
Mesenchymal Stem Cell-Derived Extracellular Vesicles: Seeking into Cell-Free Therapies for Bone-Affected Lysosomal Storage Disorders




Abstract
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LSD | Enzyme Deficiency | Accumulated Substrate | Symptoms | Ref. |
|---|---|---|---|---|
| GD I/III | GBA | GlcCer | osteopenia, sclerotic lesions, osteonecrosis, decrease mineralization. | [30] |
| ML II/III | GlcNAc-1-fosfo. | Mucolipids | dystosis multiplex, osteopenia, osteodystrophy, kyphosis, coarse facies. | [31] |
| MPS I | IDUA | DS/HS | dysostosis multiplex, kyphosis, coarse facies, short stature, hip dysplasia, pectus excavatum | [32] |
| MPS II | IDS | DS/HS | dysostosis multiplex, coarse facies, claw hands, kyphosis/gibbus, scoliosis, short stature, foot deformity. | [33] |
| MPS IIIA | SGSH | HS | Joint stiffness, contractures, dysostosis multiplex, scoliosis and hip dysplasia. | [34] |
| MPS IVA | GALNS | KS/C6S | dysostosis multiplex, pectus carinatum, gibbus, kyphosis, scoliosis, genu valgum, short stature, hypermobile joints, coarse facies | [35] |
| MPS VI | ARSB | DS | dysostosis multiplex, genu valgum, coarse facies, short stature | [36] |
| MPS VII | GUSB | HS, DS, and CS | dysostosis multiplex, coarse facies, joint contractures, genu valgum, short stature. | [37] |
| NPD-B | ASMase | Sphingo. | delayed skeletal maturation, osteopenia, osteoporosis. | [38] |
| Mann. | α-Mannosidase | MCO | dysostosis multiple, coarse facies. | [39,40] |
| Galacto. | Cathepsin A | Sial-Oligo | dysostosis multiple, coarse facial features. | [41] |
| Sial-II | Neuraminidase | Sial-Oligo | coarse facies, dysostosis multiplex, kyphoscoliosis. | [42] |
| Aspartyl. | AGA | GlcNAc-Asn | osteoporosis, hypermobile joints, delayed skeletal maturation. | [43,44] |
2. The Bone: Structure and Microenvironment
3. MSCs and MSC-EVs
3.1. MSC Secretome
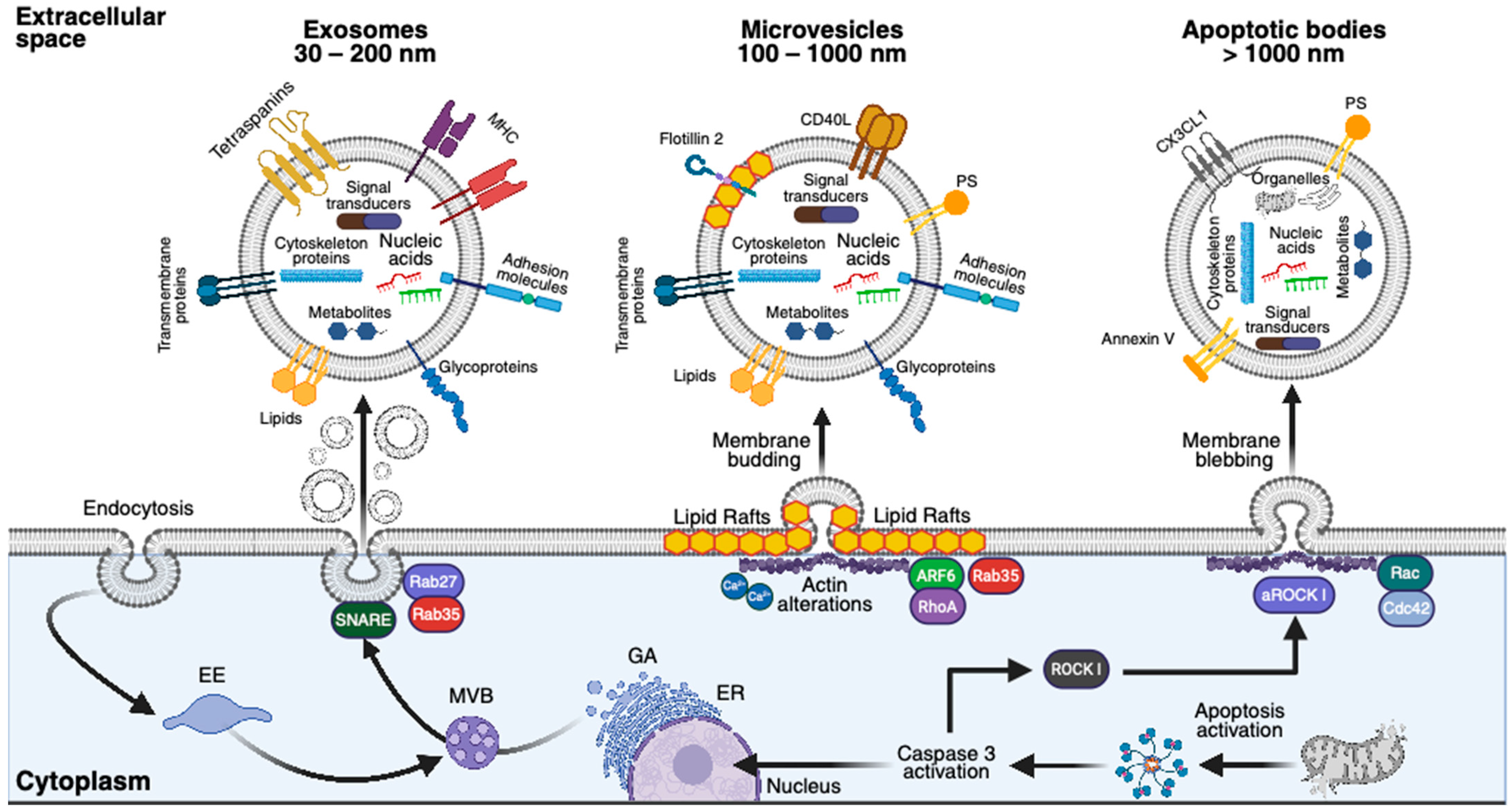
3.1.1. Exosomes
3.1.2. Microvesicles
3.1.3. Apoptotic Bodies
4. MSC-EVs and Bone-Affected LSDs
4.1. MSC-EVs and ERT
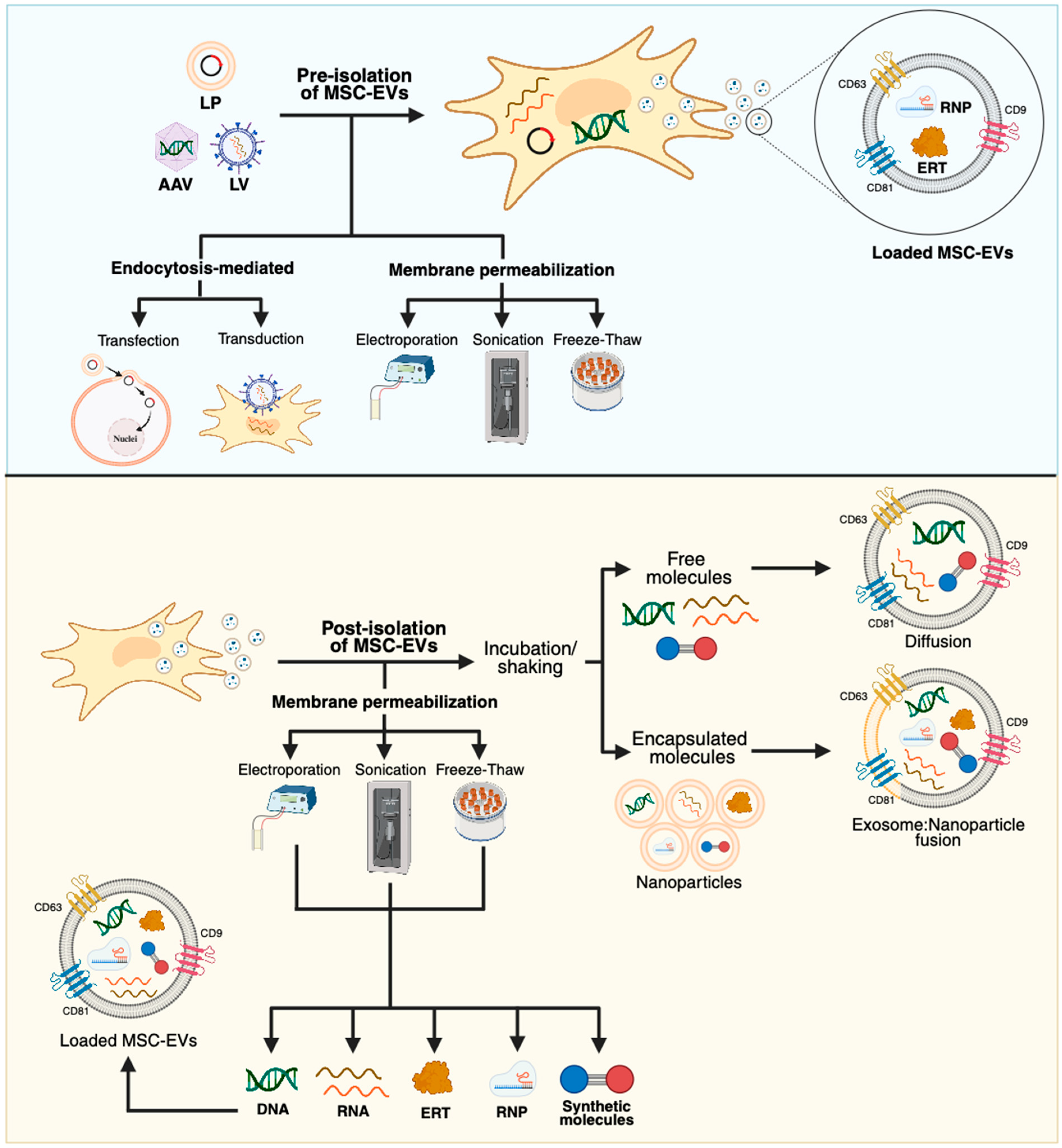
4.2. MSC-EVs and GT
5. MSC-EVs: A Perspective from Bone-Affected Non-LSDs
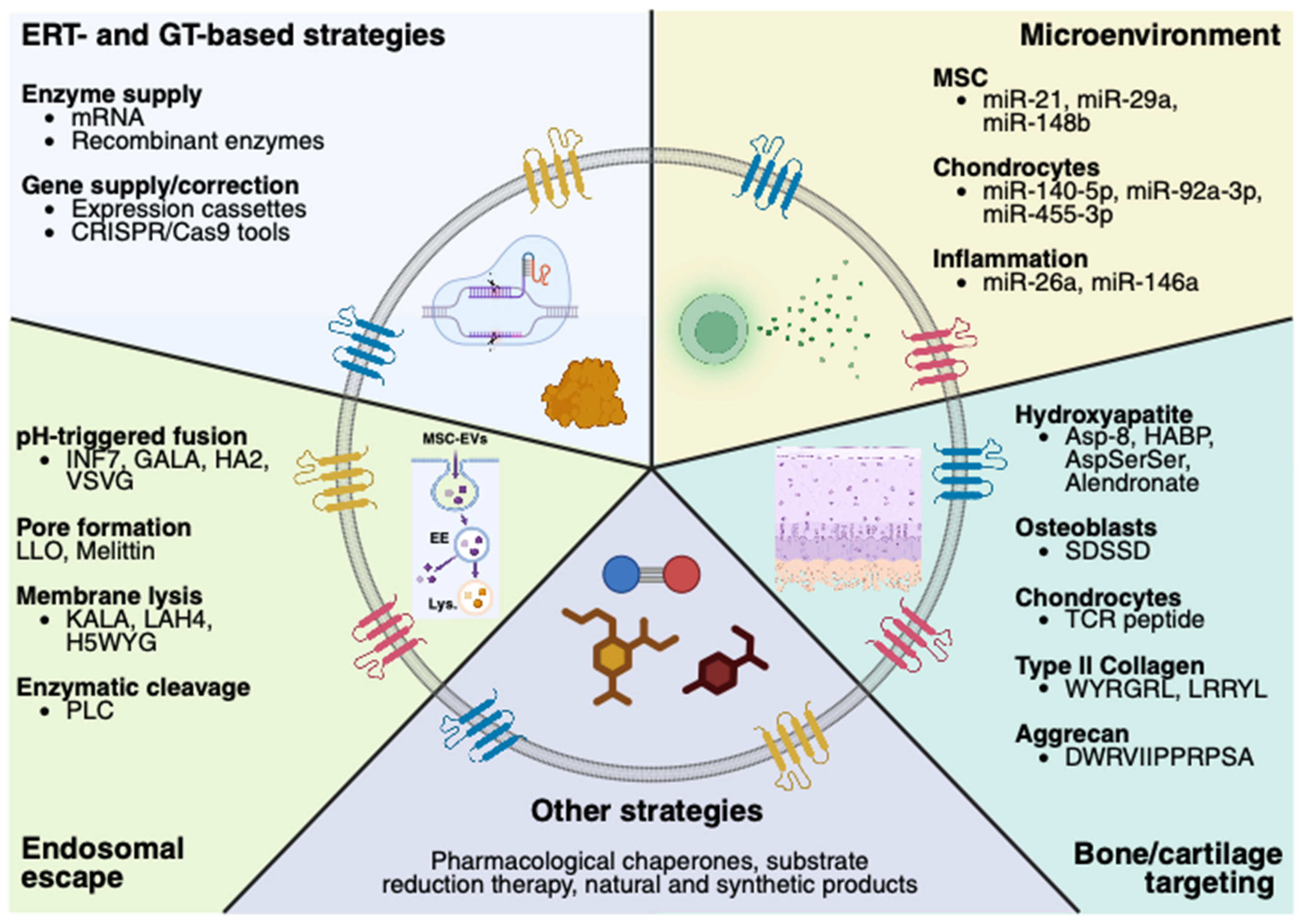
5.1. MSC-EVs’ Engineering—Therapeutic Molecules
5.2. MSC-EVs’ Engineering—Targeting Bone/Cartilage
6. Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD-MSC | Adipose-derived mesenchymal stem cells |
| BM | Bone marrow |
| BM-MSCs | Bone marrow-derived mesenchymal stem cells |
| DP-MSCs | Dental pup-derived mesenchymal stem cells |
| ERT | Enzyme replacement therapy |
| GT | Gene therapy |
| MSCs | Mesenchymal stem cells |
| P-MSCs | Placenta-derived mesenchymal stem cells |
| SM-MSCs | Skeletal muscle-derived mesenchymal stem cells |
| UC-MSCs | Umbilical cord-derived mesenchymal stem cells |
References
- Leal, A.F.; Espejo-Mojica, A.J.; Sánchez, O.F.; Ramírez, C.M.; Reyes, L.H.; Cruz, J.C.; Alméciga-Díaz, C.J. Lysosomal storage diseases: Current therapies and future alternatives. J. Mol. Med. 2020, 98, 931–946. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Medina, D.L.; Ballabio, A. The rapidly evolving view of lysosomal storage diseases. EMBO Mol. Med. 2021, 13, e12836. [Google Scholar] [CrossRef]
- Leal, A.F.; Benincore-Flórez, E.; Rintz, E.; Herreño-Pachón, A.M.; Celik, B.; Ago, Y.; Alméciga-Díaz, C.J.; Tomatsu, S. Mucopolysaccharidoses: Cellular Consequences of Glycosaminoglycans Accumulation and Potential Targets. Int. J. Mol. Sci. 2022, 24, 477. [Google Scholar] [CrossRef]
- Wang, L.; You, X.; Zhang, L.; Zhang, C.; Zou, W. Mechanical regulation of bone remodeling. Bone Res. 2022, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Salhotra, A.; Shah, H.N.; Levi, B.; Longaker, M.T. Mechanisms of bone development and repair. Nat. Rev. Mol. Cell Biol. 2020, 21, 696–711. [Google Scholar] [CrossRef]
- Callens, S.J.; né Betts, D.C.T.; Müller, R.; Zadpoor, A.A. The local and global geometry of trabecular bone. Acta Biomater. 2021, 130, 343–361. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; An, Y.Z.; Guo, Q.; Zhou, H.Y.; Luo, X.H. Energy homeostasis in the bone. Trends Endocrinol. Metab. 2024, 35, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Liguori, L.; Monticelli, M.; Allocca, M.; Hay Mele, B.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations. Int. J. Mol. Sci. 2020, 21, 489. [Google Scholar] [CrossRef]
- Olarte-Avellaneda, S.; Cepeda Del Castillo, J.; Rojas-Rodriguez, A.F.; Sánchez, O.; Rodríguez-López, A.; Suárez García, D.A.; Pulido, L.M.S.; Alméciga-Díaz, C.J. Bromocriptine as a Novel Pharmacological Chaperone for Mucopolysaccharidosis IV A. ACS Med. Chem. Lett. 2020, 11, 1377–1385. [Google Scholar] [CrossRef]
- Alméciga-Diaz, C.J.; Hidalgo, O.A.; Olarte-Avellaneda, S.; Rodríguez-López, A.; Guzman, E.; Garzón, R.; Pimentel-Vera, L.N.; Puentes-Tellez, M.A.; Rojas-Rodriguez, A.F.; Gorshkov, K.; et al. Identification of Ezetimibe and Pranlukast as Pharmacological Chaperones for the Treatment of the Rare Disease Mucopolysaccharidosis Type IVA. J. Med. Chem. 2019, 62, 6175–6189. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, M.; Faggiano, S.; Mozzarelli, A. Enzyme Replacement Therapy for Genetic Disorders Associated with Enzyme Deficiency. Curr. Med. Chem. 2022, 29, 489–525. [Google Scholar] [CrossRef]
- Cleary, M.; Davison, J.; Gould, R.; Geberhiwot, T.; Hughes, D.; Mercer, J.; Morrison, A.; Murphy, E.; Santra, S.; Jarrett, J.; et al. Impact of long-term elosulfase alfa treatment on clinical and patient-reported outcomes in patients with mucopolysaccharidosis type IVA: Results from a Managed Access Agreement in England. Orphanet J. Rare Dis. 2021, 16, 38. [Google Scholar] [CrossRef]
- Yue, W.W.; Mackinnon, S.; Bezerra, G.A. Substrate reduction therapy for inborn errors of metabolism. Emerg. Top. Life Sci. 2019, 3, 63–73. [Google Scholar]
