.B. Aniracetam: An evidence-based model for preventing the accumulation of amyloid-β plaques in Alzheimer’s Disease. J. Alzheimers Dis. 2024, 98, 1235–1241. [Google Scholar] [CrossRef]
- Hu, X.; Tian, X.; Guo, X.; He, Y.; Chen, H.; Zhou, J.; Wang, Z.J. AMPA receptor positive allosteric modulators attenuate morphine tolerance and dependence. Neuropharmacology 2018, 137, 50–58. [Google Scholar] [CrossRef]
- Wijayawardhane, N.; Shonesy, B.C.; Vaglenova, J.; Vaithianathan, T.; Carpenter, M.; Breese, C.R.; Dityatev, A.; Suppiramaniam, V. Postnatal aniracetam treatment improves prenatal ethanol induced attenuation of AMPA receptor-mediated synaptic transmission. Neurobiol. Dis. 2007, 26, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Vaglenova, J.; Pandiella, N.; Wijayawardhane, N.; Vaithianathan, T.; Birru, S.; Breese, C.; Suppiramaniam, V.; Randal, C. Aniracetam reversed learning and memory deficits following prenatal ethanol exposure by modulating functions of synaptic AMPA receptors. Neuropsychopharmacology 2008, 33, 1071–1083. [Google Scholar] [CrossRef]
- Manetti, D.; Ghelardini, C.; Bartolini, A.; Bellucci, C.; Dei, S.; Galeotti, N.; Gualtieri, F.; Romanelli, M.N.; Scapecchi, S.; Teodori, E. Design, synthesis, and preliminary pharmacological evaluation of 1,4-diazabicyclo[4.3.0]nonan-9-ones as a new class of highly potent nootropic agents. J. Med. Chem. 2000, 43, 1969–1974. [Google Scholar] [CrossRef]
- Manetti, D.; Ghelardini, C.; Bartolini, A.; Dei, S.; Galeotti, N.; Gualtieri, F.; Romanelli, M.N.; Teodori, E. Molecular simplification of 1,4-diazabicyclo[4.3.0]nonan-9-ones gives piperazine derivatives that maintain high nootropic activity. J. Med. Chem. 2000, 43, 4499–4507. [Google Scholar] [CrossRef] [PubMed]
- Romanelli, M.N.; Galeotti, N.; Ghelardini, C.; Manetti, D.; Martini, E.; Gualtieri, F. Pharmacological characterization of DM232 (unifiram) and DM235 (sunifiram), new potent cognition enhancers. CNS Drug Rev. 2006, 12, 39–52. [Google Scholar] [CrossRef]
- Gualtieri, F. Unifi nootropics from the lab to the web: A story of academic (and industrial) shortcomings. J. Enzym. Inhib. Med. Chem. 2016, 31, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Arai, A.C.; Xia, Y.-F.; Rogers, G.; Lynch, G.; Kessler, M. Benzamide-type AMPA receptor modulators form two subfamilies with distinct modes of action. J. Pharmacol. Exp. Ther. 2002, 303, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Krintel, C.; Harpsøe, K.; Zachariassen, L.G.; Peters, D.; Frydenvang, K.; Pickering, D.S.; Gajhede, M.; Kastrup, J.S. Structural analysis of the positive AMPA receptor modulators CX516 and Me-CX516 in complex with the GluA2 ligand-binding domain. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1645–1652. [Google Scholar] [CrossRef]
- Murru, L.; Vezzoli, E.; Longatti, A.; Ponzoni, L.; Falqui, A.; Folci, A.; Moretto, E.; Bianchi, V.; Braida, D.; Sala, M.; et al. Pharmacological modulation of AMPAR rescues intellectual disability-like phenotype in tm4sf2−/y mice. Cereb. Cortex 2017, 27, 5369–5384. [Google Scholar] [CrossRef]
- Tatsukawa, T.; Raveau, M.; Ogiwara, I.; Hattori, S.; Miyamoto, H.; Mazaki, E.; Itohara, S.; Miyakawa, T.; Montal, M.; Yamakawa, K. Scn2a haploinsufficient mice display a spectrum of phenotypes affecting anxiety, sociability, memory flexibility and ampakine CX516 rescues their hyperactivity. Mol. Autism 2019, 10, 15. [Google Scholar] [CrossRef]
- Yao, H.; Zhang, D.; Yu, H.; Shen, H.; Lan, X.; Liu, H.; Chen, X.; Wu, X.; Zhang, G.; Wang, X. AMPAkine CX516 alleviated chronic ethanol exposure-induced neurodegeneration and depressive-like behavior in mice. Toxicol. Appl. Pharmacol. 2022, 439, 115924. [Google Scholar] [CrossRef]
- Güvel, M.C.; Aykan, U.; Paykal, G.; Uluoğlu, C. Chronic administration of caffeine, modafinil, AVL-3288 and CX516 induces time-dependent complex effects on cognition and mood in an animal model of sleep deprivation. Pharmacol. Biochem. Behav. 2024, 241, 173793. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, K.; Martinez, E.; Dale, J.; Huang, D.; Wang, J. AMPAkines and morphine provide complementary analgesia. Behav. Brain Res. 2017, 334, 1–5. [Google Scholar] [CrossRef]
- Le, A.M.; Lee, M.; Su, C.; Zou, A.; Wang, J. AMPAkines have novel analgesic properties in rat models of persistent neuropathic and inflammatory pain. Anesthesiology 2014, 121, 1080–1090. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Poon, B.Y.; Tang, Y.; Funk, G.D.; Greer, J.J. Ampakines alleviate respiratory depression in rats. Am. J. Respir. Crit. Care Med. 2006, 174, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Cichon, J.; Ninan, I.; Yang, G. Post-anesthesia AMPA receptor potentiation prevents anesthesia-induced learning and synaptic deficits. Sci. Transl. Med. 2016, 8, 344ra385. [Google Scholar] [CrossRef]
- Rogers, G.A.; Marrs, C.M. Benzofurazan compounds for enhancing glutamatergic synaptic responses. U.S. Patent 6730677B2, 29 August 2000. [Google Scholar]
- Radin, D.P.; Cerne, R.; Smith, J.L.; Witkin, J.M.; Lippa, A. Antipsychotic-like pharmacological profile of the low impact ampakine CX691 (farampator): Implications for the use of low impact ampakines in the treatment of schizophrenia. J. Psychiatr. Res. 2025, 186, 145–153. [Google Scholar] [CrossRef]
- Mozafari, N.; Shamsizadeh, A.; Fatemi, I.; Allahtavakoli, M.; Moghadam-Ahmadi, A.; Kaviani, E.; Kaeidi, A. CX691, as an AMPA receptor positive modulator, improves the learning and memory in a rat model of Alzheimer’s disease. Iran. J. Basic. Med. Sci. 2018, 21, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Radin, D.P.; Zhong, S.; Cerne, R.; Smith, J.L.; Witkin, J.M.; Lippa, A. Preclinical pharmacology of the low-impact ampakine CX717. Future Pharmacol. 2024, 4, 494–509. [Google Scholar] [CrossRef]
- Greer, J.J.; Ren, J. Ampakine therapy to counter fentanyl-induced respiratory depression. Respir. Physiol. Neurobiol. 2009, 168, 153–157. [Google Scholar] [CrossRef]
- Turner, S.M.; ElMallah, M.K.; Hoyt, A.K.; Greer, J.J.; Fuller, D.D. Ampakine CX717 potentiates intermittent hypoxia-induced hypoglossal long-term facilitation. J. Neurophysiol. 2016, 116, 1232–1238. [Google Scholar] [CrossRef]
- Rana, S.; Sunshine, M.D.; Greer, J.J.; Fuller, D.D. Ampakines stimulate diaphragm activity after spinal cord injury. J. Neurotrauma 2021, 38, 3467–3482. [Google Scholar] [CrossRef]
- Rana, S.; Thakre, P.P.; Fuller, D.D. Ampakines increase diaphragm activation following mid-cervical contusion injury in rats. Exp. Neurol. 2024, 376, 114769. [Google Scholar] [CrossRef]
- Thakre, P.P.; Sunshine, M.D.; Fuller, D.D. Spinally delivered ampakine CX717 increases phrenic motor output in adult rats. Respir. Physiol. Neurobiol. 2022, 296, 103814. [Google Scholar] [CrossRef] [PubMed]
- Thakre, P.P.; Fuller, D.D. Pattern sensitivity of ampakine-hypoxia interactions for evoking phrenic motor facilitation in anesthetized rat. J. Neurophysiol. 2024, 131, 216–224. [Google Scholar] [CrossRef]
- Gordillo-Salas, M.; Pascual-Antón, R.; Ren, J.; Greer, J.; Adell, A. Antidepressant-like effects of CX717, a positive allosteric modulator of AMPA receptors. Mol. Neurobiol. 2020, 57, 3498–3507. [Google Scholar] [CrossRef] [PubMed]
- Radin, D.P.; Zhong, S.; Cerne, R.; Shoaib, M.; Witkin, J.M.; Lippa, A. Low-impact ampakine CX1739 exerts pro-cognitive effects and reverses opiate-induced respiratory depression in rodents. Future Pharmacol. 2024, 4, 173–187. [Google Scholar] [CrossRef]
- Xiao, D.; Xie, F.; Xu, X.; Zhou, X. The impact and mechanism of ampakine CX1739 on protection against respiratory depression in rats. Future Med. Chem. 2020, 12, 2093–2104. [Google Scholar] [CrossRef]
- Dai, W.; Gao, X.; Xiao, D.; Li, Y.L.; Zhou, X.B.; Yong, Z.; Su, R.B. The impact and mechanism of a novel allosteric AMPA receptor modulator LCX001 on protection against respiratory depression in rodents. Front. Pharmacol. 2019, 10, 105. [Google Scholar] [CrossRef]
