
Shorter Telomere Length in Individuals with Neurocognitive Disorder and APOE ε4 Genotype
 ,
,
Abstract
1. Introduction
2. Results and Discussion
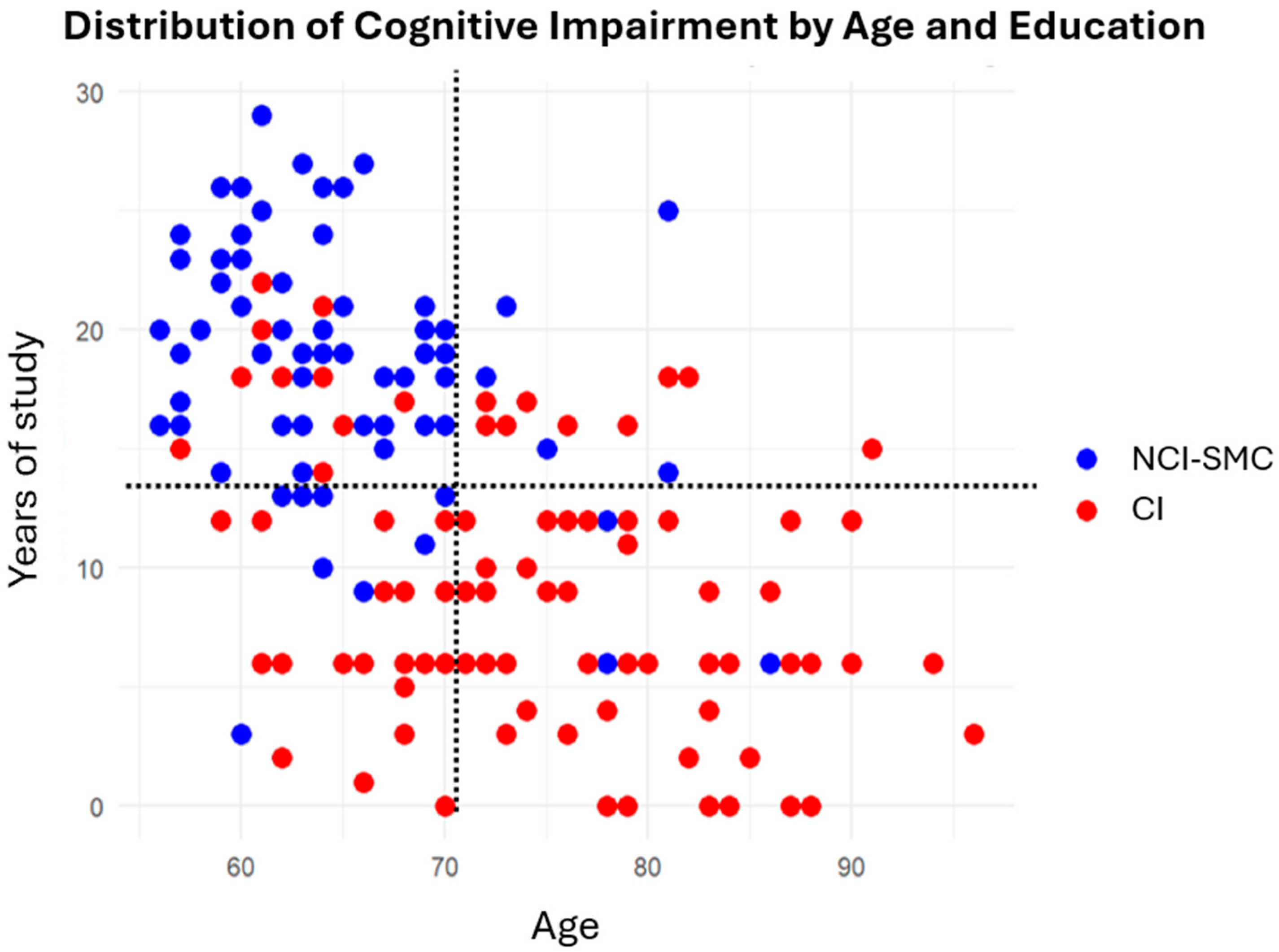
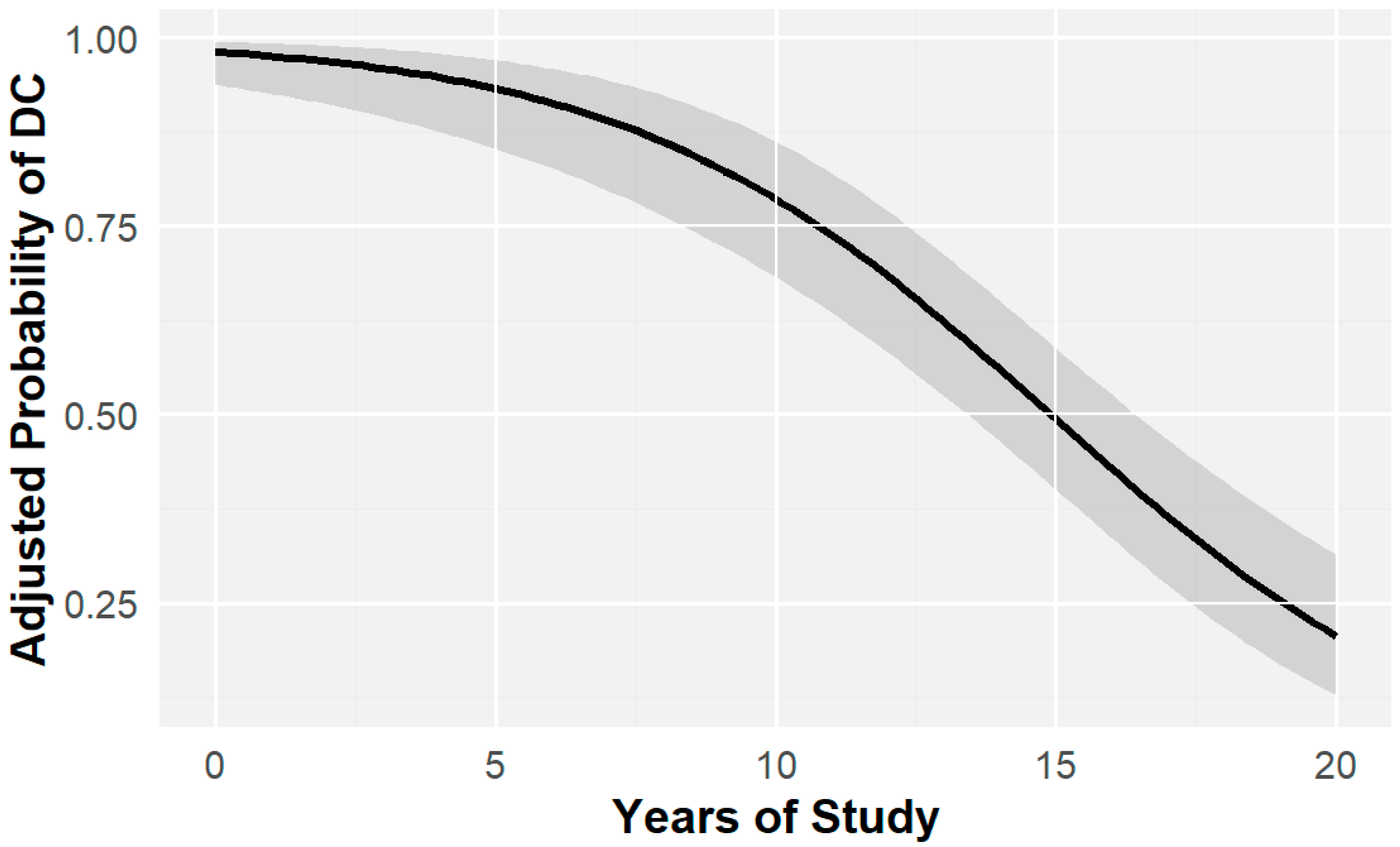
2.1. Education and Cognitive Impairment
2.2. Age and Cognitive Impairment
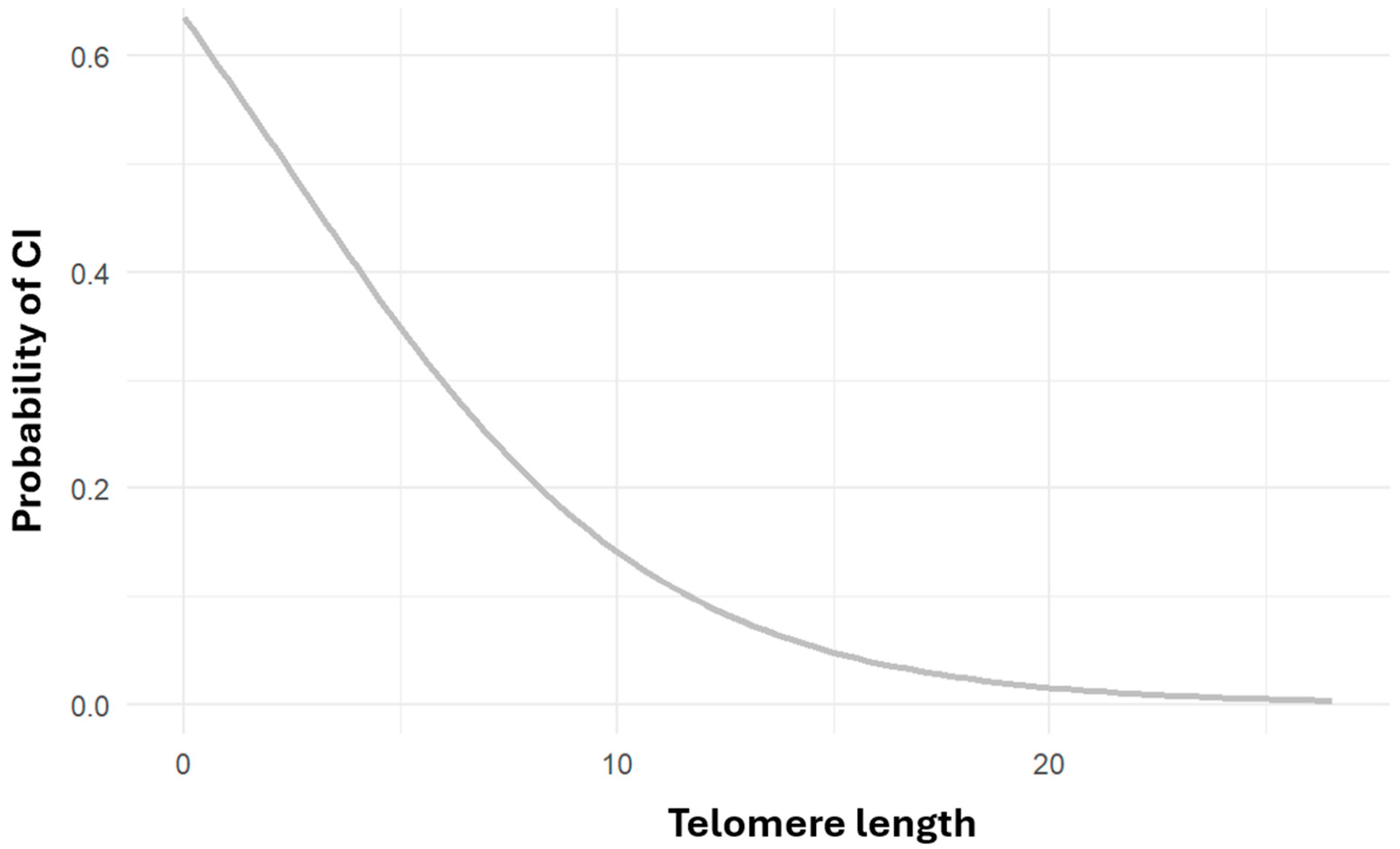
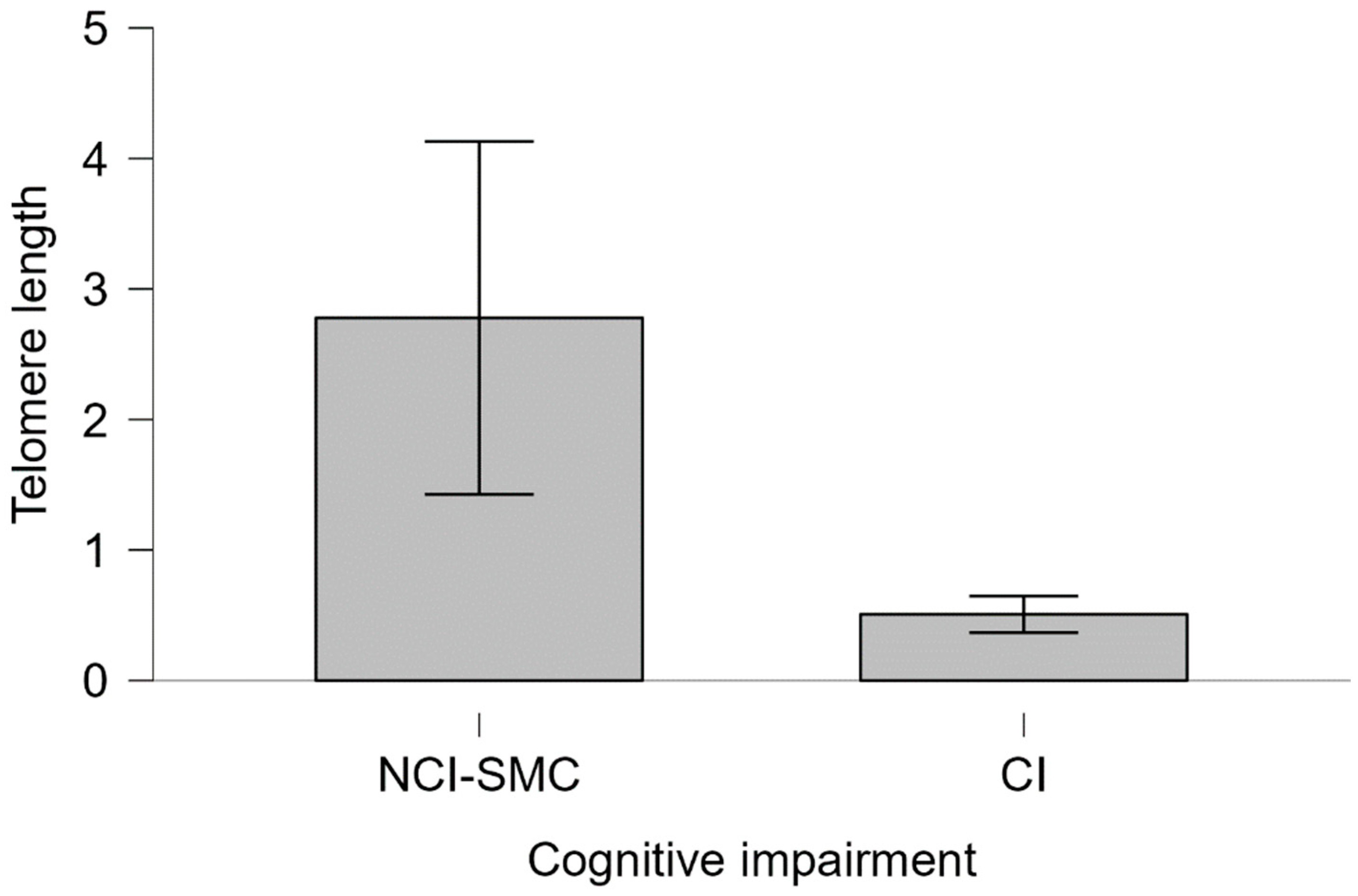
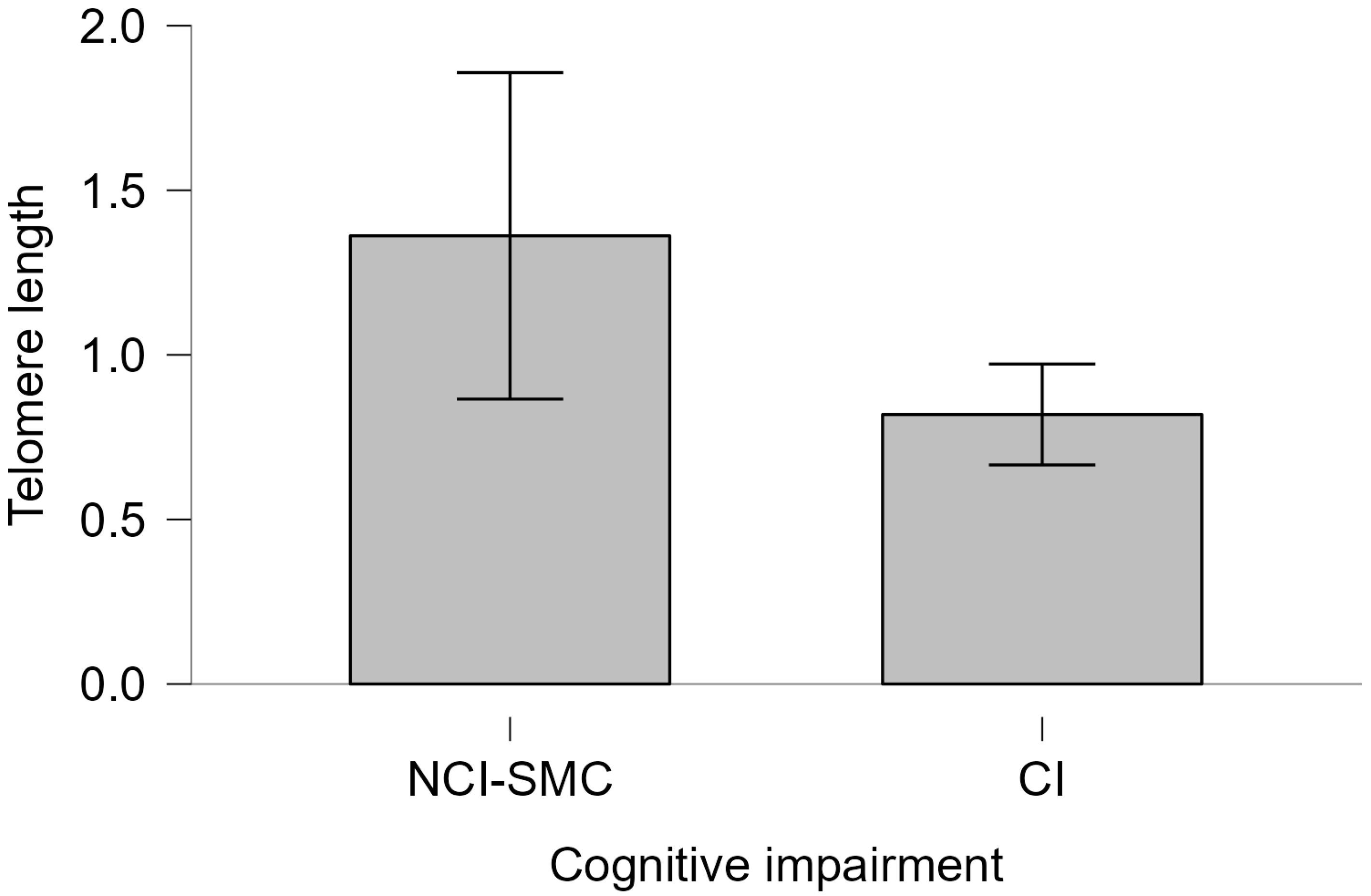
2.3. Telomere Length and Cognitive Impairment
2.4. Telomere Length and APOE Genotype
3. Materials and Methods
- Cognitive impairment group (CI): Individuals with cognitive impairment. All individuals with final scores below 25 points on the MoCA test and/or below 26 points on the MMSE test were included. Individuals with cognitive impairment due to non-neurodegenerative causes as established by the DSM-V (infections, delirium, and vitamin deficiency) were excluded. This study did not exclude participants with vascular or mixed neurocognitive disorder.
- Non-cognitive impairment with subjective memory complaint group (NCI-SMC): Individuals with subjective memory complaints but no cognitive impairment: Individuals who scored above the designated threshold for cognitive impairment on cognitive tests. It is important to consider that scores above 26 on the cognitive tests do not exclude the possibility of an initial decline in some cognitive domain reported by the participants; however, clinicians identified these cases as non-cognitive impairment. However, clinicians identified these cases as non-cognitive impairment.
Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Washington, DC, USA, 2013. [Google Scholar]
- Sachdev, P.S.; Blacker, D.; Blazer, D.G.; Ganguli, M.; Jeste, D.V.; Paulsen, J.S.; Petersen, R.C. Classifying neurocognitive disorders: The DSM-5 approach. Nat. Rev. Neurol. 2014, 10, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Castro-Chavira, S.; Fernandez, T.; Nicolini, H.; Diaz-Cintra, S.; Prado-Alcala, R. Genetic markers in biological fluids for aging-related major neurocognitive disorder. Curr. Alzheimer Res. 2015, 12, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Choreño-Parra, J.A.; Rosa-Arredondo, T.D.L.; Guadarrama-Ortíz, P. Abordaje diagnóstico del paciente con deterioro cognitivo en el primer nivel de atención. Med. Interna México 2020, 36, 807–824. [Google Scholar] [CrossRef]
- Instituto Nacional de Estadística y Geografía. (2023, 6 de julio). Encuesta Nacional Sobre Salud y Envejecimiento en México (ENASEM) 2018; Encuesta Nacional Sobre Salud y Envejecimiento en México (ENASEM) y Encuesta de Evaluación Cognitiva. 2021. Available online: https://www.inegi.org.mx/app/saladeprensa/noticia/8294 (accessed on 30 November 2024).
- Organización Mundial de la Salud. (2024, 1 de octubre). Envejecimiento y Salud. Available online: https://www.who.int (accessed on 30 November 2024).
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef]