- Sawamoto, K.; Tomatsu, S. Development of Substrate Degradation Enzyme Therapy for Mucopolysaccharidosis IVA Murine Model. Int. J. Mol. Sci. 2019, 20, 4139. [Google Scholar] [CrossRef]
- Consiglieri, G.; Bernardo, M.E.; Brunetti-Pierri, N.; Aiuti, A. Ex Vivo and In Vivo Gene Therapy for Mucopolysaccharidoses: State of the Art. Hematol. Oncol. Clin. N. Am. 2022, 36, 865–878. [Google Scholar] [CrossRef]
- Akyol, M.U.; Alden, T.D.; Amartino, H.; Ashworth, J.; Belani, K.; Berger, K.I.; Borgo, A.; Braunlin, E.; Eto, Y.; Gold, J.I.; et al. Recommendations for the management of MPS IVA: Systematic evidence- and consensus-based guidance. Orphanet J. Rare Dis. 2019, 14, 137. [Google Scholar] [CrossRef]
- Sawamoto, K.; Karumuthil-Melethil, S.; Khan, S.; Stapleton, M.; Bruder, J.T.; Danos, O.; Tomatsu, S. Liver-Targeted AAV8 Gene Therapy Ameliorates Skeletal and Cardiovascular Pathology in a Mucopolysaccharidosis IVA Murine Model. Mol. Ther. Methods Clin. Dev. 2020, 18, 50–61. [Google Scholar] [CrossRef]
- Leal, A.F.; Celik, B.; Fnu, N.; Khan, S.; Tomatsu, S.; Alméciga-Díaz, C.J. Iron oxide-coupled CRISPR/nCas9-based genome editing assessment in mucopolysaccharidosis IVA mice. Mol. Ther. Methods Clin. Dev. 2023, 31, 101153. [Google Scholar] [CrossRef]
- Chen, H.H.; Sawamoto, K.; Mason, R.W.; Kobayashi, H.; Yamaguchi, S.; Suzuki, Y.; Orii, K.; Orii, T.; Tomatsu, S. Enzyme replacement therapy for mucopolysaccharidoses; past, present, and future. J. Hum. Genet. 2019, 64, 1153–1171. [Google Scholar] [CrossRef]
- Concolino, D.; Deodato, F.; Parini, R. Enzyme replacement therapy: Efficacy and limitations. Ital. J. Pediatr. 2018, 44 (Suppl. S2), 120. [Google Scholar] [CrossRef]
- Ago, Y.; Rintz, E.; Musini, K.S.; Ma, Z.; Tomatsu, S. Molecular Mechanisms in Pathophysiology of Mucopolysaccharidosis and Prospects for Innovative Therapy. Int. J. Mol. Sci. 2024, 25, 1113. [Google Scholar] [CrossRef]
- Mandolfo, O.; Parker, H.; Bigger, B. Innate Immunity in Mucopolysaccharide Diseases. Int. J. Mol. Sci. 2022, 23, 1999. [Google Scholar] [CrossRef]
- Squillaro, T.; Antonucci, I.; Alessio, N.; Esposito, A.; Cipollaro, M.; Melone, M.A.B.; Peluso, G.; Stuppia, L.; Galderisi, U. Impact of lysosomal storage disorders on biology of mesenchymal stem cells: Evidences from in vitro silencing of glucocerebrosidase (GBA) and alpha-galactosidase A (GLA) enzymes. J. Cell. Physiol. 2017, 232, 3454–3467. [Google Scholar] [CrossRef]
- Lecourt, S.; Mouly, E.; Freida, D.; Cras, A.; Ceccaldi, R.; Heraoui, D.; Chomienne, C.; Marolleau, J.P.; Arnulf, B.; Porcher, R.; et al. A prospective study of bone marrow hematopoietic and mesenchymal stem cells in type 1 Gaucher disease patients. PLoS ONE 2013, 8, e69293. [Google Scholar] [CrossRef]
- Clarke, L.A.; Hollak, C.E. The clinical spectrum and pathophysiology of skeletal complications in lysosomal storage disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 219–235. [Google Scholar] [CrossRef]
- Reed, M.C.; Schiffer, C.; Heales, S.; Mehta, A.B.; Hughes, D.A. Impact of sphingolipids on osteoblast and osteoclast activity in Gaucher disease. Mol. Genet. Metab. 2018, 124, 278–286. [Google Scholar] [CrossRef]
- Ormazabal, M.E.; Pavan, E.; Vaena, E.; Ferino, D.; Biasizzo, J.; Mucci, J.M.; Serra, F.; Cifù, A.; Scarpa, M.; Rozenfeld, P.A.; et al. Exploring the Pathophysiologic Cascade Leading to Osteoclastogenic Activation in Gaucher Disease Monocytes Generated via CRISPR/Cas9 Technology. Int. J. Mol. Sci. 2023, 24, 11204. [Google Scholar] [CrossRef]
- Jiang, Z.; Derrick-Roberts, A.L.; Reichstein, C.; Byers, S. Cell cycle progression is disrupted in murine MPS VII growth plate leading to reduced chondrocyte proliferation and transition to hypertrophy. Bone 2020, 132, 115195. [Google Scholar] [CrossRef]
- Fujitsuka, H.; Sawamoto, K.; Peracha, H.; Mason, R.W.; Mackenzie, W.; Kobayashi, H.; Yamaguchi, S.; Suzuki, Y.; Orii, K.; Orii, T.; et al. Biomarkers in patients with mucopolysaccharidosis type II and IV. Mol. Genet. Metab. Rep. 2019, 19, 100455. [Google Scholar] [CrossRef]
- Minervini, G.; Franco, R.; Marrapodi, M.M.; Mehta, V.; Fiorillo, L.; Badnjević, A.; Cervino, G.; Cicciù, M. Gaucher: A Systematic Review on Oral and Radiological Aspects. Medicina 2023, 59, 670. [Google Scholar] [CrossRef]
- David-Vizcarra, G.; Briody, J.; Ault, J.; Fietz, M.; Fletcher, J.; Savarirayan, R.; Wilson, M.; McGill, J.; Edwards, M.; Munns, C.; et al. The natural history and osteodystrophy of mucolipidosis types II and III. J. Paediatr. Child Health 2010, 46, 316–322. [Google Scholar] [CrossRef]
- De Ponti, G.; Donsante, S.; Frigeni, M.; Pievani, A.; Corsi, A.; Bernardo, M.E.; Riminucci, M.; Serafini, M. MPSI Manifestations and Treatment Outcome: Skeletal Focus. Int. J. Mol. Sci. 2022, 23, 11168. [Google Scholar] [CrossRef]
- Link, B.; Botha, J.; Giugliani, R. Characterization of orthopedic manifestations in patients with mucopolysaccharidosis II using data from 15 years of the Hunter Outcome Survey. JIMD Rep. 2024, 65, 17–24. [Google Scholar] [CrossRef]
- White, K.K.; Karol, L.A.; White, D.R.; Hale, S. Musculoskeletal manifestations of Sanfilippo Syndrome (mucopolysaccharidosis type III). J. Pediatr. Orthop. 2011, 31, 594–598. [Google Scholar] [CrossRef]
- Sawamoto, K.; Álvarez González, J.V.; Piechnik, M.; Otero, F.J.; Couce, M.L.; Suzuki, Y.; Tomatsu, S. Mucopolysaccharidosis IVA: Diagnosis, Treatment, and Management. Int. J. Mol. Sci. 2020, 21, 1517. [Google Scholar] [CrossRef]
- Hwang-Wong, E.; Amar, G.; Das, N.; Zhang, X.; Aaron, N.; Gale, K.; Rothman, N.; Fante, M.; Baik, A.; Bhargava, A.; et al. Skeletal phenotype amelioration in mucopolysaccharidosis VI requires intervention at the earliest stages of postnatal development. JCI Insight 2023, 8, e171312. [Google Scholar] [CrossRef]