- Su, X.W.; Li, X.-Y.; Banasr, M.; Koo, J.W.; Shahid, M.; Henry, B.; Duman, R.S. Chronic treatment with AMPA receptor potentiator Org 26576 increases neuronal cell proliferation and survival in adult rodent hippocampus. Psychopharmacology 2009, 206, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, F.; Calabrese, F.; Luoni, A.; Shahid, M.; Racagni, G.; Riva, M.A. The AMPA receptor potentiator Org 26576 modulates stress-induced transcription of BDNF isoforms in rat hippocampus. Pharmacol. Res. 2012, 65, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Hamlyn, E.; Brand, L.; Shahid, M.; Harvey, B.H. The ampakine, Org 26576, bolsters early spatial reference learning and retrieval in the Morris water maze: A subchronic, dose-ranging study in rats. Behav. Pharmacol. 2009, 20, 662–667. [Google Scholar] [CrossRef]
- Arai, A.C.; Kessler, M.; Rogers, G.; Lynch, G. Effects of the potent ampakine CX614 on hippocampal and recombinant AMPA receptors: Interactions with cyclothiazide and GYKI 52466. Mol. Pharmacol. 2000, 58, 802–813. [Google Scholar] [CrossRef]
- Lauterborn, J.C.; Lynch, G.; Vanderklish, P.; Arai, A.; Gall, C.M. Positive modulation of AMPA receptors increases neurotrophin expression by hippocampal and cortical neurons. J. Neurosci. 2000, 20, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Wolak, M.; Siwek, A.; Szewczyk, B.; Poleszak, E.; Pilc, A.; Popik, P.; Nowak, G. Involvement of NMDA and AMPA receptors in the antidepressant-like activity of antidepressant drugs in the forced swim test. Pharmacol. Rep. 2013, 65, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.M.; Severs, L.; Moreira, T.S.; Ramirez, J.-M.; Takakura, A.C. Ampakine CX614 increases respiratory rate in a mouse model of Parkinson’s disease. Brain Res. 2023, 1815, 148448. [Google Scholar] [CrossRef] [PubMed]
- Radin, D.P.; Purcell, R.; Lippa, A.S. Oncolytic properties of ampakines in vitro. Anticancer. Res. 2018, 38, 265–269. [Google Scholar] [CrossRef]
- Cordi, A. 3-Substituted-[1,2,3]-benzotriazinone compound for enhancing glutamatergic synaptic responses. U.S. Patent 2010137295A1, 3 June 2010. [Google Scholar]
- Mueller, R.; Li, Y.-X.; Hampson, A.; Zhong, S.; Harris, C.; Marrs, C.; Rachwal, S.; Ulas, J.; Nielsson, L.; Rogers, G. Benzoxazinones as potent positive allosteric AMPA receptor modulators: Part I. Bioorg Med. Chem. Lett. 2011, 21, 3923–3926. [Google Scholar] [CrossRef]
- Mueller, R.; Rachwal, S.; Tedder, M.E.; Li, Y.-X.; Zhong, S.; Hampson, A.; Ulas, J.; Varney, M.; Nielsson, L.; Rogers, G. Substituted benzoxazinones as potent positive allosteric AMPA receptor modulators: Part II. Bioorg Med. Chem. Lett. 2011, 21, 3927–3930. [Google Scholar] [CrossRef]
- Mueller, R.; Rachwal, S.; Lee, S.; Zhong, S.; Li, Y.-X.; Haroldsen, P.; Herbst, T.; Tanimura, S.; Varney, M.; Johnson, S.; et al. Benzotriazinone and benzopyrimidinone derivatives as potent positive allosteric AMPA receptor modulators. Bioorg Med. Chem. Lett. 2011, 21, 6170–6175. [Google Scholar] [CrossRef]
- Bretin, S.; Louis, C.; Seguin, L.; Wagner, S.; Thomas, J.-Y.; Challal, S.; Rogez, N.; Albinet, K.; Iop, F.; Villain, N.; et al. Pharmacological characterisation of S 47445, a novel positive allosteric modulator of AMPA receptors. PLoS ONE 2017, 12, e0184429. [Google Scholar] [CrossRef]
- Mueller, R.; Rachwal, S.; Varney, M.A.; Johnson, S.A.; Alisala, K.; Zhong, S.; Li, Y.-X.; Haroldsen, P.; Herbst, T.; Street, L.J. Benzobistriazinones and related heterocyclic ring systems as potent, orally bioavailable positive allosteric AMPA receptor modulators. Bioorg Med. Chem. Lett. 2011, 21, 7455–7459. [Google Scholar] [CrossRef]
- Giralt, A.; Gómez-Climent, M.Á.; Alcalá, R.; Bretin, S.; Bertrand, D.; María Delgado-García, J.; Pérez-Navarro, E.; Alberch, J.; Gruart, A. The AMPA receptor positive allosteric modulator S 47445 rescues in vivo CA3-CA1 long-term potentiation and structural synaptic changes in old mice. Neuropharmacology 2017, 123, 395–409. [Google Scholar] [CrossRef]
- Calabrese, F.; Savino, E.; Mocaer, E.; Bretin, S.; Racagni, G.; Riva, M.A. Upregulation of neurotrophins by S 47445, a novel positive allosteric modulator of AMPA receptors in aged rats. Pharmacol. Res. 2017, 121, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Wang, Y.; Jiang, M.; Warren, P.; Chen, G. Cyclothiazide induces robust epileptiform activity in rat hippocampal neurons both in vitro and in vivo. J. Physiol. 2006, 571, 605–618. [Google Scholar] [CrossRef]
- Kong, S.; Qian, B.; Liu, J.; Fan, M.; Chen, G.; Wang, Y. Cyclothiazide induces seizure behavior in freely moving rats. Brain Res. 2010, 1355, 207–213. [Google Scholar] [CrossRef]
- Deng, L.; Chen, G. Cyclothiazide potently inhibits gamma-aminobutyric acid type A receptors in addition to enhancing glutamate responses. Proc. Natl. Acad. Sci. USA 2003, 100, 13025–13029. [Google Scholar] [CrossRef] [PubMed]
- Surin, A.; Pshenichkin, S.; Grajkowska, E.; Surina, E.; Wroblewski, J.T. Cyclothiazide selectively inhibits mGluR1 receptors interacting with a common allosteric site for non-competitive antagonists. Neuropharmacology 2007, 52, 744–754. [Google Scholar] [CrossRef]
- Yamada, K.A.; Covey, D.F.; Hsu, C.Y.; Hu, R.; Hu, Y.; He, Y.Y. The diazoxide derivative IDRA 21 enhances ischemic hippocampal neuron injury. Ann. Neurol. 1998, 43, 664–669. [Google Scholar] [CrossRef]
- Lockhart, B.; Iop, F.; Closier, M.; Lestage, P. (S)-2,3-dihydro-[3,4]cyclopentano-1,2,4-benzothiadiazine-1,1-dioxide: (S18986-1) a positive modulator of AMPA receptors enhances (S)-AMPA-mediated [3H]noradrenaline release from rat hippocampal and frontal cortex slices. Eur. J. Pharmacol. 2000, 401, 145–153. [Google Scholar] [CrossRef]
- Lebrun, C.; Pillière, E.; Lestage, P. Effects of S 18986-1, a novel cognitive enhancer, on memory performances in an object recognition task in rats. Eur. J. Pharmacol. 2000, 401, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Francotte, P.; de Tullio, P.; Goffin, E.; Dintilhac, G.; Graindorge, E.; Fraikin, P.; Lestage, P.; Danober, L.; Thomas, J.-Y.; Caignard, D.-H.; et al. Design, synthesis, and pharmacology of novel 7-substituted 3,4-dihydro-2H-1,2,4-benzothiadiazine 1,1-dioxides as positive allosteric modulators of AMPA receptors. J. Med. Chem. 2007, 50, 3153–3157. [Google Scholar] [CrossRef]
- Francotte, P.; Goffin, E.; Fraikin, P.; Lestage, P.; Van Heugen, J.-C.; Gillotin, F.; Danober, L.; Thomas, J.-Y.; Chiap, P.; Caignard, D.-H.; et al. New fluorinated 1,2,4-benzothiadiazine 1,1-dioxides: Discovery of an orally active cognitive enhancer acting through potentiation of the 2-amino-3-(3-hydroxy-5-methylisoxazol-4-yl)propionic acid receptors. J. Med. Chem. 2010, 53, 1700–1711. [Google Scholar] [CrossRef]
- Nørholm, A.-B.; Francotte, P.; Olsen, L.; Krintel, C.; Frydenvang, K.; Goffin, E.; Challal, S.; Danober, L.; Botez-Pop, I.; Lestage, P.; et al. Synthesis, pharmacological and structural characterization, and thermodynamic aspects of GluA2-positive allosteric modulators with a 3,4-dihydro-2H-1,2,4-benzothiadiazine 1,1-dioxide scaffold. J. Med. Chem. 2013, 56, 8736–8745. [Google Scholar] [CrossRef] [PubMed]
- Etsè, K.S.; Dorosz, J.; McLain Christensen, K.; Thomas, J.-Y.; Botez Pop, I.; Goffin, E.; Colson, T.; Lestage, P.; Danober, L.; Pirotte, B.; et al. Development of thiochroman dioxide analogues of benzothiadiazine dioxides as new positive allosteric modulators of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. ACS Chem. Neurosci. 2021, 12, 2679–2692. [Google Scholar] [CrossRef] [PubMed]
- Francotte, P.; Bay, Y.; Goffin, E.; Colson, T.; Lesenfants, C.; Dorosz, J.; Laulumaa, S.; Fraikin, P.; de Tullio, P.; Beaufour, C.; et al. Exploring thienothiadiazine dioxides as isosteric analogues of benzo- and pyridothiadiazine dioxides in the search of new AMPA and kainate receptor positive allosteric modulators. Eur. J. Med. Chem. 2024, 264, 116036. [Google Scholar] [CrossRef]
