
Molecular Insights into Neurological Regression with a Focus on Rett Syndrome—A Narrative Review


Abstract
1. Background
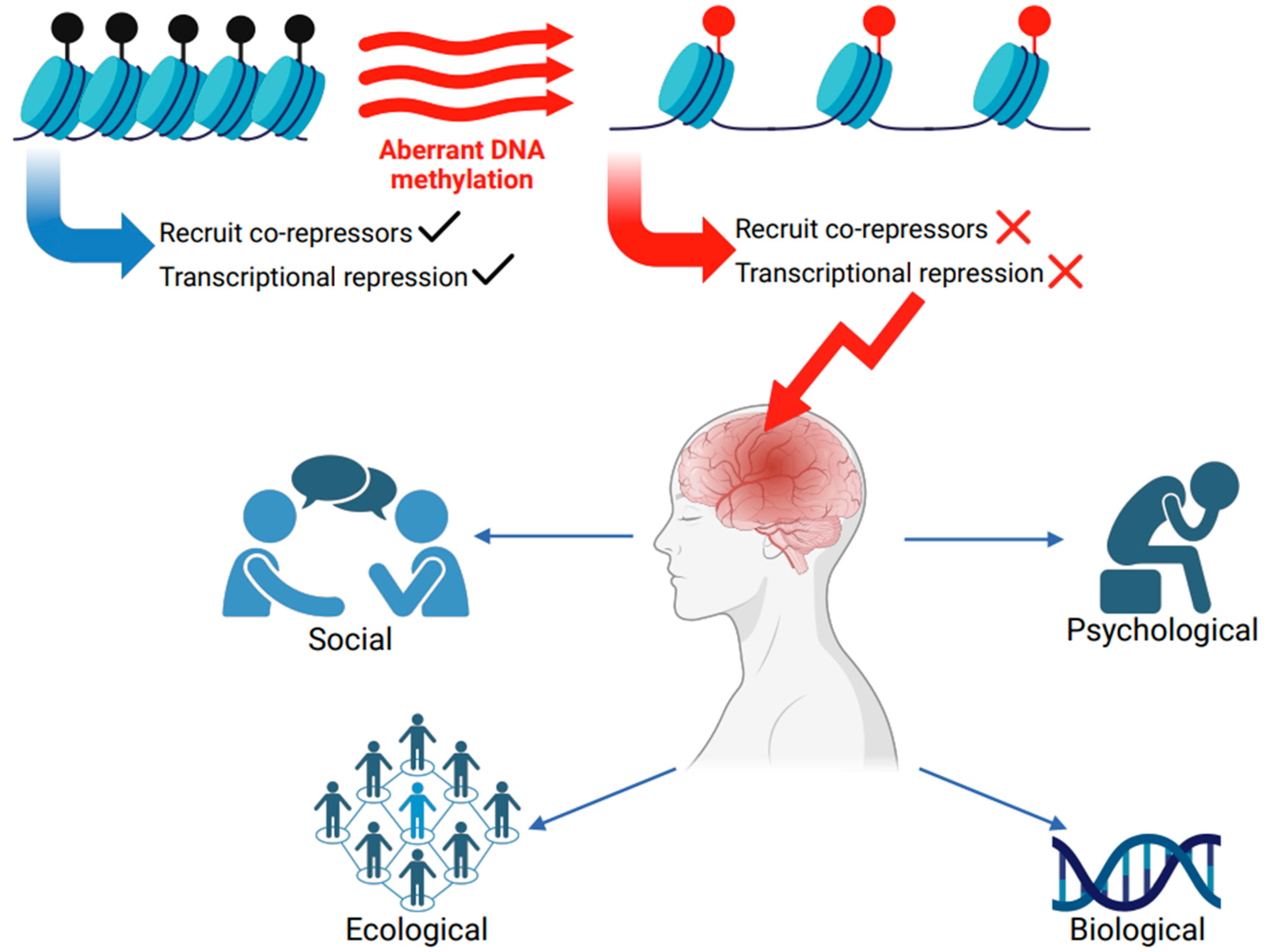
Impaired Methylation and Neurological Disorders
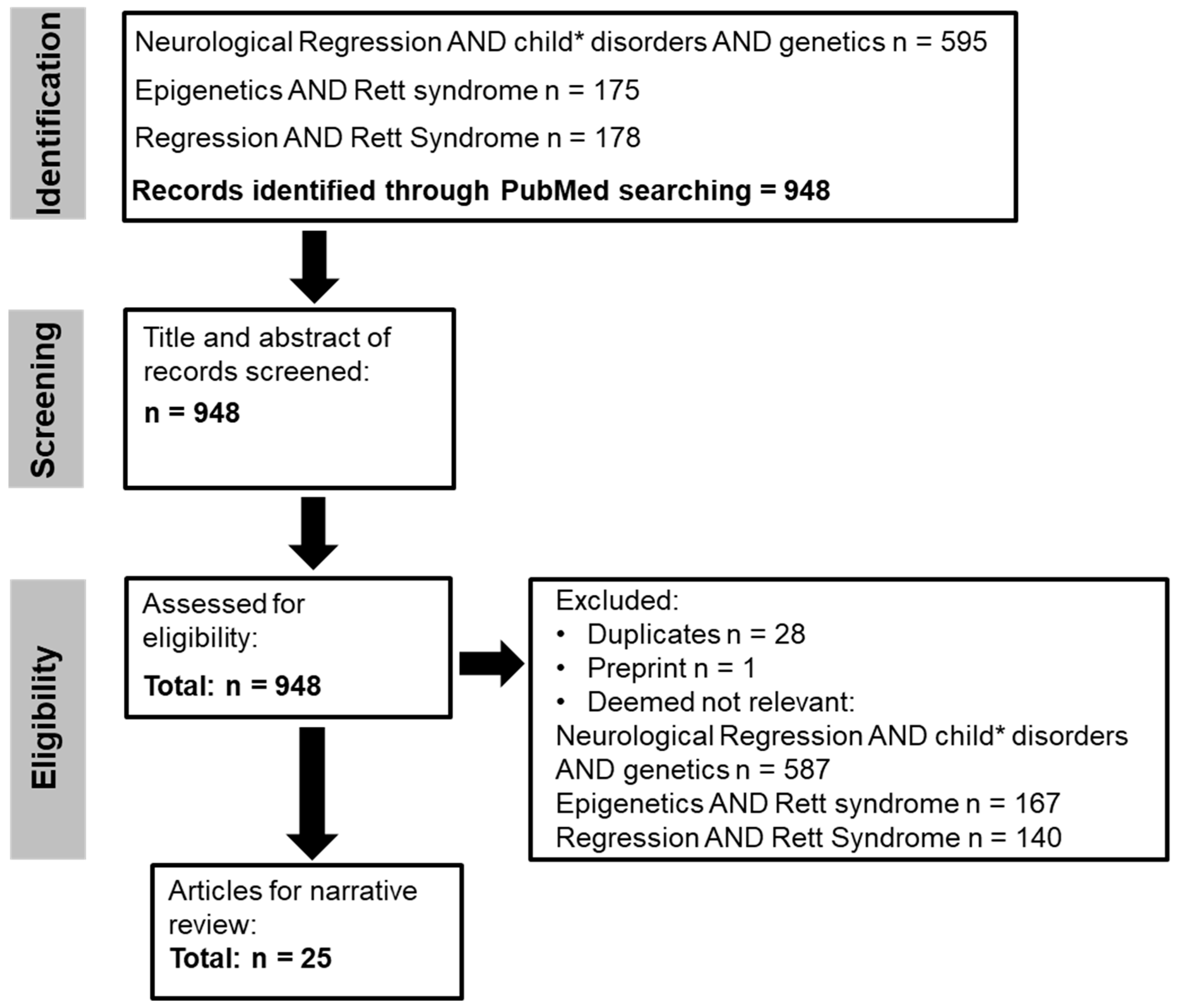
2. Methods
2.1. Search Strategy
2.2. Search Terms
2.3. Secondary Searching
2.4. Eligibility Criteria
2.4.1. Inclusion Criteria
2.4.2. Exclusion Criteria
3. Key Findings
Neurological Regression
4. What Causes Neurological Regression in RTT?
4.1. Impact of MECP2 Across the Genomic Landscape
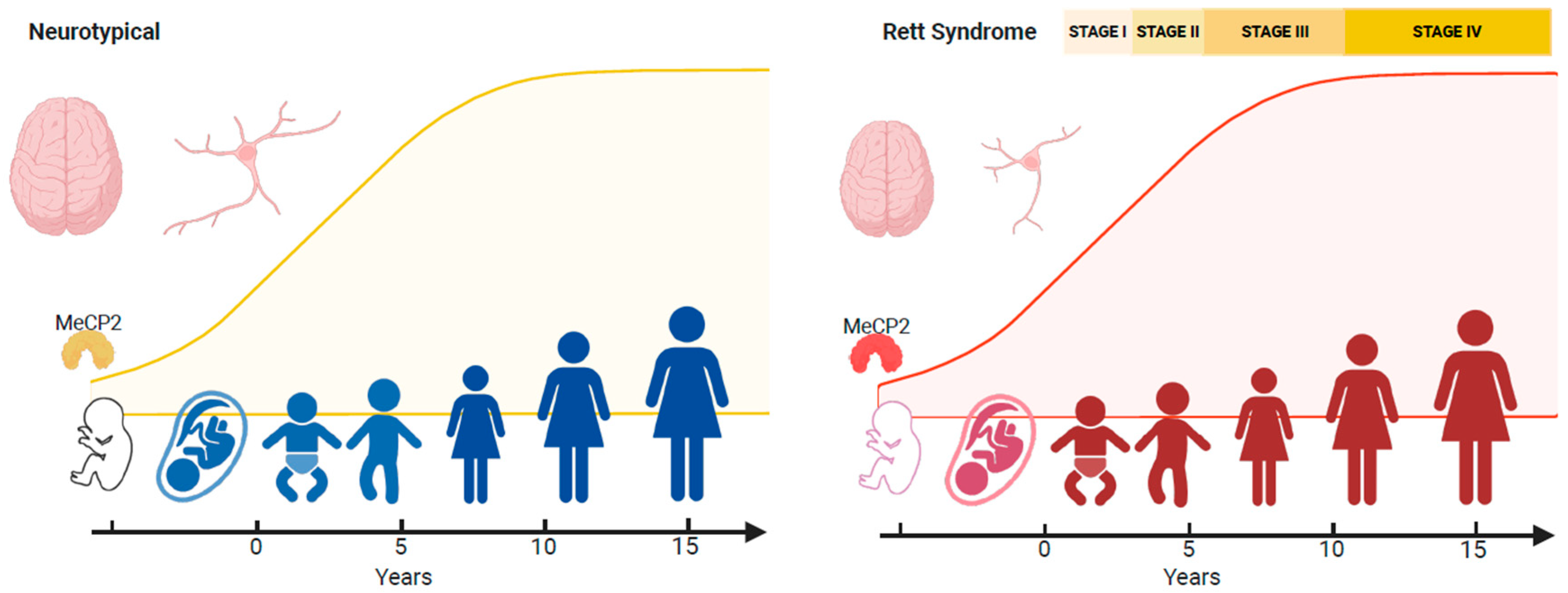
4.2. Neurological Regression
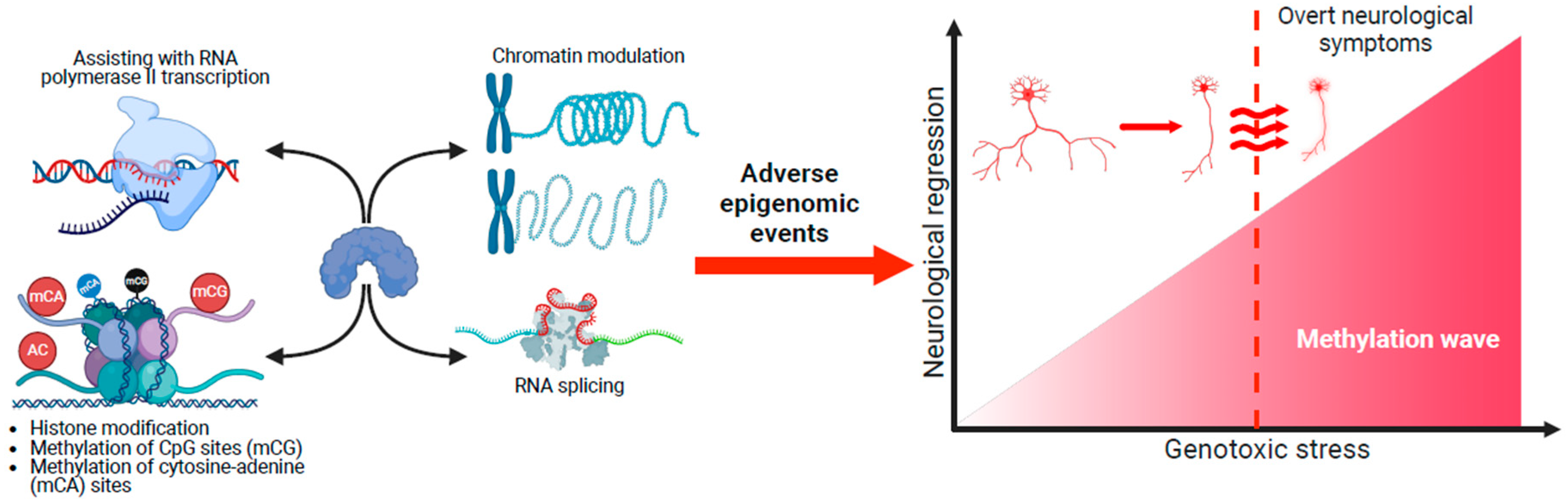
4.3. Genotoxic Stress and Neurological Regression
4.4. Therapeutic Approaches
5. Summary
Limitations
6. Concluding Remarks
- Expression patterns of MeCP2 coincide with brain maturation, and MECP2 pathogenic/likely pathogenic variants result in adverse epigenetic events that accumulate across the RTT neural epigenome. This accumulation of genotoxic stress depends on different spatial methylated gradients across the brain. Some brain regions could be more sensitive to the aberrant gradients. The cerebellum is particularly vulnerable, and motor deficits such as hand stereotypies are the first symptoms.
- These gradients operate across the disorder’s lifespan, but MeCP2’s genotoxic impact on the epigenome is too small postnatally for obvious clinical symptoms to appear. Nevertheless, deleterious changes in the neural epigenome have already started, and the concept of a seemingly typical neurodevelopmental period before 6 months in RTT should be reframed.