- Poswar, F.D.O.; Henriques Nehm, J.; Kubaski, F.; Poletto, E.; Giugliani, R. Diagnosis and Emerging Treatment Strategies for Mucopolysaccharidosis VII (Sly Syndrome). Ther. Clin. Risk Manag. 2022, 18, 1143–1155. [Google Scholar] [CrossRef]
- Wasserstein, M.; Godbold, J.; McGovern, M.M. Skeletal manifestations in pediatric and adult patients with Niemann Pick disease type B. J. Inherit. Metab. Dis. 2013, 36, 123–127. [Google Scholar] [CrossRef]
- Adam, J.; Malone, R.; Lloyd, S.; Lee, J.; Hendriksz, C.J.; Ramaswami, U. Disease progression of alpha-mannosidosis and impact on patients and carers—A UK natural history survey. Mol. Genet. Metab. Rep. 2019, 20, 100480. [Google Scholar] [CrossRef]
- Hennermann, J.B.; Raebel, E.M.; Donà, F.; Jacquemont, M.L.; Cefalo, G.; Ballabeni, A.; Malm, D. Mortality in patients with alpha-mannosidosis: A review of patients’ data and the literature. Orphanet J. Rare Dis. 2022, 17, 287. [Google Scholar] [CrossRef]
- Alsahlawi, Z.; Aljishi, E.; Kheyami, A.; Alekri, A.; Alwedaie, S.M.J. Clinical spectrum and outcome of nine patients with a novel genetic variant of galactosialidosis in the Kingdom of Bahrain. JIMD Rep. 2022, 63, 614–620. [Google Scholar] [CrossRef]
- Arora, V.; Setia, N.; Dalal, A.; Vanaja, M.C.; Gupta, D.; Razdan, T.; Phadke, S.R.; Saxena, R.; Rohtagi, A.; Verma, I.C.; et al. Sialidosis type II: Expansion of phenotypic spectrum and identification of a common mutation in seven patients. Mol. Genet. Metab. Rep. 2020, 22, 100561. [Google Scholar] [CrossRef]
- Arvio, M.; Mononen, I. Aspartylglycosaminuria: A review. Orphanet J. Rare Dis. 2016, 11, 162. [Google Scholar] [CrossRef]
- Goodspeed, K.; Feng, C.; Laine, M.; Lund, T.C. Aspartylglucosaminuria: Clinical Presentation and Potential Therapies. J. Child Neurol. 2021, 36, 403–414. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, J.; Liu, B.; Shao, C.; Shi, Y. Reciprocal regulation of mesenchymal stem cells and immune responses. Cell Stem Cell 2022, 29, 1515–1530. [Google Scholar] [CrossRef]
- Liu, J.; Gao, J.; Liang, Z.; Gao, C.; Niu, Q.; Wu, F.; Zhang, L. Mesenchymal stem cells and their microenvironment. Stem Cell Res. Ther. 2022, 13, 429. [Google Scholar] [CrossRef]
- Zhidu, S.; Ying, T.; Rui, J.; Chao, Z. Translational potential of mesenchymal stem cells in regenerative therapies for human diseases: Challenges and opportunities. Stem Cell Res. Ther. 2024, 15, 266. [Google Scholar] [CrossRef]
- Chouaib, B.; Haack-Sørensen, M.; Chaubron, F.; Cuisinier, F.; Collart-Dutilleul, P.Y. Towards the Standardization of Mesenchymal Stem Cell Secretome-Derived Product Manufacturing for Tissue Regeneration. Int. J. Mol. Sci. 2023, 24, 12594. [Google Scholar] [CrossRef]
- Giovannelli, L.; Bari, E.; Jommi, C.; Tartara, F.; Armocida, D.; Garbossa, D.; Cofano, F.; Torre, M.L.; Segale, L. Mesenchymal stem cell secretome and extracellular vesicles for neurodegenerative diseases: Risk-benefit profile and next steps for the market access. Bioact. Mater. 2023, 29, 16–35. [Google Scholar] [CrossRef]
- Wang, J.; Barr, M.M.; Wehman, A.M. Extracellular vesicles. Genetics 2024, 227, iyae088. [Google Scholar] [CrossRef]
- You, B.; Zhou, C.; Yang, Y. MSC-EVs alleviate osteoarthritis by regulating microenvironmental cells in the articular cavity and maintaining cartilage matrix homeostasis. Ageing Res. Rev. 2023, 85, 101864. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, T.; Xue, Y.; Zhan, B.; Lai, Z.; Huang, W.; Peng, X.; Zhou, Y. Research progress of extracellular vesicles and exosomes derived from mesenchymal stem cells in the treatment of oxidative stress-related diseases. Front. Immunol. 2023, 14, 1238789. [Google Scholar] [CrossRef] [PubMed]
- Amsar, R.M.; Wijaya, C.H.; Ana, I.D.; Hidajah, A.C.; Notobroto, H.B.; Kencana Wungu, T.D.; Barlian, A. Extracellular vesicles: A promising cell-free therapy for cartilage repair. Future Sci. OA 2022, 8, FSO774. [Google Scholar] [CrossRef]
- Deo, D.; Marchioni, M.; Rao, P. Mesenchymal Stem/Stromal Cells in Organ Transplantation. Pharmaceutics 2022, 14, 791. [Google Scholar] [CrossRef]
- Bhat, S.; Viswanathan, P.; Chandanala, S.; Prasanna, S.J.; Seetharam, R.N. Expansion and characterization of bone marrow derived human mesenchymal stromal cells in serum-free conditions. Sci. Rep. 2021, 11, 3403. [Google Scholar] [CrossRef]
- Parfejevs, V.; Sagini, K.; Buss, A.; Sobolevska, K.; Llorente, A.; Riekstina, U.; Abols, A. Adult Stem Cell-Derived Extracellular Vesicles in Cancer Treatment: Opportunities and Challenges. Cells 2020, 9, 1171. [Google Scholar] [CrossRef] [PubMed]
- Kou, M.; Huang, L.; Yang, J.; Chiang, Z.; Chen, S.; Liu, J.; Guo, L.; Zhang, X.; Zhou, X.; Xu, X.; et al. Mesenchymal stem cell-derived extracellular vesicles for immunomodulation and regeneration: A next generation therapeutic tool? Cell Death Dis. 2022, 13, 580. [Google Scholar] [CrossRef]
- Cosenza, S.; Toupet, K.; Maumus, M.; Luz-Crawford, P.; Blanc-Brude, O.; Jorgensen, C.; Noël, D. Mesenchymal stem cells-derived exosomes are more immunosuppressive than microparticles in inflammatory arthritis. Theranostics 2018, 8, 1399–1410. [Google Scholar] [CrossRef]
- Zhang, S.; Teo, K.Y.W.; Chuah, S.J.; Lai, R.C.; Lim, S.K.; Toh, W.S. MSC exosomes alleviate temporomandibular joint osteoarthritis by attenuating inflammation and restoring matrix homeostasis. Biomaterials 2019, 200, 35–47. [Google Scholar] [CrossRef]
- Qi, X.; Zhang, J.; Yuan, H.; Xu, Z.; Li, Q.; Niu, X.; Hu, B.; Wang, Y.; Li, X. Exosomes Secreted by Human-Induced Pluripotent Stem Cell-Derived Mesenchymal Stem Cells Repair Critical-Sized Bone Defects through Enhanced Angiogenesis and Osteogenesis in Osteoporotic Rats. Int. J. Biol. Sci. 2016, 12, 836–849. [Google Scholar] [CrossRef]