- Martínez, S.; Ochoa, B.; Pérez, M.R.; Torrico, F.; García, I.; Garcia, C.C. Apolipoprotein E polymorphisms in adults over 60 years of age with mild cognitive impairment and Alzheimer’s disease in different Venezuelan populations. Polimorfismos del gen de la apolipoproteína E en adultos mayores de 60 años con disminución de la memoria cognitiva y enfermedad de Alzheimer en diferentes poblaciones venezolanas. Biomédica 2022, 42 (Suppl. S1), 116–129. [Google Scholar] [CrossRef]
- Andrews, S.J.; Fulton-Howard, B.; Goate, A. Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet Neurol. 2020, 19, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-Analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Namba, Y.; Tomonaga, M.; Kawasaki, H.; Otomo, E.; Ikeda, K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991, 541, 163–166. [Google Scholar] [CrossRef]
- Roses, A.D.; Strittmatter, W.J.; Pericak-Vance, M.A.; Corder, E.H.; Saunders, A.M.; Schmechel, D.E. Clinical application of apolipoprotein E genotyping to Alzheimer’s disease. Lancet 1994, 343, 1564–1565. [Google Scholar] [CrossRef]
- Belloy, M.E.; Napolioni, V.; Greicius, M.D. A Quarter Century of APOE and Alzheimer’s Disease: Progress to Date and the Path Forward. Neuron 2019, 101, 820–838. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Pimenova, A.A.; Raj, T.; Goate, A.M. Untangling Genetic Risk for Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 300–310. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cacciaglia, R.; Molinuevo, J.L.; Salvadó, G.; Suárez-Calvet, M.; Fauria, K.; Minguillón, C.; Shekari, M.; Farrar, G.; Buckley, C.; Blennow, K.; et al. Impact of the APOE gene on amyloid deposition in participants with abnormal soluble amyloid levels. Alzheimer’s Dement. 2020, 16, e042955. [Google Scholar] [CrossRef]
- Orrego, N.T.; Medina, M.; Sanchez, J.P.; Londono, N.; A Parra-Rodriguez, M.; Aguirre-Acevedo, D.C.; Lopera, F. Association of the cognitive reserve with the age of onset of mild cognitive impairment and dementia in patients with or Alzheimer’s disease. Antioquia, Colombia. Alzheimer’s Dement. 2023, 19, e071071. [Google Scholar] [CrossRef]
- Koutsodendris, N.; Nelson, M.R.; Rao, A.; Huang, Y. Apolipoprotein E and Alzheimer’s Disease: Findings, Hypotheses, and Potential Mechanisms. Annu. Rev. Pathol. Mech. Dis. 2022, 17, 73–99. [Google Scholar] [CrossRef] [PubMed]
- Coon, K.D.; Myers, A.J.; Craig, D.W.; Webster, J.A.; Pearson, J.V.; Lince, D.H.; Zismann, V.L.; Beach, T.G.; Leung, D.; Bryden, L.; et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J. Clin. Psychiatry 2007, 68, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; Van Der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef]