- Epilepsy and recurrent infections could lead to the onset of regression in RTT; however, the trajectory of regression in this population is variable. The epigenetic factors that modify the onset (very early or adolescent onset) of neurological regression in RTT remain to be identified.
- A phased treatment approach would be needed. Neuroprotectants could be used at the early stages of regression. During progression, treatments that target symptom reduction and maintenance of function can be used. Biomarker-specific treatment personalisation may be required during regression to reduce impact.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Younesian, S.; Yousefi, A.M.; Momeny, M.; Ghaffari, S.H.; Bashash, D. The DNA Methylation in Neurological Diseases. Cells 2022, 11, 3439. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Liu, X.; Deng, Y.; Qing, H. DNA methylation, a hand behind neurodegenerative diseases. Front. Aging Neurosci. 2013, 5, 85. [Google Scholar]
- Vuu, Y.M.; Roberts, C.T.; Rastegar, M. MeCP2 Is an Epigenetic Factor That Links DNA Methylation with Brain Metabolism. Int. J. Mol. Sci. 2023, 24, 4218. [Google Scholar] [CrossRef]
- Lewis, J.D.; Meehan, R.R.; Henzel, W.J.; Maurer-Fogy, I.; Jeppesen, P.; Klein, F.; Bird, A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992, 69, 905–914. [Google Scholar]
- Zhu, H.; Wang, G.; Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef]
- Roberts, C.T.; Arezoumand, K.S.; Kadar Shahib, A.; Davie, J.R.; Rastegar, M. Epigenetics in rare neurological diseases. Front. Cell Dev. Biol. 2024, 12, 1413248. [Google Scholar]