- Zuo, R.; Liu, M.; Wang, Y.; Li, J.; Wang, W.; Wu, J.; Sun, C.; Li, B.; Wang, Z.; Lan, W.; et al. BM-MSC-derived exosomes alleviate radiation-induced bone loss by restoring the function of recipient BM-MSCs and activating Wnt/β-catenin signaling. Stem Cell Res. Ther. 2019, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T.; Miyaki, S.; Ishitobi, H.; Ogura, T.; Kato, Y.; Kamei, N.; Miyado, K.; Higashi, Y.; Ochi, M. Mesenchymal Stem Cell-Derived Exosomes Promote Fracture Healing in a Mouse Model. Stem Cells Transl. Med. 2016, 5, 1620–1630. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hao, Z.; Wang, P.; Xia, Y.; Wu, J.; Xia, D.; Fang, S.; Xu, S. Exosomes from human umbilical cord mesenchymal stem cells enhance fracture healing through HIF-1α-mediated promotion of angiogenesis in a rat model of stabilized fracture. Cell Prolif. 2019, 52, e12570. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Dooner, M.; Papa, E.; Del Tatto, M.; Pereira, M.; Borgovan, T.; Cheng, Y.; Goldberg, L.; Liang, O.; Camussi, G.; et al. Biodistribution of Mesenchymal Stem Cell-Derived Extracellular Vesicles in a Radiation Injury Bone Marrow Murine Model. Int. J. Mol. Sci. 2019, 20, 5468. [Google Scholar] [CrossRef]
- Matsuzaka, Y.; Yashiro, R. Current Strategies and Therapeutic Applications of Mesenchymal Stem Cell-Based Drug Delivery. Pharmaceuticals 2024, 17, 707. [Google Scholar] [CrossRef]
- Wang, Y.; Wen, J.; Lu, T.; Han, W.; Jiao, K.; Li, H. Mesenchymal Stem Cell-Derived Extracellular Vesicles in Bone-Related Diseases: Intercellular Communication Messengers and Therapeutic Engineering Protagonists. Int. J. Nanomed. 2024, 19, 3233–3257. [Google Scholar] [CrossRef]
- Qin, B.; Zhang, Q.; Chen, D.; Yu, H.Y.; Luo, A.X.; Suo, L.P.; Cai, Y.; Cai, D.Y.; Luo, J.; Huang, J.F.; et al. Extracellular vesicles derived from mesenchymal stem cells: A platform that can be engineered. Histol. Histopathol. 2021, 36, 615–632. [Google Scholar]
- Li, Q.; Xu, R.; Lei, K.; Yuan, Q. Insights into skeletal stem cells. Bone Res. 2022, 10, 61. [Google Scholar] [CrossRef]
- Celik, B.; Leal, A.F.; Tomatsu, S. Potential Targeting Mechanisms for Bone-Directed Therapies. Int. J. Mol. Sci. 2024, 25, 8339. [Google Scholar] [CrossRef]
- Hallett, S.A.; Ono, W.; Ono, N. Growth Plate Chondrocytes: Skeletal Development, Growth and Beyond. Int. J. Mol. Sci. 2019, 20, 6009. [Google Scholar] [CrossRef]
- Koosha, E.; Eames, B.F. Two Modulators of Skeletal Development: BMPs and Proteoglycans. J. Dev. Biol. 2022, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Patil, S.; Gao, Y.G.; Qian, A. The Bone Extracellular Matrix in Bone Formation and Regeneration. Front. Pharmacol. 2020, 11, 757. [Google Scholar] [CrossRef] [PubMed]
- Dalle Carbonare, L.; Cominacini, M.; Trabetti, E.; Bombieri, C.; Pessoa, J.; Romanelli, M.G.; Valenti, M.T. The bone microenvironment: New insights into the role of stem cells and cell communication in bone regeneration. Stem Cell Res. Ther. 2025, 16, 169. [Google Scholar] [CrossRef]
- Xu, X.; Zheng, L.; Yuan, Q.; Zhen, G.; Crane, J.L.; Zhou, X.; Cao, X. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res. 2018, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Safari, B.; Davaran, S.; Aghanejad, A. Osteogenic potential of the growth factors and bioactive molecules in bone regeneration. Int. J. Biol. Macromol. 2021, 175, 544–557. [Google Scholar] [CrossRef]
- de Araújo Farias, V.; Carrillo-Gálvez, A.B.; Martin, F.; Anderson, P. TGF-β and mesenchymal stromal cells in regenerative medicine, autoimmunity and cancer. Cytokine Growth Factor. Rev. 2018, 43, 25–37. [Google Scholar] [CrossRef]
- Zhao, C.; Jiang, W.; Zhou, N.; Liao, J.; Yang, M.; Hu, N.; Liang, X.; Xu, W.; Chen, H.; Liu, W.; et al. Sox9 augments BMP2-induced chondrogenic differentiation by downregulating Smad7 in mesenchymal stem cells (MSCs). Genes Dis. 2017, 4, 229–239. [Google Scholar] [CrossRef]
- Dong, D.L.; Jin, G.Z. Targeting Chondrocyte Hypertrophy as Strategies for the Treatment of Osteoarthritis. Bioengineering 2025, 12, 77. [Google Scholar] [CrossRef]
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv. Pharmacol. 2018, 81, 241–330. [Google Scholar]
- Komori, T. Bone development by Hedgehog and Wnt signaling, Runx2, and Sp7. J. Bone Miner. Metab. 2025, 43, 33–38. [Google Scholar] [CrossRef]
- Hu, K.; Olsen, B.R. The roles of vascular endothelial growth factor in bone repair and regeneration. Bone 2016, 91, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Adam, M.S.; Zhuang, H.; Ren, X.; Zhang, Y.; Zhou, P. The metabolic characteristics and changes of chondrocytes. Front. Endocrinol. 2024, 15, 1393550. [Google Scholar] [CrossRef]
- de Oliveira, P.G.; Baldo, G.; Mayer, F.Q.; Martinelli, B.; Meurer, L.; Giugliani, R.; Matte, U.; Xavier, R.M. Characterization of joint disease in mucopolysaccharidosis type I mice. Int. J. Exp. Pathol. 2013, 94, 305–311. [Google Scholar] [CrossRef]
- Tomatsu, S.; Orii, K.O.; Vogler, C.; Nakayama, J.; Levy, B.; Grubb, J.H.; Gutierrez, M.A.; Shim, S.; Yamaguchi, S.; Nishioka, T.; et al. Mouse model of N-acetylgalactosamine-6-sulfate sulfatase deficiency (Galns-/-) produced by targeted disruption of the gene defective in Morquio A disease. Hum. Mol. Genet. 2003, 12, 3349–3358. [Google Scholar] [CrossRef] [PubMed]
- Broeders, M.; van Rooij, J.; Oussoren, E.; van Gestel, T.; Smith, C.; Kimber, S.; Verdijk, R.; Wagenmakers, M.; van den Hout, J.; van der Ploeg, A.; et al. Modeling cartilage pathology in mucopolysaccharidosis VI using iPSCs reveals early dysregulation of chondrogenic and metabolic gene expression. Front. Bioeng. Biotechnol. 2022, 10, 949063. [Google Scholar] [CrossRef]
- Rowan, D.J.; Tomatsu, S.; Grubb, J.H.; Montaño, A.M.; Sly, W.S. Assessment of bone dysplasia by micro-CT and glycosaminoglycan levels in mouse models for mucopolysaccharidosis type I, IIIA, IVA, and VII. J. Inherit. Metab. Dis. 2013, 36, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Lui, J.C.; Yue, S.; Lee, A.; Kikani, B.; Temnycky, A.; Barnes, K.M.; Baron, J. Persistent Sox9 expression in hypertrophic chondrocytes suppresses transdifferentiation into osteoblasts. Bone 2019, 125, 169–177. [Google Scholar] [CrossRef]