- Larsen, A.P.; Fièvre, S.; Frydenvang, K.; Francotte, P.; Pirotte, B.; Kastrup, J.S.; Mulle, C. Identification and structure-function study of positive allosteric modulators of kainate receptors. Mol. Pharmacol. 2017, 91, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Bay, Y.; Egeberg Jeppesen, M.; Frydenvang, K.; Francotte, P.; Pirotte, B.; Pickering, D.S.; Kristensen, A.S.; Kastrup, J.S. The positive allosteric modulator BPAM344 and L-glutamate introduce an active-like structure of the ligand-binding domain of GluK2. FEBS Lett. 2024, 598, 743–757. [Google Scholar] [CrossRef]
- Bay, Y.; Venskutonytė, R.; Frantsen, S.M.; Thorsen, T.S.; Musgaard, M.; Frydenvang, K.; Francotte, P.; Pirotte, B.; Biggin, P.C.; Kristensen, A.S.; et al. Small-molecule positive allosteric modulation of homomeric kainate receptors GluK1-3: Development of screening assays and insight into GluK3 structure. FEBS J. 2024, 291, 1506–1529. [Google Scholar] [CrossRef]
- Gangwar, S.P.; Yen, L.Y.; Yelshanskaya, M.V.; Sobolevsky, A.I. Positive and negative allosteric modulation of GluK2 kainate receptors by BPAM344 and antiepileptic perampanel. Cell Rep. 2023, 42, 112124. [Google Scholar] [CrossRef]
- Chen, J.; Ran, W.; Huang, Y.; Wei, J.; Rong, J.; Wei, H.; Li, Y.; Li, G.; Chen, Z.; Collier, L.; et al. Evaluation of thiadiazine-based PET radioligands for imaging the AMPA receptor. Biomed. Pharmacother. 2023, 168, 115842. [Google Scholar] [CrossRef]
- Goffin, E.; Drapier, T.; Larsen, A.P.; Geubelle, P.; Ptak, C.P.; Laulumaa, S.; Rovinskaja, K.; Gilissen, J.; de Tullio, P.; Olsen, L.; et al. 7-phenoxy-substituted 3,4-dihydro-2h-1,2,4-benzothiadiazine 1,1-dioxides as positive allosteric modulators of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors with nanomolar potency. J. Med. Chem. 2018, 61, 251–264. [Google Scholar] [CrossRef]
- Krintel, C.; Francotte, P.; Pickering, D.S.; Juknaitė, L.; Pøhlsgaard, J.; Olsen, L.; Frydenvang, K.; Goffin, E.; Pirotte, B.; Kastrup, J.S. Enthalpy-entropy compensation in the binding of modulators at ionotropic glutamate receptor GluA2. Biophys. J. 2016, 110, 2397–2406. [Google Scholar] [CrossRef]
- Bay, Y.; Cabello, F.J.M.; Koens, C.C.; Frantsen, S.M.; Pickering, D.S.; Frydenvang, K.; Francotte, P.; Pirotte, B.; Kristensen, A.S.; Bowie, D.; et al. Crystal structure of the GluK1 ligand-binding domain with kainate and the full-spanning positive allosteric modulator BPAM538. J. Struct. Biol. 2024, 216, 108113. [Google Scholar] [CrossRef]
- Drapier, T.; Geubelle, P.; Bouckaert, C.; Nielsen, L.; Laulumaa, S.; Goffin, E.; Dilly, S.; Francotte, P.; Hanson, J.; Pochet, L.; et al. Enhancing action of positive allosteric modulators through the design of dimeric compounds. J. Med. Chem. 2018, 61, 5279–5291. [Google Scholar] [CrossRef]
- Chen, J.; Gan, J.; Sun, J.; Chen, Z.; Fu, H.; Rong, J.; Deng, X.; Shang, J.; Gong, J.; Shao, T.; et al. Radiosynthesis and preliminary evaluation of (11)C-labeled 4-cyclopropyl-7-(3-methoxyphenoxy)-3,4-dihydro-2H-benzo[e] [1,2,4] thiadiazine 1,1-dioxide for PET imaging AMPA receptors. Tetrahedron Lett. 2020, 61, 151635. [Google Scholar] [CrossRef]
- Battisti, U.M.; Jozwiak, K.; Cannazza, G.; Puia, G.; Stocca, G.; Braghiroli, D.; Parenti, C.; Brasili, L.; Carrozzo, M.M.; Citti, C.; et al. 5-arylbenzothiadiazine type compounds as positive allosteric modulators of AMPA/kainate receptors. ACS Med. Chem. Lett. 2012, 3, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Puja, G.; Ravazzini, F.; Losi, G.; Bardoni, R.; Battisti, U.M.; Citti, C.; Cannazza, G. Benzothiadiazines derivatives as novel allosteric modulators of kainic acid receptors. J. Physiol. Pharmacol. 2022, 73, 9–28I. [Google Scholar] [CrossRef]
- Citti, C.; Battisti, U.M.; Cannazza, G.; Jozwiak, K.; Stasiak, N.; Puja, G.; Ravazzini, F.; Ciccarella, G.; Braghiroli, D.; Parenti, C.; et al. 7-Chloro-5-(furan-3-yl)-3-methyl-4H-benzo[e][1,2,4]thiadiazine 1,1-dioxide as positive allosteric modulator of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor. The end of the unsaturated-inactive paradigm? ACS Chem. Neurosci. 2016, 7, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Carrozzo, M.M.; Battisti, U.M.; Cannazza, G.; Puia, G.; Ravazzini, F.; Falchicchio, A.; Perrone, S.; Citti, C.; Jozwiak, K.; Braghiroli, D.; et al. Design, stereoselective synthesis, configurational stability and biological activity of 7-chloro-9-(furan-3-yl)-2,3,3a,4-tetrahydro-1H-benzo[e]pyrrolo[2,1-c][1,2,4]thiadiazine 5,5-dioxide. Bioorg Med. Chem. 2014, 22, 4667–4676. [Google Scholar] [CrossRef]
- Kunugi, A.; Tanaka, M.; Suzuki, A.; Tajima, Y.; Suzuki, N.; Suzuki, M.; Nakamura, S.; Kuno, H.; Yokota, A.; Sogabe, S.; et al. TAK-137, an AMPA-R potentiator with little agonistic effect, has a wide therapeutic window. Neuropsychopharmacology 2019, 44, 961–970. [Google Scholar] [CrossRef]
- Suzuki, A.; Kunugi, A.; Tajima, Y.; Suzuki, N.; Suzuki, M.; Toyofuku, M.; Kuno, H.; Sogabe, S.; Kosugi, Y.; Awasaki, Y.; et al. Strictly regulated agonist-dependent activation of AMPA-R is the key characteristic of TAK-653 for robust synaptic responses and cognitive improvement. Sci. Rep. 2021, 11, 14532. [Google Scholar] [CrossRef]
- Tanaka, M.; Kunugi, A.; Suzuki, A.; Suzuki, N.; Suzuki, M.; Kimura, H. Preclinical characterization of AMPA receptor potentiator TAK-137 as a therapeutic drug for schizophrenia. Pharmacol. Res. Perspect. 2019, 7, e479. [Google Scholar] [CrossRef]
- Suzuki, A.; Murakami, K.; Tajima, Y.; Hara, H.; Kunugi, A.; Kimura, H. TAK-137, an AMPA receptor potentiator with little agonistic effect, produces antidepressant-like effect without causing psychotomimetic effects in rats. Pharmacol. Biochem. Behav. 2019, 183, 80–86. [Google Scholar] [CrossRef]
- Hara, H.; Suzuki, A.; Kunugi, A.; Tajima, Y.; Yamada, R.; Kimura, H. TAK-653, an AMPA receptor potentiator with minimal agonistic activity, produces an antidepressant-like effect with a favorable safety profile in rats. Pharmacol. Biochem. Behav. 2021, 211, 173289. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Hara, H.; Kimura, H. Role of the AMPA receptor in antidepressant effects of ketamine and potential of AMPA receptor potentiators as a novel antidepressant. Neuropharmacology 2023, 222, 109308. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, M.; Nishikawa, K.; Aoki, S.; Wada, K. A desensitization-selective potentiator of AMPA-type glutamate receptors. Br. J. Pharmacol. 2002, 136, 1033–1041. [Google Scholar] [CrossRef]
- Zushida, K.; Sakurai, M.; Wada, K.; Sekiguchi, M. Facilitation of extinction learning for contextual fear memory by PEPA: A potentiator of AMPA receptors. J. Neurosci. 2007, 27, 158–166. [Google Scholar] [CrossRef]
- Yamada, D.; Wada, K.; Sekiguchi, M. Facilitating actions of an AMPA receptor potentiator upon extinction of contextually conditioned fear response in stressed mice. Neurosci. Lett. 2011, 488, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Miu, P.; Jarvie, K.R.; Radhakrishnan, V.; Gates, M.R.; Ogden, A.; Ornstein, P.L.; Zarrinmayeh, H.; Ho, K.; Peters, D.; Grabell, J.; et al. Novel AMPA receptor potentiators LY392098 and LY404187: Effects on recombinant human AMPA receptors in vitro. Neuropharmacology 2001, 40, 976–983. [Google Scholar] [CrossRef]
- Gates, M.; Ogden, A.; Bleakman, D. Pharmacological effects of AMPA receptor potentiators LY392098 and LY404187 on rat neuronal AMPA receptors in vitro. Neuropharmacology 2001, 40, 984–991. [Google Scholar] [CrossRef]
- Baumbarger, P.; Muhlhauser, M.; Yang, C.R.; Nisenbaum, E.S. LY392098, a novel AMPA receptor potentiator: Electrophysiological studies in prefrontal cortical neurons. Neuropharmacology 2001, 40, 992–1002. [Google Scholar] [CrossRef]
- Quirk, J.C.; Nisenbaum, E.S. LY404187: A novel positive allosteric modulator of AMPA receptors. CNS Neurosci. Ther. 2002, 8, 255–282. [Google Scholar] [CrossRef]
- Deutschenbaur, L.; Beck, J.; Kiyhankhadiv, A.; Mühlhauser, M.; Borgwardt, S.; Walter, M.; Hasler, G.; Sollberger, D.; Lang, U.E. Role of calcium, glutamate and NMDA in major depression and therapeutic application. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tizzano, J.P.; Griffey, K.; Clay, M.; Lindstrom, T.; Skolnick, P. Antidepressant-like actions of an AMPA receptor potentiator (LY392098). Neuropharmacology 2001, 40, 1028–1033. [Google Scholar] [CrossRef]