- Li, L.; Chen, R.; Zhang, H.; Li, J.; Huang, H.; Weng, J.; Tan, H.; Guo, T.; Wang, M.; Xie, J. The epigenetic modification of DNA methylation in neurological diseases. Front. Immunol. 2024, 15, 1401962. [Google Scholar]
- Xu, G.L.; Bestor, T.H.; Bourc’his, D.; Hsieh, C.L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Péquignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef]
- Koch, M.W.; Metz, L.M.; Kovalchuk, O. Epigenetics and miRNAs in the diagnosis and treatment of multiple sclerosis. Trends Mol. Med. 2013, 19, 23–30. [Google Scholar] [CrossRef]
- Ng, C.W.; Yildirim, F.; Yap, Y.S.; Dalin, S.; Matthews, B.J.; Velez, P.J.; Labadorf, A.; Housman, D.E.; Fraenkel, E. Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc. Natl. Acad. Sci. USA 2013, 110, 2354–2359. [Google Scholar] [CrossRef] [PubMed]
- De Souza, R.A.; Islam, S.A.; McEwen, L.M.; Mathelier, A.; Hill, A.; Mah, S.M.; Wasserman, W.W.; Kobor, M.S.; Leavitt, B.R. DNA methylation profiling in human Huntington’s disease brain. Hum. Mol. Genet. 2016, 25, 2013–2030. [Google Scholar] [CrossRef] [PubMed]
- Eggermann, T.; Monk, D.; de Nanclares, G.P.; Kagami, M.; Giabicani, E.; Riccio, A.; Tümer, Z.; Kalish, J.M.; Tauber, M.; Duis, J.; et al. Imprinting disorders. Nat. Rev. Dis. Primers 2023, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Angulo, M.A.; Butler, M.G.; Cataletto, M.E. Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J. Endocrinol. Investig. 2015, 38, 1249–1263. [Google Scholar] [CrossRef]
- Edwards, M.M.; Wang, N.; Sagi, I.; Kinreich, S.; Benvenisty, N.; Gerhardt, J.; Egli, D.; Koren, A. Parent-of-origin-specific DNA replication timing is confined to large imprinted regions. Cell Rep. 2024, 43, 114700. [Google Scholar] [CrossRef]