- Khoswanto, C. Role of matrix metalloproteinases in bone regeneration: Narrative review. J. Oral. Biol. Craniofac. Res. 2023, 13, 539–543. [Google Scholar] [CrossRef]
- Zhou, J.; Shi, Y. Mesenchymal stem/stromal cells (MSCs): Origin, immune regulation, and clinical applications. Cell. Mol. Immunol. 2023, 20, 555–557. [Google Scholar] [CrossRef]
- Ripoll, C.B.; Flaat, M.; Klopf-Eiermann, J.; Fisher-Perkins, J.M.; Trygg, C.B.; Scruggs, B.A.; McCants, M.L.; Leonard, H.P.; Lin, A.F.; Zhang, S.; et al. Mesenchymal lineage stem cells have pronounced anti-inflammatory effects in the twitcher mouse model of Krabbe’s disease. Stem Cells 2011, 29, 67–77. [Google Scholar] [CrossRef]
- Meuleman, N.; Vanhaelen, G.; Tondreau, T.; Lewalle, P.; Kwan, J.; Bennani, J.; Martiat, P.; Lagneaux, L.; Bron, D. Reduced intensity conditioning haematopoietic stem cell transplantation with mesenchymal stromal cells infusion for the treatment of metachromatic leukodystrophy: A case report. Haematologica 2008, 93, e11–e13. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Bae, J.S.; Jin, H.K. Human umbilical cord blood-derived mesenchymal stem cells improve neurological abnormalities of Niemann-Pick type C mouse by modulation of neuroinflammatory condition. J. Vet. Med. Sci. 2010, 72, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Taketani, T.; Oyama, C.; Mihara, A.; Tanabe, Y.; Abe, M.; Hirade, T.; Yamamoto, S.; Bo, R.; Kanai, R.; Tadenuma, T.; et al. Ex Vivo Expanded Allogeneic Mesenchymal Stem Cells With Bone Marrow Transplantation Improved Osteogenesis in Infants With Severe Hypophosphatasia. Cell Transplant. 2015, 24, 1931–1943. [Google Scholar] [CrossRef]
- Baranovskii, D.S.; Klabukov, I.D.; Arguchinskaya, N.V.; Yakimova, A.O.; Kisel, A.A.; Yatsenko, E.M.; Ivanov, S.A.; Shegay, P.V.; Kaprin, A.D. Adverse events, side effects and complications in mesenchymal stromal cell-based therapies. Stem Cell Investig. 2022, 9, 7. [Google Scholar] [CrossRef]
- Hoang, V.T.; Le, D.S.; Hoang, D.M.; Phan, T.T.K.; Ngo, L.A.T.; Nguyen, T.K.; Bui, V.A.; Nguyen Thanh, L. Impact of tissue factor expression and administration routes on thrombosis development induced by mesenchymal stem/stromal cell infusions: Re-evaluating the dogma. Stem Cell Res. Ther. 2024, 15, 56. [Google Scholar] [CrossRef] [PubMed]
- Večerić-Haler, Ž.; Kojc, N.; Sever, M.; Zver, S.; Švajger, U.; Poženel, P.; Hartman, K.; Urdih, T.; Mlinšek, G.; Oblak, M.; et al. Case Report: Capillary Leak Syndrome With Kidney Transplant Failure Following Autologous Mesenchymal Stem Cell Therapy. Front. Med. 2021, 8, 708744. [Google Scholar] [CrossRef]
- Issa, S.S.; Shaimardanova, A.A.; Valiullin, V.V.; Rizvanov, A.A.; Solovyeva, V.V. Mesenchymal Stem Cell-Based Therapy for Lysosomal Storage Diseases and Other Neurodegenerative Disorders. Front. Pharmacol. 2022, 13, 859516. [Google Scholar] [CrossRef]
- Műzes, G.; Sipos, F. Mesenchymal Stem Cell-Derived Secretome: A Potential Therapeutic Option for Autoimmune and Immune-Mediated Inflammatory Diseases. Cells 2022, 11, 2300. [Google Scholar] [CrossRef]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: From biogenesis to uptake and intracellular signalling. Cell Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef]
- Mizenko, R.R.; Feaver, M.; Bozkurt, B.T.; Lowe, N.; Nguyen, B.; Huang, K.W.; Wang, A.; Carney, R.P. A critical systematic review of extracellular vesicle clinical trials. J. Extracell. Vesicles 2024, 13, e12510. [Google Scholar] [CrossRef]
- Liang, Y.; Duan, L.; Lu, J.; Xia, J. Engineering exosomes for targeted drug delivery. Theranostics 2021, 11, 3183–3195. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrügger, U.; et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 2024, 13, e12404. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, A.L.; Johansson, K.; Mossberg, M.; Kahn, R.; Karpman, D. Exosomes and microvesicles in normal physiology, pathophysiology, and renal diseases. Pediatr. Nephrol. 2019, 34, 11–30. [Google Scholar] [CrossRef]
- Xunian, Z.; Kalluri, R. Biology and therapeutic potential of mesenchymal stem cell-derived exosomes. Cancer Sci. 2020, 111, 3100–3110. [Google Scholar] [CrossRef] [PubMed]
- Ovčar, A.; Kovačič, B. Biogenesis of Extracellular Vesicles (EVs) and the Potential Use of Embryo-Derived EVs in Medically Assisted Reproduction. Int. J. Mol. Sci. 2024, 26, 42. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Shin, K.J.; Chae, Y.C. Regulation of cargo selection in exosome biogenesis and its biomedical applications in cancer. Exp. Mol. Med. 2024, 56, 877–889. [Google Scholar] [CrossRef]
- Leal, A.F.; Inci, O.K.; Seyrantepe, V.; Rintz, E.; Celik, B.; Ago, Y.; León, D.; Suarez, D.A.; Alméciga-Díaz, C.J.; Tomatsu, S. Molecular Trojan Horses for treating lysosomal storage diseases. Mol. Genet. Metab. 2023, 140, 107648. [Google Scholar] [CrossRef]
- Lu, B.; Ku, J.; Flojo, R.; Olson, C.; Bengford, D.; Marriott, G. Exosome- and extracellular vesicle-based approaches for the treatment of lysosomal storage disorders. Adv. Drug Deliv. Rev. 2022, 188, 114465. [Google Scholar] [CrossRef]
- Fedele, A.O.; Isenmann, S.; Kamei, M.; Snel, M.F.; Trim, P.J.; Proud, C.G.; Hopwood, J.J. Lysosomal N-acetyltransferase interacts with ALIX and is detected in extracellular vesicles. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1451–1464. [Google Scholar] [CrossRef]
- Seras-Franzoso, J.; Díaz-Riascos, Z.V.; Corchero, J.L.; González, P.; García-Aranda, N.; Mandaña, M.; Riera, R.; Boullosa, A.; Mancilla, S.; Grayston, A.; et al. Extracellular vesicles from recombinant cell factories improve the activity and efficacy of enzymes defective in lysosomal storage disorders. J. Extracell. Vesicles 2021, 10, e12058. [Google Scholar] [CrossRef]