- Andreasen, J.T.; Fitzpatrick, C.M.; Larsen, M.; Skovgaard, L.; Nielsen, S.D.; Clausen, R.P.; Troelsen, K.; Pickering, D.S. Differential role of AMPA receptors in mouse tests of antidepressant and anxiolytic action. Brain Res. 2015, 1601, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, C.M.; Larsen, M.; Madsen, L.H.; Caballero-Puntiverio, M.; Pickering, D.S.; Clausen, R.P.; Andreasen, J.T. Positive allosteric modulation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid glutamate receptors differentially modulates the behavioural effects of citalopram in mouse models of antidepressant and anxiolytic action. Behav. Pharmacol. 2016, 27, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, C.L.; Hurst, R.S.; Scialis, R.J.; Osgood, S.M.; Bryce, D.K.; Hoffmann, W.E.; Lazzaro, J.T.; Hanks, A.N.; Lotarski, S.; Weber, M.L.; et al. Positive allosteric modulation of AMPA receptors from efficacy to toxicity: The interspecies exposure-response continuum of the novel potentiator PF-4778574. J. Pharmacol. Exp. Ther. 2013, 347, 212–224. [Google Scholar] [CrossRef]
- Shaffer, C.L.; Patel, N.C.; Schwarz, J.; Scialis, R.J.; Wei, Y.; Hou, X.J.; Xie, L.; Karki, K.; Bryce, D.K.; Osgood, S.M.; et al. The discovery and characterization of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor potentiator N-((3 S,4 S)-4-[4-(5-cyano-2-thienyl)phenoxy]tetrahydrofuran-3-yl)propane-2-sulfonamide (PF-04958242). J. Med. Chem. 2015, 58, 4291–4308. [Google Scholar] [CrossRef]
- Ward, S.E.; Beswick, P.; Calcinaghi, N.; Dawson, L.A.; Gartlon, J.; Graziani, F.; Jones, D.N.C.; Lacroix, L.; Selina Mok, M.H.; Oliosi, B.; et al. Pharmacological characterization of N-[(2S)-5-(6-fluoro-3-pyridinyl)-2, 3-dihydro-1H-inden-2-yl]-2-propanesulfonamide: A novel, clinical AMPA receptor positive allosteric modulator. Br. J. Pharmacol. 2017, 174, 370–385. [Google Scholar] [CrossRef]
- Patel, N.C.; Schwarz, J.; Hou, X.J.; Hoover, D.J.; Xie, L.; Fliri, A.J.; Gallaschun, R.J.; Lazzaro, J.T.; Bryce, D.K.; Hoffmann, W.E.; et al. Discovery and characterization of a novel dihydroisoxazole class of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor potentiators. J. Med. Chem. 2013, 56, 9180–9191. [Google Scholar] [CrossRef]
- Estep, K.G.; Fliri, A.F.J.; O’Donnell, C.J. Sulfonamides and pharmaceutical compositions thereof. WO Patent 2008120093A1, 9 August 2008. [Google Scholar]
- Estep, K.G.; Fliri, A.F.J.; Gallaschun, R.J.; O’Donnell, C.J.; Patel, N.C.; Schwarz, J.B.; Xie, L. Oxopiperdinyl and pyranyl sulfonamides and pharmaceutical compositions thereof. U.S. Patent 20110172297, 14 July 2011. [Google Scholar]
- Estep, K.G.; O’Donnell, C.J.; Xie, L. Tetrahydrofuranyl sulfonamides and pharmaceutical compositions thereof. U.S. Patent 20110178165A1, 21 July 2011. [Google Scholar]
- Reuillon, T.; Ward, S.E.; Beswick, P. AMPA receptor positive allosteric modulators: Potential for the treatment of neuropsychiatric and neurological disorders. Curr. Top. Med. Chem. 2016, 16, 3536–3565. [Google Scholar] [CrossRef]
- Shen, M.; Lv, D.; Li, S.; Zhang, Y.; Wang, Z.; Zhao, C.; Chen, X.; Wang, C. Positive allosteric modulation of AMPAR by PF-4778574 produced rapid onset antidepressant actions in mice. Cereb. Cortex 2019, 29, 4438–4451. [Google Scholar] [CrossRef]
- Guo, L.; Wang, S.; Tian, H.; Shang, M.; Xu, J.; Wang, C. Novel synergistic treatment for depression: Involvement of GSK3β-regulated AMPA receptors in the prefrontal cortex of mice. Cereb. Cortex 2023, 33, 10504–10513. [Google Scholar] [CrossRef]
- Sindi, M.; Dietrich, M.; Klees, D.; Gruchot, J.; Hecker, C.; Silbereis, J.; Issberner, A.; Hartung, H.P.; Ruck, T.; Stark, H.; et al. Positive allosteric modulation of AMPA receptors via PF4778574 leads to reduced demyelination and clinical disability in experimental models of multiple sclerosis. Front. Immunol. 2025, 16, 1532877. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Jia, K.; Arakawa, R.; Datta, P.; Scott, D.; Shaffer, C.; Moein, M.M.; Hutchison, M.; Kaliszczak, M.; Halldin, C. Synthesis of [(11)C]BIIB104, an α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic-acid-positive allosteric modulator, and evaluation of the bio-distribution in non-human primate brains using positron emission tomography. Molecules 2024, 29, 427. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Huang, J.; Muste, C.; Zhong, J.; Walker, G.S.; Obach, R.S.; Shaffer, C.L. Radiolabel uncovers nonintuitive metabolites of BIIB104: Novel release of [(14)C]cyanide from 2-cyanothiophene and subsequent formation of [(14)C]thiocyanate. Drug Metab. Dispos. 2024, 52, 323–336. [Google Scholar] [CrossRef]
- Ward, S.E.; Harries, M.; Aldegheri, L.; Andreotti, D.; Ballantine, S.; Bax, B.D.; Harris, A.J.; Harker, A.J.; Lund, J.; Melarange, R.; et al. Discovery of N -[(2 S)-5-(6-fluoro-3-pyridinyl)-2,3-dihydro-1 H -inden-2-yl]-2-propanesulfonamide, a novel clinical AMPA receptor positive modulator. J. Med. Chem. 2010, 53, 5801–5812. [Google Scholar] [CrossRef]
- Knobelsdorf, J.A.; Shepherd, T.A.; Tromiczak, E.; Zarrinmayeh, H.; Zimmerman, D.M. Sulfonamide derivatives. U.S. Patent 2003229102A1, 11 December 2003. [Google Scholar]
- Kunugi, A.; Tajima, Y.; Kuno, H.; Sogabe, S.; Kimura, H. HBT1, a novel AMPA receptor potentiator with lower agonistic effect, avoided bell-shaped response in in vitro BDNF productions. J. Pharmacol. Exp. Ther. 2018, 364, 377–389. [Google Scholar] [CrossRef]
- Miraucourt, L.S.; Accardi, M.V.; Asin, K.E.; Pugsley, M.K.; Curtis, M.J.; Authier, S. The application of electrophysiological methods to characterize AMPA receptors in dissociated adult rat and non-human primate cerebellar neurons for use in neuronal safety pharmacology assessments of the central nervous system. J. Pharmacol. Toxicol. Methods 2020, 105, 106883. [Google Scholar] [CrossRef]
- Kaae, B.H.; Harpsøe, K.; Kastrup, J.S.; Sanz, A.C.; Pickering, D.S.; Metzler, B.; Clausen, R.P.; Gajhede, M.; Sauerberg, P.; Liljefors, T.; et al. Structural proof of a dimeric positive modulator bridging two identical AMPA receptor-binding sites. Cell Chem. Biol. 2007, 14, 1294–1303. [Google Scholar] [CrossRef]
- Timm, D.E.; Benveniste, M.; Weeks, A.M.; Nisenbaum, E.S.; Partin, K.M. Structural and functional analysis of two new positive allosteric modulators of GluA2 desensitization and deactivation. Mol. Pharmacol. 2011, 80, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Malá, H.; Chen, Y.; Worm, V.H.; Kure, J.; Kaae, B.H.; Madsen, U.; Badolo, L.; Pickering, D.S.; Mogensen, J. Cognitive enhancing effects of an AMPA receptor positive modulator on place learning in mice. Behav. Brain Res. 2012, 226, 18–25. [Google Scholar] [CrossRef]
- Weeks, A.M.; Harms, J.E.; Partin, K.M.; Benveniste, M. Functional insight into development of positive allosteric modulators of AMPA receptors. Neuropharmacology 2014, 85, 57–66. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, H.; Johnson, K.M.; Wang, C.Z. Bivalent AMPA receptor positive allosteric modulators. U.S. Patent 9328125B2, 3 May 2016. [Google Scholar]
- Jamieson, C.; Basten, S.; Campbell, R.A.; Cumming, I.A.; Gillen, K.J.; Gillespie, J.; Kazemier, B.; Kiczun, M.; Lamont, Y.; Lyons, A.J.; et al. A novel series of positive modulators of the AMPA receptor: Discovery and structure based hit-to-lead studies. Bioorg Med. Chem. Lett. 2010, 20, 5753–5756. [Google Scholar] [CrossRef]