- Anisimova, A.S.; Alexandrov, A.I.; Makarova, N.E.; Gladyshev, V.N.; Dmitriev, S.E. Protein synthesis and quality control in aging. Aging 2018, 10, 4269–4288. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- da Silva, P.F.; Schumacher, B. Principles of the Molecular and Cellular Mechanisms of Aging. J. Investig. Dermatol. 2021, 141, 951–960. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Polsky, L.R.; Rentscher, K.E.; Carroll, J.E. Stress-induced biological aging: A review and guide for research priorities. Brain, Behav. Immun. 2022, 104, 97–109. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dziechciaż, M.; Filip, R. Biological psychological and social determinants of old age: Bio-psycho-social aspects of human aging. Ann. Agric. Environ. Med. 2014, 21, 835–838. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; di Fagagna, F.D. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wagner, K.-H.; Cameron-Smith, D.; Wessner, B.; Franzke, B. Biomarkers of Aging: From Function to Molecular Biology. Nutrients 2016, 8, 338. [Google Scholar] [CrossRef]
- Scarabino, D.; Broggio, E.; Gambina, G.; Corbo, R.M. Leukocyte telomere length in mild cognitive impairment and Alzheimer’s disease patients. Exp. Gerontol. 2017, 98, 143–147. [Google Scholar] [CrossRef]
- Fernández, R.A.H. Telómeros y telomerasas. Rev. Cuba. Investig. Biomédicas 2000, 18, 121–129. Available online: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-03001999000200009&lng=es&tlng=es (accessed on 30 December 2024).
- Ferrucci, L.; Gonzalez-Freire, M.; Fabbri, E.; Simonsick, E.; Tanaka, T.; Moore, Z.; Salimi, S.; Sierra, F.; de Cabo, R. Measuring biological aging in humans: A quest. Aging Cell 2020, 19, e13080. [Google Scholar] [CrossRef]
- Rodríguez-Fernández, B.; Vilor-Tejedor, N.; Arenaza-Urquijo, E.M.; Sánchez-Benavides, G.; Suárez-Calvet, M.; Operto, G.; Minguillón, C.; Fauria, K.; Kollmorgen, G.; Suridjan, I.; et al. Genetically predicted telomere length and Alzheimer’s disease endophenotypes: A Mendelian randomization study; ALFA study. Transl. Psychiatry 2022, 12, 9. [Google Scholar] [CrossRef]
- Suchy-Dicey, A.M.; Muller, C.J.; Madhyastha, T.M.; Shibata, D.; A Cole, S.; Zhao, J.; Longstreth, W.T.; Buchwald, D. Telomere Length and Magnetic Resonance Imaging Findings of Vascular Brain Injury and Central Brain Atrophy. Am. J. Epidemiol. 2018, 187, 1231–1239. [Google Scholar] [CrossRef]
- King, K.S.; Kozlitina, J.; Rosenberg, R.N.; Peshock, R.M.; McColl, R.W.; Garcia, C.K. Effect of leukocyte telomere length on total and regional brain volumes in a large population-based cohort. JAMA Neurol. 2014, 71, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Gampawar, P.; Schmidt, R.; Schmidt, H. Telomere length and brain aging: A systematic review and meta-analysis. Ageing Res. Rev. 2022, 80, 101679. [Google Scholar] [CrossRef] [PubMed]
- Demanelis, K.; Jasmine, F.; Chen, L.S.; Chernoff, M.; Tong, L.; Delgado, D.; Zhang, C.; Shinkle, J.; Sabarinathan, M.; Lin, H.; et al. Determinants of telomere length across human tissues. Science 2020, 369, eaaz6876. [Google Scholar] [CrossRef]
- Ska, B.; Joanette, Y. Vieillissement normal et cognition. M S-Med. Sci. 2006, 22, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, N.; Tosto, G. Genetics of Alzheimer’s Disease: The Importance of Polygenic and Epistatic Components. Curr. Neurol. Neurosci. Rep. 2017, 17, 78. [Google Scholar] [CrossRef]