- Akbari, V.; Dada, S.; Shen, Y.; Dixon, K.; Hejla, D.; Galbraith, A.; Choufani, S.; Weksberg, R.; Boerkoel, C.F.; Stewart, L.; et al. Long-read sequencing for detection and subtyping of Prader-Willi and Angelman syndromes. J. Med. Genet. 2024, 62, 32–36. [Google Scholar] [CrossRef]
- Van Gils, J.; Magdinier, F.; Fergelot, P.; Lacombe, D. Rubinstein-Taybi Syndrome: A Model of Epigenetic Disorder. Genes 2021, 12, 968. [Google Scholar] [CrossRef] [PubMed]
- Malta, T.M.; de Souza, C.F.; Sabedot, T.S.; Silva, T.C.; Mosella, M.S.; Kalkanis, S.N.; Snyder, J.; Castro, A.V.B.; Noushmehr, H. Glioma CpG island methylator phenotype (G-CIMP): Biological and clinical implications. Neuro-Oncology 2018, 20, 608–620. [Google Scholar] [CrossRef]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef]
- Li, C.H.; Coffey, E.L.; Dall’Agnese, A.; Hannett, N.M.; Tang, X.; Henninger, J.E.; Platt, J.M.; Oksuz, O.; Zamudio, A.V.; Afeyan, L.K.; et al. MeCP2 links heterochromatin condensates and neurodevelopmental disease. Nature 2020, 586, 440–444. [Google Scholar] [CrossRef]
- Liu, Y.; Whitfield, T.W.; Bell, G.W.; Guo, R.; Flamier, A.; Young, R.A.; Jaenisch, R. Exploring the complexity of MECP2 function in Rett syndrome. Nat. Rev. Neurosci. 2025, 1–20. [Google Scholar] [CrossRef]
- Halbach, N.S.; Smeets, E.E.; van den Braak, N.; van Roozendaal, K.E.; Blok, R.M.; Schrander-Stumpel, C.T.; Frijns, J.; Maaskant, M.A.; Curfs, L.M. Genotype-phenotype relationships as prognosticators in Rett syndrome should be handled with care in clinical practice. Am. J. Med. Genet. Part A 2012, 158A, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Santosh, P. The Newborn Screening Programme Revisited: An Expert Opinion on the Challenges of Rett Syndrome. Genes 2024, 15, 1570. [Google Scholar] [CrossRef] [PubMed]