- Jamieson, C.; Campbell, R.A.; Cumming, I.A.; Gillen, K.J.; Gillespie, J.; Kazemier, B.; Kiczun, M.; Lamont, Y.; Lyons, A.J.; MacLean, J.K.F.; et al. A novel series of positive modulators of the AMPA receptor: Structure-based lead optimization. Bioorg Med. Chem. Lett. 2010, 20, 6072–6075. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, C.; MacLean, J.K.F.; Brown, C.I.; Campbell, R.A.; Gillen, K.J.; Gillespie, J.; Kazemier, B.; Kiczun, M.; Lamont, Y.; Lyons, A.J.; et al. Structure based evolution of a novel series of positive modulators of the AMPA receptor. Bioorg Med. Chem. Lett. 2011, 21, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Harms, J.E.; Benveniste, M.; MacLean, J.K.F.; Partin, K.M.; Jamieson, C. Functional analysis of a novel positive allosteric modulator of AMPA receptors derived from a structure-based drug design strategy. Neuropharmacology 2013, 64, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.E.; Harries, M.; Aldegheri, L.; Austin, N.E.; Ballantine, S.; Ballini, E.; Bradley, D.M.; Bax, B.D.; Clarke, B.P.; Harris, A.J.; et al. Integration of lead optimization with crystallography for a membrane-bound ion channel target: Discovery of a new class of AMPA receptor positive allosteric modulators. J. Med. Chem. 2011, 54, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.E.; Harries, M.H.; Aldegheri, L.; Bradford, A.M.; Ballini, E.; Dawson, L.; Lacroix, L.; Pardoe, J.; Starr, K.; Weil, A.; et al. Pharmacological characterisation of MDI-222, a novel AMPA receptor positive allosteric modulator with an improved safety profile. J. Psychopharmacol. 2020, 34, 93–102. [Google Scholar] [CrossRef]
- Ward, S.E.; Bax, B.D.; Harries, M. Challenges for and current status of research into positive modulators of AMPA receptors. Br. J. Pharmacol. 2010, 160, 181–190. [Google Scholar] [CrossRef]
- Ward, S.; Beswick, P.; Pennicott, L.; Reuillon, T. AMPA receptor potentiators. U.S. Patent 201816482791A, 30 November 2021. [Google Scholar]
- Lavrov, M.I.; Grigor’ev, V.V.; Bachurin, S.O.; Palyulin, V.A.; Zefirov, N.S. Novel bivalent positive allosteric modulators of AMPA receptor. Dokl. Biochem. Biophys. 2015, 464, 322–324. [Google Scholar] [CrossRef]
- Nazarova, A.A.; Sedenkova, K.N.; Karlov, D.S.; Lavrov, M.I.; Grishin, Y.K.; Kuznetsova, T.S.; Zamoyski, V.L.; Grigoriev, V.V.; Averina, E.B.; Palyulin, V.A. Bivalent AMPA receptor positive allosteric modulators of the bis(pyrimidine) series. MedChemComm 2019, 10, 1615–1619. [Google Scholar] [CrossRef]
- Temnyakova, N.S.; Vasilenko, D.A.; Lavrov, M.I.; Karlov, D.S.; Grishin, Y.K.; Zamoyski, V.L.; Grigoriev, V.V.; Averina, E.B.; Palyulin, V.A. Novel bivalent positive allosteric AMPA receptor modulator of bis-amide series. Mendeleev Commun. 2021, 31, 216–218. [Google Scholar] [CrossRef]
- Sedenkova, K.N.; Zverev, D.V.; Nazarova, A.A.; Lavrov, M.I.; Radchenko, E.V.; Grishin, Y.K.; Gabrel’yan, A.V.; Zamoyski, V.L.; Grigoriev, V.V.; Averina, E.B.; et al. Novel nanomolar allosteric modulators of AMPA receptor of bis(pyrimidine) series: Synthesis, biotesting and SAR analysis. Molecules 2022, 27, 8252. [Google Scholar] [CrossRef]
- Vasilenko, D.A.; Temnyakova, N.S.; Dronov, S.E.; Radchenko, E.V.; Grishin, Y.K.; Gabrel’yan, A.V.; Zamoyski, V.L.; Grigoriev, V.V.; Averina, E.B.; Palyulin, V.A. 5-Nitroisoxazoles in SNAr reactions: A novel chemo- and regioselective approach to isoxazole-based bivalent ligands of AMPA receptors. Int. J. Mol. Sci. 2023, 24, 16135. [Google Scholar] [CrossRef] [PubMed]
- Karlov, D.S.; Lavrov, M.I.; Palyulin, V.A.; Zefirov, N.S. Pharmacophore analysis of positive allosteric modulators of AMPA receptors. Izv. Akad. Nauk Ser. Khim 2016, 65, 581–587. [Google Scholar] [CrossRef]
- Vasilenko, D.A.; Sadovnikov, K.S.; Sedenkova, K.N.; Karlov, D.S.; Radchenko, E.V.; Grishin, Y.K.; Rybakov, V.B.; Kuznetsova, T.S.; Zamoyski, V.L.; Grigoriev, V.V.; et al. A facile approach to bis(isoxazoles), promising ligands of the AMPA receptor. Molecules 2021, 26, 6411. [Google Scholar] [CrossRef] [PubMed]
- Vyunova, T.V.; Andreeva, L.A.; Shevchenko, K.V.; Grigoriev, V.V.; Palyulin, V.A.; Lavrov, M.I.; Bondarenko, E.V.; Kalashnikova, E.E.; Myasoedov, N.F. Characterization of a new positive allosteric modulator of AMPA receptors-PAM-43: Specific binding of the ligand and its ability to potentiate AMPAR currents. Curr. Mol. Pharmacol. 2020, 13, 216–223. [Google Scholar] [CrossRef]
- Lavrov, M.I.; Karlov, D.S.; Palyulin, V.A.; Grigoriev, V.V.; Zamoyski, V.L.; Brkich, G.E.; Pyatigorskaya, N.V.; Zapolskiy, M.E. Novel positive allosteric modulator of AMPA-receptors based on tricyclic scaffold. Mendeleev Commun. 2018, 28, 311–313. [Google Scholar] [CrossRef]
- Lavrov, M.I.; Veremeeva, P.N.; Karlov, D.S.; Zamoyski, V.L.; Grigoriev, V.V.; Palyulin, V.A. Tricyclic derivatives of bispidine as AMPA receptor allosteric modulators. Mendeleev Commun. 2019, 29, 619–621. [Google Scholar] [CrossRef]
- Lavrov, M.I.; Veremeeva, P.N.; Golubeva, E.A.; Radchenko, E.V.; Zamoyski, V.L.; Grigoriev, V.V.; Palyulin, V.A. Positive and negative AMPA receptor modulators based on tricyclic bispidine derivative: Minor structural change inverts the type of activity. Mendeleev Commun. 2022, 32, 360–363. [Google Scholar] [CrossRef]
- Lavrov, M.I.; Karlov, D.S.; Voronina, T.A.; Grigoriev, V.V.; Ustyugov, A.A.; Bachurin, S.O.; Palyulin, V.A. Novel positive allosteric modulators of AMPA receptors based on 3,7-diazabicyclo[3.3.1]nonane scaffold. Mol. Neurobiol. 2020, 57, 191–199. [Google Scholar] [CrossRef]
- Golubeva, E.A.; Lavrov, M.I.; Veremeeva, P.N.; Vyunova, T.V.; Shevchenko, K.V.; Topchiy, M.A.; Asachenko, A.F.; Palyulin, V.A. New allosteric modulators of AMPA receptors: Synthesis and study of their functional activity by radioligand-receptor binding analysis. Int. J. Mol. Sci. 2023, 24, 10293. [Google Scholar] [CrossRef]
- Bachurin, S.O.; Grigorev, V.V.; Palyulin, V.A.; Lavrov, M.I.; Zefirov, N.S.; Garibova, T.L.; Voronina, T.A.; Roziev, R.A. N,N′-Substituted 3,7-diazabicyclo[3.3.1]nonanes, pharmaceutical compositions based thereon and use thereof. RU Patent 2613071, 29 October 2017. [Google Scholar]
- Grigoriev, V.V.; Lavrov, M.I.; Zamoyski, V.L.; Garibova, T.L.; Palyulin, V.A.; Bachurin, S.O. New positive allosteric modulator of AMPA receptors: In vitro and in vivo studies. Dokl. Biochem. Biophys. 2019, 488, 304–306. [Google Scholar] [CrossRef] [PubMed]
- Hasui, T.; Nakamura, S.; Mikami, S.; Yamashita, T. Heterocyclic compounds. U.S. Patent 20200247801A1, 6 August 2020. [Google Scholar]
- Nakamura, S.; Mikami, S.; Hasui, T.; Yamashita, T.; Morimoto, S.; Tokuhara, H.; Oyabu, N.; Yamada, M.; Ochida, A.; Takami, K.; et al. Substituted pyrido[1,2-a]pyrimidines and pyrazino[1,2-a]pyrimidines for enhancing AMPA receptor function. U.S. Patent 11407748B2, 9 August 2022. [Google Scholar]
- Ward, S.; Beswick, P.; Pennicott, L.; Reuillon, T.; Chuckowree, I.; Villalonga-Barber, C.; Porter, R.A. Compounds that modulate AMPA receptor function. U.S. Patent 11298345B2, 12 April 2022. [Google Scholar]
- Azumaya, C.M.; Days, E.L.; Vinson, P.N.; Stauffer, S.; Sulikowski, G.; Weaver, C.D.; Nakagawa, T. Screening for AMPA receptor auxiliary subunit specific modulators. PLoS ONE 2017, 12, e0174742. [Google Scholar] [CrossRef]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef] [PubMed]
- Sudewi, A.A.R.; Susilawathi, N.M.; Mahardika, B.K.; Mahendra, A.N.; Pharmawati, M.; Phuong, M.A.; Mahardika, G.N. Selecting potential neuronal drug leads from Conotoxins of various venomous marine Cone snails in Bali, Indonesia. ACS Omega 2019, 4, 19483–19490. [Google Scholar] [CrossRef]
- Partin, K.M.; Patneau, D.K.; Winters, C.A.; Mayer, M.L.; Buonanno, A. Selective modulation of desensitization at AMPA versus kainate receptors by cyclothiazide and concanavalin A. Neuron 1993, 11, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Everts, I.; Villmann, C.; Hollmann, M. N-Glycosylation is not a prerequisite for glutamate receptor function but is essential for lectin modulation. Mol. Pharmacol. 1997, 52, 861–873. [Google Scholar] [CrossRef]
- Gangwar, S.P.; Yelshanskaya, M.V.; Nadezhdin, K.D.; Yen, L.Y.; Newton, T.P.; Aktolun, M.; Kurnikova, M.G.; Sobolevsky, A.I. Kainate receptor channel opening and gating mechanism. Nature 2024, 630, 762–768. [Google Scholar] [CrossRef]
- Copits, B.A.; Vernon, C.G.; Sakai, R.; Swanson, G.T. Modulation of ionotropic glutamate receptor function by vertebrate galectins. J. Physiol. 2014, 592, 2079–2096. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ (accessed on 24 May 2025).