- Au, B.; Dale-McGrath, S.; Tierney, M.C. Sex differences in the prevalence and incidence of mild cognitive impairment: A meta-analysis. Ageing Res. Rev. 2017, 35, 176–199. [Google Scholar] [CrossRef]
- Mutchie, H.L.; Albrecht, J.S.; Orwig, D.L.; Huang, Y.; Boscardin, W.J.; Hochberg, M.C.; Magaziner, J.S.; Gruber-Baldini, A.L. Differential misclassification of cognitive impairment by sex among hip fracture patients. J. Am. Geriatr. Soc. 2022, 70, 838–845. [Google Scholar] [CrossRef]
- Katabathula, S.; Davis, P.B.; Xu, R.; Alzheimer’s Disease Neuroimaging Initiative. Sex-Specific Heterogeneity of Mild Cognitive Impairment Identified Based on Multi-Modal Data Analysis. J. Alzheimer’s Dis. 2023, 91, 233–243. [Google Scholar] [CrossRef]
- Calatayud, E.; Marcén-Román, Y.; Rodríguez-Roca, B.; Salavera, C.; Gasch-Gallen, A.; Gómez-Soria, I. Sex differences on anxiety and depression in older adults and their relationship with cognitive impairment. Semergen 2023, 49, 101923. [Google Scholar] [CrossRef]
- Stern, Y.; Arenaza-Urquiljo, E.M.; Bartrés-Faz, D.; Belleville, S.; Cantillon, M.; Chetelat, G.; Ewers, M.; Franzmeier, N.; Kempermann, G.; Kremen, W.S.; et al. Whitepaper: Defining and investigating cognitive reserve, brain reserve, and brain maintenance. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2020, 16, 1305–1311. [Google Scholar] [CrossRef]
- Lizarraga, A.; Kassinopoulos, M.; González-De-Echávarri, J.M.; Huguet, J.; Sánchez-Benavides, G.; Brugulat-Serrat, A.; Suarez-Calvet, M.; Milà-Alomà, M.; Blennow, K.; Zetterberg, H.; et al. Cognitive reserve is associated with the recruitment of compensatory brain networks in individuals at risk for Alzheimer’s disease. Alzheimer’s Dement. 2024, 20, e093813. [Google Scholar] [CrossRef]
- Nelson, M.E.; Jester, D.J.; Petkus, A.J.; Andel, R. Cognitive Reserve, Alzheimer’s Neuropathology, and Risk of Dementia: A Systematic Review and Meta-Analysis. Neuropsychol. Rev. 2021, 31, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Soldan, A.; Pettigrew, C.; Cai, Q.; Wang, J.; Wang, M.-C.; Moghekar, A.; Miller, M.I.; Albert, M.; BIOCARD Research Team. Cognitive reserve and long-term change in cognition in aging and preclinical Alzheimer’s disease. Neurobiol. Aging 2017, 60, 164–172. [Google Scholar] [CrossRef]
- Wilson, R.S.; Yu, L.; Lamar, M.; Schneider, J.A.; Boyle, P.A.; Bennett, D.A. Education and cognitive reserve in old age. Neurology 2019, 92, e1041–e1050. [Google Scholar] [CrossRef]
- Yang, W.; Wang, J.; Guo, J.; Dove, A.; Qi, X.; Xu, W. Association of cognitive reserve indicator with cognitive decline and structural brain differences in mid- and late life: A large population-based cohort study. Alzheimer’s Dement. 2023, 19, e075561. [Google Scholar] [CrossRef]
- Chen, X.; Xue, B.; Hu, Y. Cognitive reserve over life course and 7-year trajectories of cognitive decline: Results from China health and retirement longitudinal study. BMC Public Health 2022, 22, 231. [Google Scholar] [CrossRef]