- Haney, M.J.; Klyachko, N.L.; Harrison, E.B.; Zhao, Y.; Kabanov, A.V.; Batrakova, E.V. TPP1 Delivery to Lysosomes with Extracellular Vesicles and their Enhanced Brain Distribution in the Animal Model of Batten Disease. Adv. Healthc. Mater. 2019, 8, e1801271. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, D.M.; El-Kares, R.; Taranta, A.; Bellomo, F.; Emma, F.; Besouw, M.; Levtchenko, E.; Toelen, J.; van den Heuvel, L.; Chu, L.; et al. Stem cell microvesicles transfer cystinosin to human cystinotic cells and reduce cystine accumulation in vitro. PLoS ONE 2012, 7, e42840. [Google Scholar] [CrossRef]
- Flanagan, M.; Pathak, I.; Gan, Q.; Winter, L.; Emnet, R.; Akel, S.; Montaño, A.M. Umbilical mesenchymal stem cell-derived extracellular vesicles as enzyme delivery vehicle to treat Morquio A fibroblasts. Stem Cell Res. Ther. 2021, 12, 276. [Google Scholar] [CrossRef] [PubMed]
- Lerussi, G.; Villagrasa-Araya, V.; Moltó-Abad, M.; Del Toro, M.; Pintos-Morell, G.; Seras-Franzoso, J.; Abasolo, I. Extracellular Vesicles as Tools for Crossing the Blood-Brain Barrier to Treat Lysosomal Storage Diseases. Life 2025, 15, 70. [Google Scholar] [CrossRef]
- Do, M.A.; Levy, D.; Brown, A.; Marriott, G.; Lu, B. Targeted delivery of lysosomal enzymes to the endocytic compartment in human cells using engineered extracellular vesicles. Sci. Rep. 2019, 9, 17274. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.I.; Park, J.; Zhu, Y.; Wang, X.; Han, Y.; Zhang, D. Recent advances in extracellular vesicles for therapeutic cargo delivery. Exp. Mol. Med. 2024, 56, 836–849. [Google Scholar] [CrossRef]
- Han, Y.; Jones, T.W.; Dutta, S.; Zhu, Y.; Wang, X.; Narayanan, S.P.; Fagan, S.C.; Zhang, D. Overview and Update on Methods for Cargo Loading into Extracellular Vesicles. Processes 2021, 9, 356. [Google Scholar] [CrossRef]
- Liu, Y.J.; Wang, C. A review of the regulatory mechanisms of extracellular vesicles-mediated intercellular communication. Cell Commun. Signal. 2023, 21, 77. [Google Scholar] [CrossRef]
- Leal, A.F.; Herreno-Pachón, A.M.; Benincore-Flórez, E.; Karunathilaka, A.; Tomatsu, S. Current Strategies for Increasing Knock-In Efficiency in CRISPR/Cas9-Based Approaches. Int. J. Mol. Sci. 2024, 25, 2456. [Google Scholar] [CrossRef]
- Leal, A.F.; Fnu, N.; Benincore-Flórez, E.; Herreño-Pachón, A.M.; Echeverri-Peña, O.Y.; Alméciga-Díaz, C.J.; Tomatsu, S. The landscape of CRISPR/Cas9 for inborn errors of metabolism. Mol. Genet. Metab. 2022, 138, 106968. [Google Scholar] [CrossRef] [PubMed]
- Berggreen, A.H.; Petersen, J.L.; Lin, L.; Benabdellah, K.; Luo, Y. CRISPR delivery with extracellular vesicles: Promises and challenges. J. Extracell. Biol. 2023, 2, e111. [Google Scholar] [CrossRef]
- Meyer, C.; Losacco, J.; Stickney, Z.; Li, L.; Marriott, G.; Lu, B. Pseudotyping exosomes for enhanced protein delivery in mammalian cells. Int. J. Nanomed. 2017, 12, 3153–3170. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; Provoda, C.; Lee, K.D. Design and characterization of novel recombinant listeriolysin O-protamine fusion proteins for enhanced gene delivery. Mol. Pharm. 2015, 12, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.; Kaelin, S.; Leroux, J.C. The TFAMoplex-Conversion of the Mitochondrial Transcription Factor A into a DNA Transfection Agent. Adv. Sci. 2022, 9, e2104987. [Google Scholar] [CrossRef]
- Klipp, A.; Burger, M.; Leroux, J.C. Get out or die trying: Peptide- and protein-based endosomal escape of RNA therapeutics. Adv. Drug Deliv. Rev. 2023, 200, 115047. [Google Scholar] [CrossRef]
- Gomez-Ospina, N.; Scharenberg, S.G.; Mostrel, N.; Bak, R.O.; Mantri, S.; Quadros, R.M.; Gurumurthy, C.B.; Lee, C.; Bao, G.; Suarez, C.J.; et al. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 2019, 10, 4045. [Google Scholar] [CrossRef]
- Huang, C.C.; Kang, M.; Leung, K.; Lu, Y.; Shirazi, S.; Gajendrareddy, P.; Ravindran, S. Micro RNA based MSC EV engineering: Targeting the BMP2 cascade for bone repair. Front. Cell Dev. Biol. 2023, 11, 1127594. [Google Scholar] [CrossRef]
- Li, N. CircTBL1XR1/miR-424 axis regulates Smad7 to promote the proliferation and metastasis of colorectal cancer. J. Gastrointest. Oncol. 2020, 11, 918–931. [Google Scholar] [CrossRef]
- Nakamura-Utsunomiya, A. Bone Biomarkers in Mucopolysaccharidoses. Int. J. Mol. Sci. 2021, 22, 12651. [Google Scholar] [CrossRef]
- Khan, S.A.; Nelson, M.S.; Pan, C.; Gaffney, P.M.; Gupta, P. Endogenous heparan sulfate and heparin modulate bone morphogenetic protein-4 signaling and activity. Am. J. Physiol. Cell Physiol. 2008, 294, C1387–C1397. [Google Scholar] [CrossRef] [PubMed]
- Rintz, E.; Herreño-Pachón, A.M.; Celik, B.; Nidhi, F.; Khan, S.; Benincore-Flórez, E.; Tomatsu, S. Bone Growth Induction in Mucopolysaccharidosis IVA Mouse. Int. J. Mol. Sci. 2023, 24, 9890. [Google Scholar] [CrossRef] [PubMed]
- Sanmartin, M.C.; Borzone, F.R.; Giorello, M.B.; Yannarelli, G.; Chasseing, N.A. Mesenchymal Stromal Cell-Derived Extracellular Vesicles as Biological Carriers for Drug Delivery in Cancer Therapy. Front. Bioeng. Biotechnol. 2022, 10, 882545. [Google Scholar] [CrossRef]
- Choi, W.; Park, D.J.; Eliceiri, B.P. Defining tropism and activity of natural and engineered extracellular vesicles. Front. Immunol. 2024, 15, 1363185. [Google Scholar] [CrossRef]
- Cui, Y.; Guo, Y.; Kong, L.; Shi, J.; Liu, P.; Li, R.; Geng, Y.; Gao, W.; Zhang, Z.; Fu, D. A bone-targeted engineered exosome platform delivering siRNA to treat osteoporosis. Bioact. Mater. 2022, 10, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Wein, M.N.; Jones, D.C.; Shim, J.H.; Aliprantis, A.O.; Sulyanto, R.; Lazarevic, V.; Poliachik, S.L.; Gross, T.S.; Glimcher, L.H. Control of bone resorption in mice by Schnurri-3. Proc. Natl. Acad. Sci. USA 2012, 109, 8173–8178. [Google Scholar] [CrossRef]