- Johnson, S.A.; Simmon, V.F. Randomized, double-blind, placebo-controlled international clinical trial of the Ampakine® CX516 in elderly participants with mild cognitive impairment: A progress report. J. Mol. Neurosci. 2002, 19, 197–200. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Krause, S.E.; Block, S.S.; Guter, S.; Wuu, J.; Leurgans, S.; Decle, P.; Potanos, K.; Cook, E.; Salt, J.; et al. Effect of CX516, an AMPA-modulating compound, on cognition and behavior in fragile X syndrome: A controlled trial. J. Child. Adolesc. Psychopharmacol. 2006, 16, 525–540. [Google Scholar] [CrossRef]
- Wezenberg, E.; Jan Verkes, R.; Ruigt, G.S.F.; Hulstijn, W.; Sabbe, B.G.C. Acute effects of the ampakine farampator on memory and information processing in healthy elderly volunteers. Neuropharmacology 2007, 32, 1272–1283. [Google Scholar] [CrossRef] [PubMed]
- Goff, D.C.; Lamberti, J.S.; Leon, A.C.; Green, M.F.; Miller, A.L.; Patel, J.; Manschreck, T.; Freudenreich, O.; Johnson, S.A. A placebo-controlled add-on trial of the Ampakine, CX516, for cognitive deficits in schizophrenia. Neuropsychopharmacology 2008, 33, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Nations, K.R.; Dogterom, P.; Bursi, R.; Schipper, J.; Greenwald, S.; Zraket, D.; Gertsik, L.; Johnstone, J.; Lee, A.; Pande, Y.; et al. Examination of Org 26576, an AMPA receptor positive allosteric modulator, in patients diagnosed with major depressive disorder: An exploratory, randomized, double-blind, placebo-controlled trial. J. Psychopharmacol. 2012, 26, 1525–1539. [Google Scholar] [CrossRef] [PubMed]
- Adler, L.A.; Kroon, R.A.; Stein, M.; Shahid, M.; Tarazi, F.I.; Szegedi, A.; Schipper, J.; Cazorla, P. A translational approach to evaluate the efficacy and safety of the novel AMPA receptor positive allosteric modulator org 26576 in adult attention-deficit/hyperactivity disorder. Biol. Psychiatry 2012, 72, 971–977. [Google Scholar] [CrossRef]
- Bernard, K.; Gouttefangeas, S.; Bretin, S.; Galtier, S.; Robert, P.; Holthoff-Detto, V.; Cummings, J.; Pueyo, M. A 24-week double-blind placebo-controlled study of the efficacy and safety of the AMPA modulator S47445 in patients with mild to moderate Alzheimer’s disease and depressive symptoms. Alzheimers Dement. (N Y) 2019, 5, 231–240. [Google Scholar] [CrossRef]
- Efficacy of 15 mg and 50 mg of S 18986 on Cognitive Symptoms in Mild Cognitive Impairment Patients Treated over a 12-Month Oral Administration Period. An International Multicentre, 3 Parallel Groups, Randomised, Double Blind, Placebo-Controlled Phase II Study. Protocol No. CL2-18986-009. Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2004-004327-35/results (accessed on 24 May 2025).
- Dijkstra, F.; O’Donnell, P.; Klaassen, E.; Buhl, D.; Asgharnejad, M.; Rosen, L.; Zuiker, R.; van Gerven, J.; Jacobs, G. Central nervous system effects of TAK-653, an investigational alpha-amino-3-hydroxy-5-methyl-4-isoxazole receptor (AMPAR) positive allosteric modulator in healthy volunteers. Transl. Psychiatry 2022, 12, 408. [Google Scholar] [CrossRef]
- Bednar, M.M.; De Martinis, N.; Banerjee, A.; Bowditch, S.; Gaudreault, F.; Zumpano, L.; Lin, F.R. The safety and efficacy of PF-04958242 in age-related sensorineural hearing loss a randomized clinical trial. JAMA Otolaryngol. Head Neck Surg. 2015, 141, 607–613. [Google Scholar] [CrossRef]
- Ranganathan, M.; DeMartinis, N.; Huguenel, B.; Gaudreault, F.; Bednar, M.M.; Shaffer, C.L.; Gupta, S.; Cahill, J.; Sherif, M.A.; Mancuso, J.; et al. Attenuation of ketamine-induced impairment in verbal learning and memory in healthy volunteers by the AMPA receptor potentiator PF-04958242. Mol. Psychiatry 2017, 22, 1633–1640. [Google Scholar] [CrossRef]
- A Study to Evaluate the Safety and Efficacy of BIIB104 in Participants with Cognitive Impairment Associated with Schizophrenia (CIAS) (NCT03745820). Biogen. Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2018-003825-27/results (accessed on 24 May 2025).
- Trzepacz, P.T.; Cummings, J.; Konechnik, T.; Forrester, T.D.; Chang, C.; Dennehy, E.B.; Willis, B.A.; Shuler, C.; Tabas, L.B.; Lyketsos, C. Mibampator (LY451395) randomized clinical trial for agitation/aggression in Alzheimer’s disease. Int. Psychogeriatr. 2013, 25, 707–719. [Google Scholar] [CrossRef]
- Radin, D.P.; Cerne, R.; Witkin, J.M.; Lippa, A. Safety, tolerability, and pharmacokinetic profile of the low-impact ampakine CX1739 in young healthy volunteers. Clin. Pharmacol. Drug Dev. 2025, 14, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Oertel, B.G.; Felden, L.; Tran, P.V.; Bradshaw, M.H.; Angst, M.S.; Schmidt, H.; Johnson, S.; Greer, J.J.; Geisslinger, G.; Varney, M.A.; et al. Selective antagonism of opioid-induced ventilatory depression by an ampakine molecule in humans without loss of opioid analgesia. Clin. Pharmacol. Ther. 2010, 87, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Wesensten, N.J.; Reichardt, R.M.; Balkin, T.J. Ampakine (CX717) effects on performance and alertness during simulated night shift work. Aerosp. Med. Hum. Perform. 2007, 78, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J.; Stanley, N.; James, L.M.; Wright, N.; Johnsen, S.; Arbon, E.L.; Dijk, D.J. Acute sleep deprivation: The effects of the AMPAKINE compound CX717 on human cognitive performance, alertness and recovery sleep. J. Psychopharmacol. 2012, 26, 1047–1057. [Google Scholar] [CrossRef]
- Dean, R.L.; Hurducas, C.; Hawton, K.; Spyridi, S.; Cowen, P.J.; Hollingsworth, S.; Marquardt, T.; Barnes, A.; Smith, R.; McShane, R.; et al. Ketamine and other glutamate receptor modulators for depression in adults with unipolar major depressive disorder. Cochrane Database Syst. Rev. 2021, 9, Cd011612. [Google Scholar] [CrossRef]
- Efficacy and Safety of 3 Doses of S 47445 Versus Placebo in Patients with Alzheimer’s Disease at Mild to Moderate Stages with Depressive Symptoms (NCT02626572). Protocol No. CL2-47445-011. Available online: https://clinicaltrials.servier.com/wp-content/uploads/CL2-47445-011-synopsis-report.pdf (accessed on 24 May 2025).