- Kleineidam, L.; Wolfsgruber, S.; Weyrauch, A.-S.; Zulka, L.E.; Forstmeier, S.; Roeske, S.; Bussche, H.v.D.; Kaduszkiewicz, H.; Wiese, B.; Weyerer, S.; et al. Midlife occupational cognitive requirements protect cognitive function in old age by increasing cognitive reserve. Front. Psychol. 2022, 13, 957308. [Google Scholar] [CrossRef]
- Kobayashi, L.C.; O’Shea, B.Q.; Wixom, C.; Jones, R.N.; Langa, K.M.; Weir, D.; Lee, J.; Wong, R.; Gross, A.L. Lifetime occupational skill and later-life cognitive function among older adults in the United States, Mexico, India, and South Africa. Alzheimer’s Dement. 2024, 20, 1933–1943. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.P.; Windsor, T.D.; Andel, R.; Luszcz, M.A. Is Occupational Complexity Associated with Cognitive Performance or Decline? Results from the Australian Longitudinal Study of Ageing. Gerontology 2017, 63, 550–559. [Google Scholar] [CrossRef]
- Cisneros-Franco, J.M.; Voss, P.; Thomas, M.E.; de Villers-Sidani, E. Critical periods of brain development. Handb. Clin. Neurol. 2020, 173, 75–88. [Google Scholar] [CrossRef]
- Huang, L.C.; Lee, M.Y.; Chien, C.F.; Chang, Y.P.; Li, K.Y.; Yang, Y.H. Age and sex differences in the association between APOE genotype and Alzheimer’s disease in a Taiwan Chinese population. Front. Aging Neurosci. 2023, 15, 1246592. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.S.; Boyle, P.A.; Yu, L.; Barnes, L.L.; Schneider, J.A.; Bennett, D.A. Life-span cognitive activity, neuropathologic burden, and cognitive aging. Neurology 2013, 81, 314–321. [Google Scholar] [CrossRef]
- Pettigrew, C.; Soldan, A. Defining Cognitive Reserve and Implications for Cognitive Aging. Curr. Neurol. Neurosci. Rep. 2019, 19, 1. [Google Scholar] [CrossRef]
- Niu, H.; Álvarez-Álvarez, I.; Guillén-Grima, F.; Aguinaga-Ontoso, I. Prevalence and incidence of Alzheimer’s disease in Europe: A meta-analysis. Prevalencia e incidencia de la enfermedad de Alzheimer en Europa: Metaanálisis. Neurologia 2017, 32, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Molero, A.E.; Pino-Ramírez, G.; Maestre, G.E. Modulación por edad y género del riesgo de enfermedad de Alzheimer y demencia vascular asociada al alelo de la apolipoproteína E-ε4 en latinoamericanos: Hallazgos del Estudio de Envejecimiento de Maracaibo. Cartas Neurocienc. 2001, 307, 5. [Google Scholar]
- Oviedo, D.C.; Lezcano, H.; Perez, A.R.; Villarreal, A.E.; Carreira, M.B.; Isaza, B.; Wesley, L.; Grajales, S.A.; Fernandez, S.; Frank, A.; et al. Vascular biomarkers and ApoE4 expression in mild cognitive impairment and Alzheimer’s disease. AIMS Neurosci. 2018, 5, 148–161. [Google Scholar] [CrossRef]
- Kumar, V.; Bishayee, K.; Park, S.; Lee, U.; Kim, J. Oxidative stress in cerebrovascular disease and associated diseases. Front. Endocrinol. 2023, 14, 1124419. [Google Scholar] [CrossRef]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Baum, L.; Chen, L.; Ng, H.K.; Pang, C.P. Apolipoprotein E isoforms in Alzheimer’s disease pathology and etiology. Microsc. Res. Tech. 2000, 50, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Campos, M.; Edland, S.D.; Peavy, G.M. Exploratory study of apolipoprotein E ε4 genotype and risk of Alzheimer’s disease in Mexican Hispanics. J. Am. Geriatr. Soc. 2013, 61, 1038–1040. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Li, J.; Li, X.; Li, Y. Association between apolipoprotein E polymorphisms and Parkinson’s disease risk: A meta-analysis. Front. Aging Neurosci. 2018, 10, 1–9. [Google Scholar] [CrossRef]