- Luo, Z.W.; Liu, Y.W.; Rao, S.S.; Yin, H.; Huang, J.; Chen, C.Y.; Hu, Y.; Zhang, Y.; Tan, Y.J.; Yuan, L.Q.; et al. Aptamer-functionalized exosomes from bone marrow stromal cells target bone to promote bone regeneration. Nanoscale 2019, 11, 20884–20892. [Google Scholar] [CrossRef]
- Wang, Y.; Yao, J.; Cai, L.; Liu, T.; Wang, X.; Zhang, Y.; Zhou, Z.; Li, T.; Liu, M.; Lai, R.; et al. Bone-Targeted Extracellular Vesicles from Mesenchymal Stem Cells for Osteoporosis Therapy. Int. J. Nanomed. 2020, 15, 7967–7977. [Google Scholar] [CrossRef]
- You, D.G.; Lim, G.T.; Kwon, S.; Um, W.; Oh, B.H.; Song, S.H.; Lee, J.; Jo, D.G.; Cho, Y.W.; Park, J.H. Metabolically engineered stem cell-derived exosomes to regulate macrophage heterogeneity in rheumatoid arthritis. Sci. Adv. 2021, 7, eabe0083. [Google Scholar] [CrossRef]
- Deng, J.; Li, Q.; Wang, F. Novel administration strategies for tissue-specific delivery of extracellular vesicles. Extracell. Vesicle 2024, 4, 100057. [Google Scholar] [CrossRef]
- Kim, M.; Steinberg, D.R.; Burdick, J.A.; Mauck, R.L. Extracellular vesicles mediate improved functional outcomes in engineered cartilage produced from MSC/chondrocyte cocultures. Proc. Natl. Acad. Sci. USA 2019, 116, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Vonk, L.A.; van Dooremalen, S.F.; Liv, N.; Klumperman, J.; Coffer, P.J.; Saris, D.B.; Lorenowicz, M.J. Mesenchymal Stromal/stem Cell-derived Extracellular Vesicles Promote Human Cartilage Regeneration. Theranostics 2018, 8, 906–920. [Google Scholar] [CrossRef] [PubMed]
- van Buul, G.M.; Villafuertes, E.; Bos, P.K.; Waarsing, J.H.; Kops, N.; Narcisi, R.; Weinans, H.; Verhaar, J.A.N.; Bernsen, M.R.; Van Osch, G.J.V.M. Mesenchymal stem cells secrete factors that inhibit inflammatory processes in short-term osteoarthritic synovium and cartilage explant culture. Osteoarthr. Cartil. 2012, 20, 1186–1196. [Google Scholar] [CrossRef] [PubMed]
- Scalzone, A.; Sanjurjo-Rodríguez, C.; Berlinguer-Palmini, R.; Dickinson, A.M.; Jones, E.; Wang, X.N.; Crossland, R.E. Functional and Molecular Analysis of Human Osteoarthritic Chondrocytes Treated with Bone Marrow-Derived MSC-EVs. Bioengineering 2024, 11, 388. [Google Scholar] [CrossRef]
- Leal, A.F.; Khan, S.A.; Tomatsu, S. Uncovering mitochondrial disturbances in MPS IVA chondrocytes. Mol. Genet. Metab. 2025, 144, 108823. [Google Scholar] [CrossRef]
- Zhao, M.; Liu, S.; Wang, C.; Wang, Y.; Wan, M.; Liu, F.; Gong, M.; Yuan, Y.; Chen, Y.; Cheng, J.; et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles Attenuate Mitochondrial Damage and Inflammation by Stabilizing Mitochondrial DNA. ACS Nano 2021, 15, 1519–1538. [Google Scholar] [CrossRef]
- Lu, T.; Zhang, J.; Cai, J.; Xiao, J.; Sui, X.; Yuan, X.; Li, R.; Li, Y.; Yao, J.; Lv, G.; et al. Extracellular vesicles derived from mesenchymal stromal cells as nanotherapeutics for liver ischaemia-reperfusion injury by transferring mitochondria to modulate the formation of neutrophil extracellular traps. Biomaterials 2022, 284, 121486. [Google Scholar] [CrossRef]
- Stepien, K.M.; Roncaroli, F.; Turton, N.; Hendriksz, C.J.; Roberts, M.; Heaton, R.A.; Hargreaves, I. Mechanisms of Mitochondrial Dysfunction in Lysosomal Storage Disorders: A Review. J. Clin. Med. 2020, 9, 2596. [Google Scholar] [CrossRef]
- To, K.; Romain, K.; Mak, C.; Kamaraj, A.; Henson, F.; Khan, W. The Treatment of Cartilage Damage Using Human Mesenchymal Stem Cell-Derived Extracellular Vesicles: A Systematic Review of in vivo Studies. Front. Bioeng. Biotechnol. 2020, 8, 580. [Google Scholar] [CrossRef]
- Chen, P.; Tang, S.; Gao, H.; Zhang, H.; Chen, C.; Fang, Z.; Peng, G.; Weng, H.; Chen, A.; Zhang, C.; et al. Wharton’s jelly mesenchymal stem cell-derived small extracellular vesicles as natural nanoparticles to attenuate cartilage injury via microRNA regulation. Int. J. Pharm. 2022, 623, 121952. [Google Scholar] [CrossRef]
- Lener, T.; Gimona, M.; Aigner, L.; Börger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; Portillo, H.A.D.; et al. Applying extracellular vesicles based therapeutics in clinical trials—An ISEV position paper. J. Extracell. Vesicles 2015, 4, 30087. [Google Scholar] [CrossRef] [PubMed]
- Vader, P.; Mol, E.A.; Pasterkamp, G.; Schiffelers, R.M. Extracellular vesicles for drug delivery. Adv. Drug Deliv. Rev. 2016, 106 Pt A, 148–156. [Google Scholar] [CrossRef]
- Gardiner, C.; Vizio, D.D.; Sahoo, S.; Théry, C.; Witwer, K.W.; Wauben, M.; Hill, A.F. Techniques used for the isolation and characterization of extracellular vesicles: Results of a worldwide survey. J. Extracell. Vesicles 2016, 5, 32945. [Google Scholar] [CrossRef] [PubMed]
- Ev-Track Consortium; Van Deun, J.; Mestdagh, P.; Agostinis, P.; Akay, Ö.; Anand, S.; Anckaert, J.; Martinez, Z.A.; Baetens, T.; Beghein, E.; et al. EV-TRACK: Transparent reporting and centralizing knowledge in extracellular vesicle research. Nat. Methods 2017, 14, 228–232. [Google Scholar] [PubMed]
- Wiklander, O.P.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef]
- Meng, W.; He, C.; Hao, Y.; Wang, L.; Li, L.; Zhu, G. Prospects and challenges of extracellular vesicle-based drug delivery system: Considering cell source. Drug Deliv. 2020, 27, 585–598. [Google Scholar] [CrossRef]
- Xia, Y.; Zhang, J.; Liu, G.; Wolfram, J. Immunogenicity of Extracellular Vesicles. Adv. Mater. 2024, 36, e2403199. [Google Scholar] [CrossRef]
- Lins, P.M.P.; Pirlet, E.; Szymonik, M.; Bronckaers, A.; Nelissen, I. Manufacture of extracellular vesicles derived from mesenchymal stromal cells. Trends Biotechnol. 2023, 41, 965–981. [Google Scholar] [CrossRef]
- Liu, X.; Gao, M.; Bao, J. Precisely Targeted Nanoparticles for CRISPR-Cas9 Delivery in Clinical Applications. Nanomaterials 2025, 15, 540. [Google Scholar] [CrossRef]
- Liu, W.; Huang, J.; Chen, F.; Xie, D.; Wang, L.; Ye, C.; Zhu, Q.; Li, X.; Li, X.; Yang, L. MSC-derived small extracellular vesicles overexpressing miR-20a promoted the osteointegration of porous titanium alloy by enhancing osteogenesis via targeting BAMBI. Stem Cell Res. Ther. 2021, 12, 348. [Google Scholar] [CrossRef]