- Wilkinson, S.T.; Sanacora, G. A new generation of antidepressants: An update on the pharmaceutical pipeline for novel and rapid-acting therapeutics in mood disorders based on glutamate/GABA neurotransmitter systems. Drug Discov. Today 2019, 24, 606–615. [Google Scholar] [CrossRef]
- Bernard, K.; Danober, L.; Thomas, J.Y.; Lebrun, C.; Muñoz, C.; Cordi, A.; Desos, P.; Lestage, P.; Morain, P. S 18986: A positive allosteric modulator of AMPA-type glutamate receptors pharmacological profile of a novel cognitive enhancer. CNS Neurosci. Ther. 2010, 16, e193-212. [Google Scholar] [CrossRef]
- Lin, S.; Ionescu, A.; Maynard-Scott, J.; Kennedy, M.; Walling, D.P.; Furey, M.; Singh, J.B. Effects of the selective AMPA modulator NBI-1065845 on the pharmacokinetics of midazolam or ethinyl estradiol-levonorgestrel in healthy adults. Clin. Transl. Sci. 2024, 17, e13791. [Google Scholar] [CrossRef]
- Veselinović, T.; Neuner, I. Progress and pitfalls in developing agents to treat neurocognitive deficits associated with schizophrenia. CNS Drugs 2022, 36, 819–858. [Google Scholar] [CrossRef]
- Chappell, A.S.; Gonzales, C.; Williams, J.; Witte, M.M.; Mohs, R.C.; Sperling, R. AMPA potentiator treatment of cognitive deficits in Alzheimer disease. Neurology 2007, 68, 1008–1012. [Google Scholar] [CrossRef]
- Schifano, F.; Catalani, V.; Sharif, S.; Napoletano, F.; Corkery, J.M.; Arillotta, D.; Fergus, S.; Vento, A.; Guirguis, A. Benefits and harms of ‘smart drugs’ (nootropics) in healthy individuals. Drugs 2022, 82, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Ingegneri, M.; Smeriglio, E.; Zebbiche, Y.; Cornara, L.; Visalli, L.; Smeriglio, A.; Trombetta, D. The dark side of “smart drugs”: Cognitive enhancement vs. clinical concerns. Toxics 2025, 13, 247. [Google Scholar] [CrossRef] [PubMed]
- Malík, M.; Tlustoš, P. Nootropics as cognitive enhancers: Types, dosage and side effects of smart drugs. Nutrients 2022, 14, 3367. [Google Scholar] [CrossRef] [PubMed]

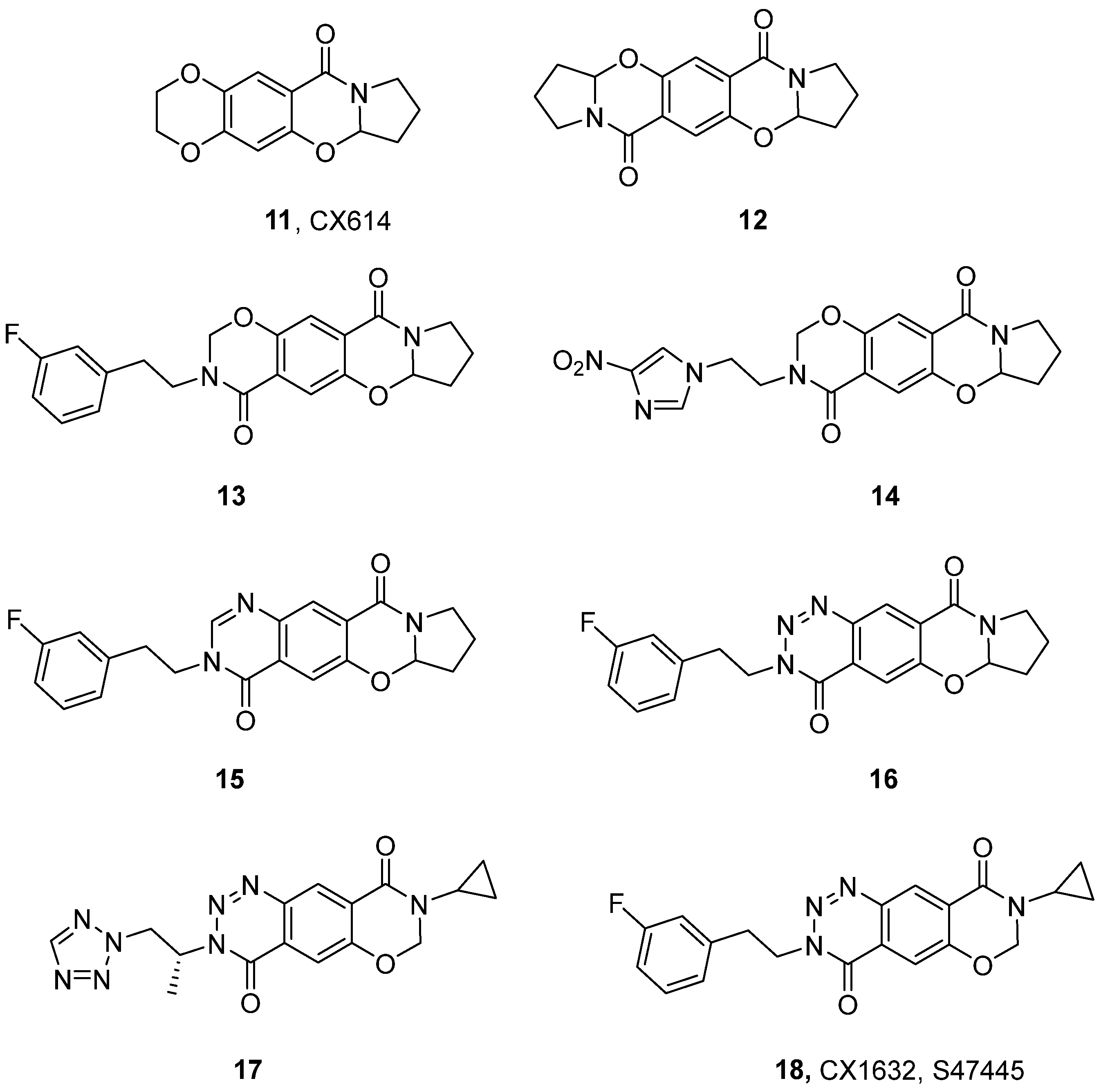
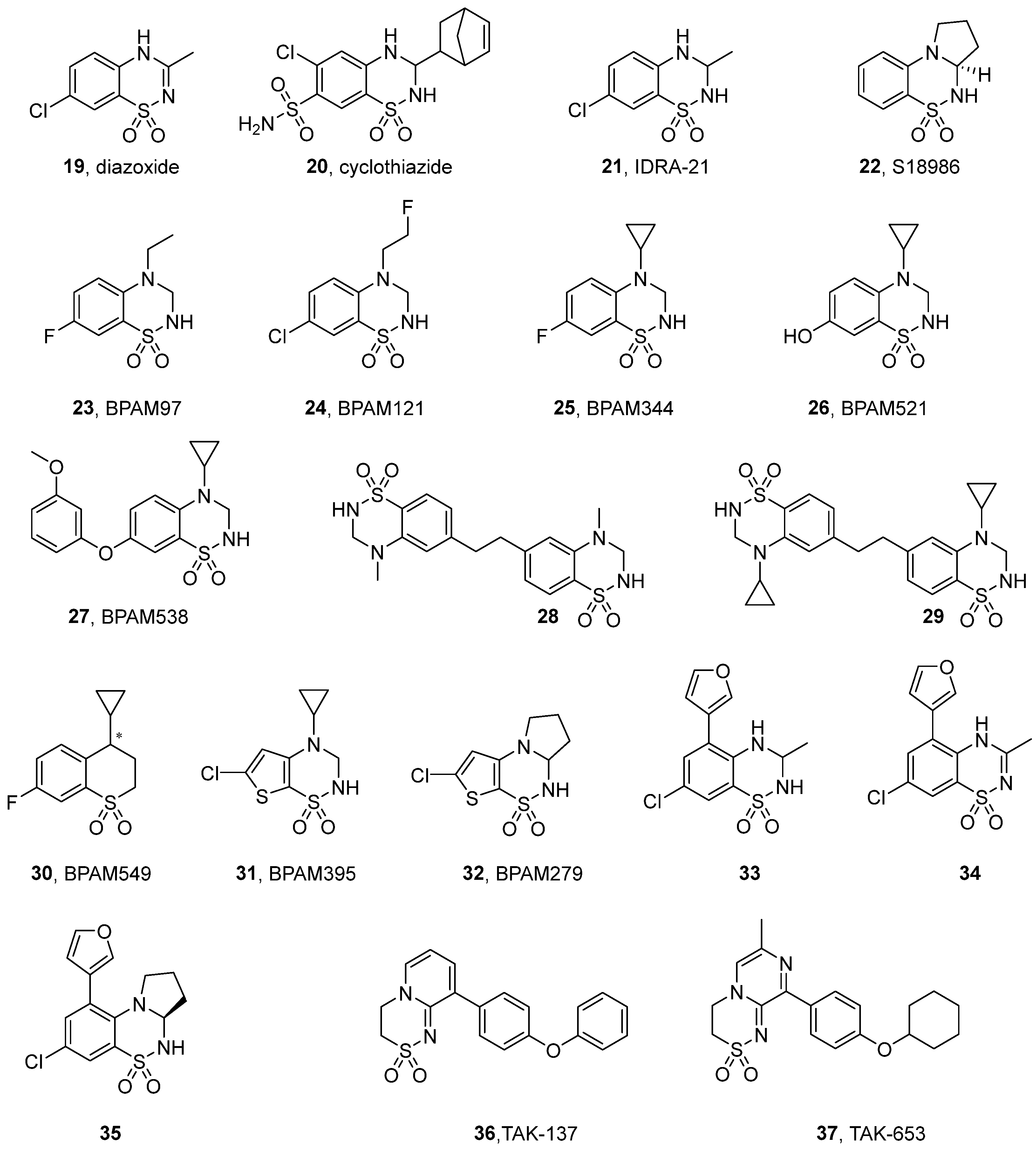
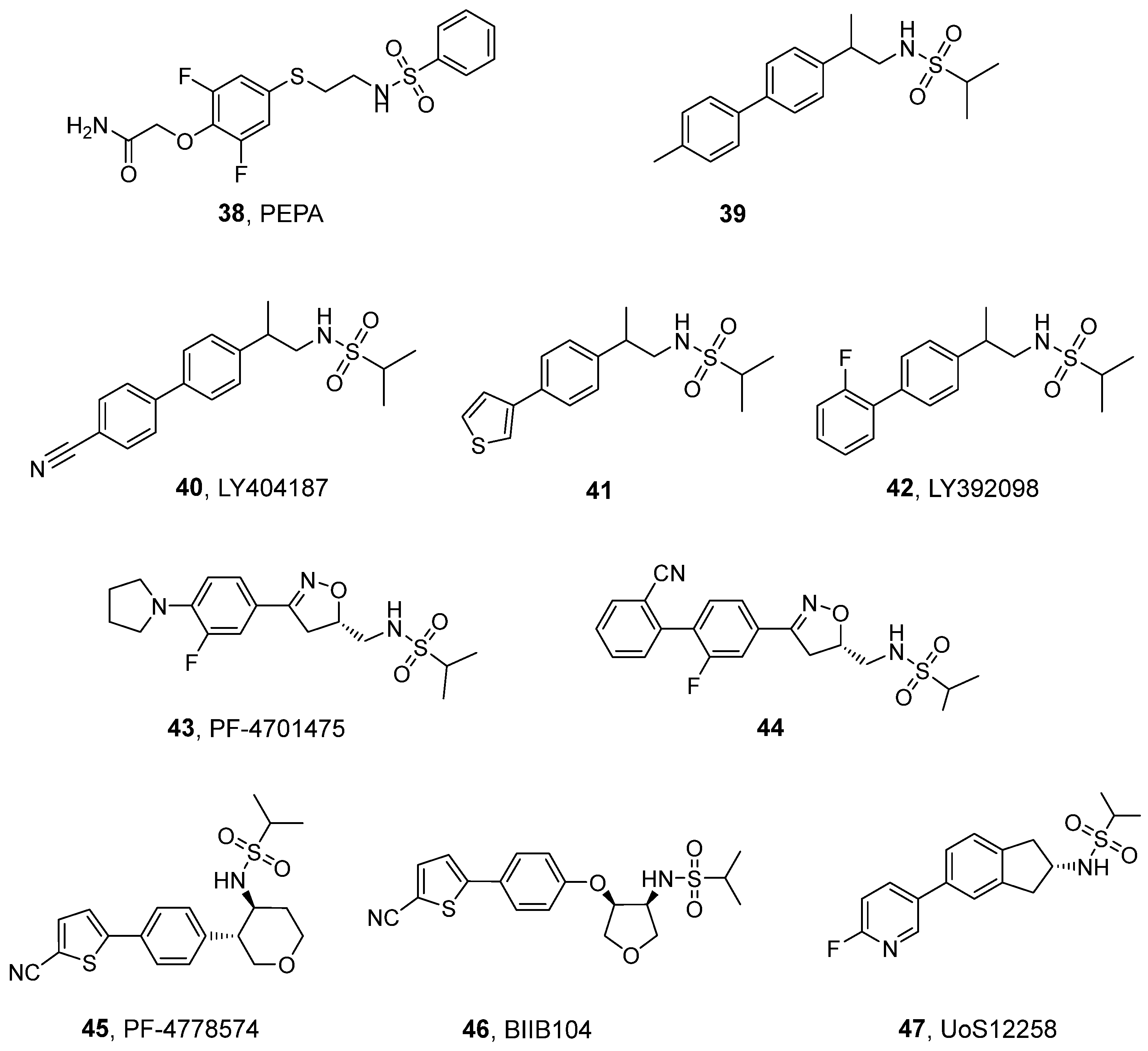

| Compound | EC2X [μM] | A [%] (Conc. [μM] a or Dose [mg/kg] b) | EC50 [μM] | Emax (Fold Increase) |
|---|---|---|---|---|
| CX614 (11) | 2.3 c,d 1.0 e 0.88 f | nd | 17 e 11 f | 30 e 23 f |
| 12 c,d | 0.06 | nd | nd | nd |
| 13 c,g | 0.011 | 16 (3) a | nd | nd |
| 14 c,g | 0.0007 | 19 (0.3) a | nd | nd |
| 15 c,h | 0.0037 | 27 (3) b | nd | nd |
| 16 c,h | 0.0031 | 24 (5) b | nd | nd |
| 17 c,h | 0.46 | 33 (5) b | nd | nd |
| CX1632 (18) | 1.5 e 4.8 f | nd | 6.5 e 7.1 f | 7.9 e 6.9 f |
| Compound | EC2X a or b [μM] | EC50 [μM] | Emax [%] |
|---|---|---|---|
| Cyclothiazide (20) | 1.6 a,c | 7.1 c | 844 c |
| IDRA-21 (21) | 134 a,c | nd | >700 c |
| S18986 (22) | 25 a,c | 130 c | 1263 c |
| BPAM97 (23) | 3.2 a,c | 33 c | 4066 c |
| BPAM121 (24) | 19 b,d 6.7 a,d | nd | nd |
| BPAM344 (25) | 0.27 b,e | 0.90 e | 1500 e |