- Rajabli, F.; Adams, L.D.; Tejada, S.; Rodriguez, V.C.; Mena, P.R.; Prough, M.; Bussies, P.; Feliciano, N.I.; Silva-Vergara, C.; Contreras, M.; et al. Southern European genetic ancestry shows reduced APOE E4 risk for Alzheimer disease in Caribbean Hispanic population. Alzheimer’s Dement. 2020, 16, e045951. [Google Scholar] [CrossRef]
- O’Bryant, S.E.; Johnson, L.; Reisch, J.; Edwards, M.; Hall, J.; Barber, R.; Devous, M.D.; Royall, D.; Singh, M. Risk factors for mild cognitive impairment among Mexican Americans. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2013, 9, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Granot-Hershkovitz, E.; Tarraf, W.; Kurniansyah, N.; Daviglus, M.; Isasi, C.R.; Kaplan, R.; Lamar, M.; Perreira, K.M.; Wassertheil-Smoller, S.; Stickel, A.; et al. APOE alleles’ association with cognitive function differs across Hispanic/Latino groups and genetic ancestry in the study of Latinos-investigation of neurocognitive aging (HCHS/SOL). Alzheimer’s Dement. 2021, 17, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Villalpando-Berumen, J.M.; Mejia-Arango, S.; Aguilar-Salinas, C.A.; Ordonez-Sanchez, M.L.; Gutierrez-Robledo, L.M. Apolipoprotein E epsilon4, Alzheimer’s disease, and cognitive performance in elderly Mexican Mestizos. J. Am. Geriatr. Soc. 2008, 56, 677–682. [Google Scholar] [CrossRef]
- Huggins, L.K.; Min, S.H.; Kaplan, S.; Wei, J.; Welsh-Bohmer, K.; Xu, H. Meta-Analysis of Variations in Association between APOE ε4 and Alzheimer’s Disease and Related Dementias Across Hispanic Regions of Origin. J. Alzheimer’s Dis. 2023, 93, 1095–1109. [Google Scholar] [CrossRef]
- Sohail, M.; Palma-Martínez, M.J.; Chong, A.Y.; Quinto-Cortés, C.D.; Barberena-Jonas, C.; Medina-Muñoz, S.G.; Ragsdale, A.; Delgado-Sánchez, G.; Cruz-Hervert, L.P.; Ferreyra-Reyes, L.; et al. Mexican Biobank advances population and medical genomics of diverse ancestries. Nature 2023, 622, 775–783. [Google Scholar] [CrossRef]
- Mimenza, A.J.; Aparacio, I.I.; Funes, J.A.A.; Navarro, S.G.A. Association between APOEε4 genotype and cardiovascular risk factors in Mexican elderly adults with amnestic and non-amnestic mild cognitive impairment. Alzheimer’s Dement. 2020, 16, e042820. [Google Scholar] [CrossRef]
- Silva-Zolezzi, I.; Hidalgo-Miranda, A.; Estrada-Gil, J.; Fernandez-Lopez, J.C.; Uribe-Figueroa, L.; Contreras, A.; Balam-Ortiz, E.; del Bosque-Plata, L.; Velazquez-Fernandez, D.; Lara, C.; et al. Analysis of genomic diversity in Mexican Mestizo populations to develop genomic medicine in Mexico. Proc. Natl. Acad. Sci. USA 2009, 106, 8611–8616. [Google Scholar] [CrossRef]