- Abbott, M.; Angione, K.; Forbes, E.; Stoecker, M.; Saenz, M. Rett syndrome diagnostic odyssey: Limitations of NextGen sequencing. Am. J. Med. Genet. Part A 2024, 194, e63725. [Google Scholar] [CrossRef]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.S.; Schanen, N.C.; Zappella, M.; et al. RettSearch Consortium. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef]
- Hagberg, B. Clinical manifestations and stages of Rett syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 2002, 8, 61–65. [Google Scholar] [CrossRef]
- Singh, J.; Santosh, P. Key issues in Rett syndrome: Emotional, behavioural and autonomic dysregulation (EBAD)—A target for clinical trials. Orphanet J. Rare Dis. 2018, 13, 128. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Singh, J.; Lanzarini, E.; Nardocci, N.; Santosh, P. Movement disorders in patients with Rett syndrome: A systematic review of evidence and associated clinical considerations. Psychiatry Clin. Neurosci. 2021, 75, 369–393. [Google Scholar] [CrossRef]
- Gulmez Karaca, K.; Brito, D.V.C.; Oliveira, A.M.M. MeCP2: A Critical Regulator of Chromatin in Neurodevelopment and Adult Brain Function. Int. J. Mol. Sci. 2019, 20, 4577. [Google Scholar] [CrossRef]
- Gold, W.A.; Percy, A.K.; Neul, J.L.; Cobb, S.R.; Pozzo-Miller, L.; Issar, J.K.; Ben-Zeev, B.; Vignoli, A.; Kaufmann, W.E. Rett syndrome. Nat. Rev. Dis. Primers 2024, 10, 84. [Google Scholar] [CrossRef]