| BPAM521 (26) | 0.5 b,f | nd | 620 f |
| BPAM549 (30) | 2.2 b,f | nd | 850 f |
| BPAM395 (31) | 0.24 b,g | 4.7 g | nd |
| BPAM279 (32) | 34 b,g | 40 g | nd |
| Receptor | EC50 [μM] | Emax [%] | |||
|---|---|---|---|---|---|
| IDRA-21 (21) | 33 | IDRA-21 (21) | 33 | ||
| Native kainate receptors | KA a | 133 | 8 | 99 | 177 |
| KA + GYKI b | 568 | 20 | 375 | 500 | |
| Recombinant homomeric and heteromeric iGluRs | GluA1 c | 585 | 70 | 300 | 1600 |
| GluA2 c | 532 | 47 | 105 | 529 | |
| GluK1 d | 590 | 105 | 180 | 275 | |
| GluK1/4 d | 690 | 550 | 290 | 660 | |
| GluK1/5 d | 340 | 338 | 283 | 710 | |
| GluK2 e | 688 | 190 | 400 | 800 | |
| GluK2/4 e | 238 | 206 | 92 | 657 | |
| GluK2/5 e | 714 | 195 | 293 | 448 | |
| Compound | Intracellular Ca2+ Influx Assay | Electrophysiological Recordings | ||
|---|---|---|---|---|
| Potentiation EC50 (μM) a | Agonistic Effect (%) c | Potentiation EC50 (μM) b | Agonistic Effect (%) c | |
| TAK-137 (36) | 0.42 | 7.6 | 1.4 | 6.4 |
| TAK-653 (37) | 0.93 | 4.8 | 4.4 | 1.7 |
| Compound | EC50 [µM] (Emax [%]) | ||||||
|---|---|---|---|---|---|---|---|
| GluA1i | GluA1o | GluA2i | GluA2o | GluA3i | GluA4i | GluA4o | |
| LY451395 (48) a | 0.5 | 2.2 | nd | nd | nd | 0.40 | 1.9 |
| PIMSD (49a) b | 1.52 (1335) | nd | 0.73 (790) | 0.64 (804) | 1.90 (2217) | 0.87 (383) | nd |
| CMPDA (50) c | nd | nd | 0.045 | 0.063 | nd | nd | nd |
| CMPDB (51) c | nd | nd | 0.12 | 0.47 | nd | nd | nd |
| Compound | Native AMPAR | GluA1i | GluA2i |
|---|---|---|---|
| EC50 [µM] | EC50 [µM] | EC50 [µM] (Emax [%]) | |
| 55 a | nd | 0.25 | nd |
| 56 a | nd | 0.79 | nd |
| JAMI1001A (57) a | nd | 0.39 | nd |
| 58 a | nd | 0.50 | nd |
| 59 a | nd | 0.50 | nd |
| HBT1 (60) | 1.3 b | 4.6 c | nd |
| 61 d,e | nd | nd | 25.1 (94) |
| MDI-222 (62) d,f | nd | 22.4 (30) | 5.0 (123) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vialko, A.; Chałupnik, P.; Szymańska, E. Positive AMPA and Kainate Receptor Modulators and Their Therapeutic Potential in CNS Diseases: A Comprehensive Review. Int. J. Mol. Sci. 2025, 26, 6450. https://doi.org/10.3390/ijms26136450
Vialko A, Chałupnik P, Szymańska E. Positive AMPA and Kainate Receptor Modulators and Their Therapeutic Potential in CNS Diseases: A Comprehensive Review. International Journal of Molecular Sciences. 2025; 26(13):6450. https://doi.org/10.3390/ijms26136450
Chicago/Turabian StyleVialko, Alina, Paulina Chałupnik, and Ewa Szymańska. 2025. "Positive AMPA and Kainate Receptor Modulators and Their Therapeutic Potential in CNS Diseases: A Comprehensive Review" International Journal of Molecular Sciences 26, no. 13: 6450. https://doi.org/10.3390/ijms26136450
APA StyleVialko, A., Chałupnik, P., & Szymańska, E. (2025). Positive AMPA and Kainate Receptor Modulators and Their Therapeutic Potential in CNS Diseases: A Comprehensive Review. International Journal of Molecular Sciences, 26(13), 6450. https://doi.org/10.3390/ijms26136450