- Hackenhaar, F.S.; Josefsson, M.; Adolfsson, A.N.; Landfors, M.; Kauppi, K.; Hultdin, M.; Adolfsson, R.; Degerman, S.; Pudas, S. Short leukocyte telomeres predict 25-year Alzheimer’s disease incidence in non-APOE ε4-carriers. Alzheimer’s Res. Ther. 2021, 13, 130. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Crocco, P.; De Rango, F.; Dato, S.; La Grotta, R.; Maletta, R.; Bruni, A.C.; Passarino, G.; Rose, G. The shortening of leukocyte telomere length contributes to Alzheimer’s disease: Further evidence from late-onset familial and sporadic cases. Biology 2023, 12, 1286. [Google Scholar] [CrossRef]
- Fani, L.; Hilal, S.; Sedaghat, S.; Broer, L.; Licher, S.; Arp, P.P.; van Meurs, J.B.J.; Ikram, M.K.; Ikram, M.A. Telomere length and the risk of Alzheimer’s disease: The Rotterdam Study. J. Alzheimer’s Dis. 2020, 73, 707–714. [Google Scholar] [CrossRef]
- Piyush, G.; Schmidt, R.; Schmidt, H. Leukocyte telomere length is related to brain parenchymal fraction and attention/speed in the elderly: Results of the Austrian Stroke Prevention Study. Front. Psychiatry 2020, 11, 100. [Google Scholar] [CrossRef]
- Zhang, S.; Zhen, K.; Su, Q.; Chen, Y.; Lv, Y.; Yu, L. The Effect of Aerobic Exercise on Cognitive Function in People with Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Int. J. Environ. Res. Public Health 2022, 19, 15700. [Google Scholar] [CrossRef]
- Song, S.; Lee, E.; Kim, H. Does Exercise Affect Telomere Length? A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Medicina 2022, 58, 242. [Google Scholar] [CrossRef] [PubMed]
- Barnes, R.P.; Fouquerel, E.; Opresko, P.L. The impact of oxidative DNA damage and stress on telomere homeostasis. Mech. Ageing Dev. 2019, 177, 37–45. [Google Scholar] [CrossRef]
- Barragán, R.; Ortega-Azorín, C.; Sorlí, J.V.; Asensio, E.M.; Coltell, O.; St-Onge, M.-P.; Portolés, O.; Corella, D. Effect of physical activity, smoking, and sleep on telomere length: A systematic review of observational and intervention studies. J. Clin. Med. 2022, 11, 76. [Google Scholar] [CrossRef]
- Chen, C.; Yang, K.; Nan, H.; Unverzagt, F.; McClure, L.A.; Irvin, M.R.; Judd, S.; Cushman, M.; Mukaz, D.K.; Klaunig, J.E.; et al. Associations of Telomere Length and Change With Cognitive Decline Were Modified by Sex and Race: The REGARDS Study. Am. J. Alzheimer’s Dis. Other Dementiasr 2023, 38, 15333175231175797. [Google Scholar] [CrossRef]
- Dhillon, V.S.; Deo, P.; Chua, A.; Thomas, P.; Fenech, M. Shorter Telomere Length in Carriers of APOE-ε4 and High Plasma Concentration of Glucose, Glyoxal and Other Advanced Glycation End Products (AGEs). J. Gerontology. Ser. A Biol. Sci. Med. Sci. 2020, 75, 1894–1898. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2020, 20, 148–160. [Google Scholar] [CrossRef]