- Yang, Y.; Yuan, L.; Cao, H.; Guo, J.; Zhou, X.; Zeng, Z. Application and Molecular Mechanisms of Extracellular Vesicles Derived from Mesenchymal Stem Cells in Osteoporosis. Curr. Issues Mol. Biol. 2022, 44, 6346–6367. [Google Scholar] [CrossRef] [PubMed]
- Man, K.; Eisenstein, N.M.; Hoey, D.A.; Cox, S.C. Bioengineering extracellular vesicles: Smart nanomaterials for bone regeneration. J. Nanobiotechnology 2023, 21, 137. [Google Scholar] [CrossRef]
- Ellison, S.; Parker, H.; Bigger, B. Advances in therapies for neurological lysosomal storage disorders. J. Inherit. Metab. Dis. 2023, 46, 874–905. [Google Scholar] [CrossRef]
- Doherty, C.; Stapleton, M.; Piechnik, M.; Mason, R.W.; Mackenzie, W.G.; Yamaguchi, S.; Kobayashi, H.; Suzuki, Y.; Tomatsu, S. Effect of enzyme replacement therapy on the growth of patients with Morquio A. J. Hum. Genet. 2019, 64, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Duanis-Assaf, T.; Hu, T.; Lavie, M.; Zhang, Z.; Reches, M. Understanding the Adhesion Mechanism of Hydroxyapatite-Binding Peptide. Langmuir 2022, 38, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.J.; Chen, H.T.; Chang, C.H. Peptides for Targeting Chondrogenic Induction and Cartilage Regeneration in Osteoarthritis. Cartilage 2024, 19476035241276406. [Google Scholar] [CrossRef]
- Fu, Z.; Zhang, X.; Zhou, X.; Ur-Rehman, U.; Yu, M.; Liang, H.; Guo, H.; Guo, X.; Kong, Y.; Su, Y.; et al. In vivo self-assembled small RNAs as a new generation of RNAi therapeutics. Cell Res. 2021, 31, 631–648. [Google Scholar] [CrossRef]
- Heath, N.; Osteikoetxea, X.; de Oliveria, T.M.; Lázaro-Ibáñez, E.; Shatnyeva, O.; Schindler, C.; Tigue, N.; Mayr, L.M.; Dekker, N.; Overman, R.; et al. Endosomal escape enhancing compounds facilitate functional delivery of extracellular vesicle cargo. Nanomedicine 2019, 14, 2799–2814. [Google Scholar] [CrossRef]
- Ewaisha, R.; Anderson, K.S. Immunogenicity of CRISPR therapeutics-Critical considerations for clinical translation. Front. Bioeng. Biotechnol. 2023, 11, 1138596. [Google Scholar] [CrossRef]
- Lopes, R.; Prasad, M.K. Beyond the promise: Evaluating and mitigating off-target effects in CRISPR gene editing for safer therapeutics. Front. Bioeng. Biotechnol. 2023, 11, 1339189. [Google Scholar] [CrossRef]

| Trial ID | Status | Dis. | Source | Phase | DM | Dose Par/Dose | Outcomes Measured | Main Findings |
|---|---|---|---|---|---|---|---|---|
| NCT06713902 | Recruiting | OA/KJ | AD | Obs. | NA | NA | Encapsulation of AD-MSCs into PRP-derived fibrin gel | NP |
| NCT05060107 | Completed | OA/KJ | NA | I | IA | Single 3–5 × 1011 | Safety, pain, and disability reduction | MSC-EVs were safe and reduced pain while improving function |
| NCT06431152 | Recruiting | OA/KJ | UC | I | IA | Single L: 2 × 109 M: 6 × 109 H: 2 × 1010 | Safety, pain, and disability reduction | NP |
| NCT06466850 | Recruiting | OA/KJ | NA | I | IA | * Double | Safety, pain, and disability reduction | NP |
| NCT06463132 | Not yet recruiting | OA/KJ | PL | I | IA | ** Single | Safety, clinical improvements after 12 months | NP |
| NCT06713902 | Recruiting | OA/KJ | AD | Obs. | NA | NA | Encapsulation of AD-MSCs into PRP-derived fibrin gel | NP |
| NCT06688318 | Active, not recruiting | OA/KJ | UC | I/II | IA | ** Single | Safety, pain reduction. | NP |
| NCT04998058 | Not yet recruiting | EM | AD | I/II | MSL | ** Single | Bone density and quantity | NP |
| NCT05261360 | Unknown | DMI | SF | II | IA | *** 1 × 106 | Safety, pain reduction, cytokine profile | NP |
| Drawback | Description | Ref. |
|---|---|---|
| Cargo heterogeneity | Cargo is greatly influenced by MSCs’ source, passage, and culture conditions. | [103,151] |
| Limited cargo loading | Low loading efficiency is often observed, mainly when passive (incubation/shaking) methods are used. | [152] |
| Non-standardized isolation protocols | Ultracentrifugation, density gradient centrifugation, size-exclusion chromatography, ultrafiltration, precipitation, and immunocapture yield different purities. Characterization procedures upon isolation are inconsistent, thereby limiting biochemical composition identification. | [153,154] |
| Leak of biodistribution assays | Although some studies use fluorescence-based MSC-EVs, this is not the case for all studies. It is also unclear how MSC-EVs are cleared. | [155,156] |
| Potential immunogenicity | Engineered MSC-EVs can increase the risk of immune response activation, thus exacerbating disease pathology. | [57,157] |
| Scalability | Large-scale production is still limited. | [158] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leal, A.F.; Pachajoa, H.; Tomatsu, S. Mesenchymal Stem Cell-Derived Extracellular Vesicles: Seeking into Cell-Free Therapies for Bone-Affected Lysosomal Storage Disorders. Int. J. Mol. Sci. 2025, 26, 6448. https://doi.org/10.3390/ijms26136448
Leal AF, Pachajoa H, Tomatsu S. Mesenchymal Stem Cell-Derived Extracellular Vesicles: Seeking into Cell-Free Therapies for Bone-Affected Lysosomal Storage Disorders. International Journal of Molecular Sciences. 2025; 26(13):6448. https://doi.org/10.3390/ijms26136448
Chicago/Turabian StyleLeal, Andrés Felipe, Harry Pachajoa, and Shunji Tomatsu. 2025. "Mesenchymal Stem Cell-Derived Extracellular Vesicles: Seeking into Cell-Free Therapies for Bone-Affected Lysosomal Storage Disorders" International Journal of Molecular Sciences 26, no. 13: 6448. https://doi.org/10.3390/ijms26136448
APA StyleLeal, A. F., Pachajoa, H., & Tomatsu, S. (2025). Mesenchymal Stem Cell-Derived Extracellular Vesicles: Seeking into Cell-Free Therapies for Bone-Affected Lysosomal Storage Disorders. International Journal of Molecular Sciences, 26(13), 6448. https://doi.org/10.3390/ijms26136448