- Ciesielska, N.; Sokołowski, R.; Mazur, E.; Podhorecka, M.; Polak-Szabela, A.; Kędziora-Kornatowska, K. Is the Montreal Cognitive Assessment (MoCA) test better suited than the Mini-Mental State Examination (MMSE) in mild cognitive impairment (MCI) detection among people aged over 60? Meta-analysis. Psychiatria Polska 2016, 50, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Delgado, C.; Araneda, A.; Behrens, M.I. Validation of the Spanish-language version of the Montreal Cognitive Assessment test in adults older than 60 years. Validación del instrumento Montreal Cognitive Assessment en español en adultos mayores de 60 años. Neurologia 2019, 34, 376–385. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean (n = 170, %) | CI (99, 58.24) | NCI-SMC (71, 41.76) | |
|---|---|---|---|
| Age a | 70.5 (±9.224) | 74.18 (±9.12) | 65.36 (±6.57) |
| Years of study a | 12.55 (±6.318) | 9.45 (±5.77) | 16.88 (±4.13) |
| Female b | 94 (55.29) | 47 (27.65) | 47 (27.65) |
| Male b | 76 (44.7) | 52 (30.58) | 24 (14.11) |
| MMSE a | 21.31 (±7.03) | 16.85 (±5.72) | 28 (±1.33) |
| MoCA a | 24.63 (±3.87) | 21.56 (±3.6) | 27.23 (±1.41) |
| Non-carriers APOE ε4 b | 131 (77.65) | 82 (48.23) | 49 (28.82) |
| Carriers APOE ε4 b | 39 (22.35) | 17 (10) | 22 (12.94) |
| Telomere length (2−ΔCT) a | 1.04 (±2.38) | 0.76 (±1.28) | 1.43 (±3.36) |
| APOE ε4 | |||
|---|---|---|---|
| With | Without | p-Value | |
| Cognitive impairment a | 17 (10) | 82 (48.23) | p < 0.05 * |
| Age b | 74.118 (±8.49) | 74.195 (±9.29) | |
| Years of study b | 7.941 (±5.55) | 9.964 (±6.01) | |
| NCI-subjetive memory complaint a | 22 (12.94) | 49 (28.8) | |
| Age b | 63.238 (±5.04) | 66.26 (±6.56) | |
| Years of study b | 18.047 (±5.8) | 18.204 (±5.53) | |
| Total a | 39 (22.94) | 132 (77.65) | |
| Age | 68.105 (±8.67) | 71.189 (±9.3) | p = 0.966 |
| Years of study | 13.52 (±7.58) | 13.023 (±7.05) | p = 0.347 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mejía-Ortiz, P.; Genis-Mendoza, A.D.; Ramírez Villanueva, R.; López Ramírez, S.; Guzmán Sánchez, R.; Fernández, T.; Sigg-Alonso, J.; Nicolini-Sánchez, H. Shorter Telomere Length in Individuals with Neurocognitive Disorder and APOE ε4 Genotype. Int. J. Mol. Sci. 2025, 26, 4577. https://doi.org/10.3390/ijms26104577
Mejía-Ortiz P, Genis-Mendoza AD, Ramírez Villanueva R, López Ramírez S, Guzmán Sánchez R, Fernández T, Sigg-Alonso J, Nicolini-Sánchez H. Shorter Telomere Length in Individuals with Neurocognitive Disorder and APOE ε4 Genotype. International Journal of Molecular Sciences. 2025; 26(10):4577. https://doi.org/10.3390/ijms26104577
Chicago/Turabian StyleMejía-Ortiz, Paola, Alma Delia Genis-Mendoza, Ramon Ramírez Villanueva, Susana López Ramírez, Rafael Guzmán Sánchez, Thalia Fernández, Jorge Sigg-Alonso, and Humberto Nicolini-Sánchez. 2025. "Shorter Telomere Length in Individuals with Neurocognitive Disorder and APOE ε4 Genotype" International Journal of Molecular Sciences 26, no. 10: 4577. https://doi.org/10.3390/ijms26104577
APA StyleMejía-Ortiz, P., Genis-Mendoza, A. D., Ramírez Villanueva, R., López Ramírez, S., Guzmán Sánchez, R., Fernández, T., Sigg-Alonso, J., & Nicolini-Sánchez, H. (2025). Shorter Telomere Length in Individuals with Neurocognitive Disorder and APOE ε4 Genotype. International Journal of Molecular Sciences, 26(10), 4577. https://doi.org/10.3390/ijms26104577