- Marano, D.; Fioriniello, S.; D’Esposito, M.; Della Ragione, F. Transcriptomic and Epigenomic Landscape in Rett Syndrome. Biomolecules 2021, 11, 967. [Google Scholar] [CrossRef] [PubMed]
- Vidal, S.; Xiol, C.; Pascual-Alonso, A.; O’Callaghan, M.; Pineda, M.; Armstrong, J. Genetic Landscape of Rett Syndrome Spectrum: Improvements and Challenges. Int. J. Mol. Sci. 2019, 20, 3925. [Google Scholar] [CrossRef]
- Zade, K.; Campbell, C.; Bach, S.; Fernandes, H.; Tropea, D. Rett syndrome in Ireland: A demographic study. Orphanet J. Rare Dis. 2024, 19, 34. [Google Scholar] [CrossRef]
- Einspieler, C.; Marschik, P.B. Regression in Rett syndrome: Developmental pathways to its onset. Neurosci. Biobehav. Rev. 2019, 98, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, L.; Vigli, D.; Franchi, F.; Laviola, G.; De Filippis, B. Rett syndrome before regression: A time window of overlooked opportunities for diagnosis and intervention. Neurosci. Biobehav. Rev. 2019, 107, 115–135. [Google Scholar] [CrossRef]
- Neul, J.L. Can Rett syndrome be diagnosed before regression? Neurosci. Biobehav. Rev. 2019, 104, 158–159. [Google Scholar] [CrossRef]
- Smeets, E.E.; Townend, G.S.; Curfs, L.M.G. Rett syndrome and developmental regression. Neurosci. Biobehav. Rev. 2019, 104, 100–101. [Google Scholar] [CrossRef]
- Müller, M. Disturbed redox homeostasis and oxidative stress: Potential players in the developmental regression in Rett syndrome. Neurosci. Biobehav. Rev. 2019, 98, 154–163. [Google Scholar] [CrossRef]
- Sigafoos, J.; O’Reilly, M.F.; Ledbetter-Cho, K.; Lim, N.; Lancioni, G.E.; Marschik, P.B. Addressing sequelae of developmental regression associated with developmental disabilities: A systematic review of behavioral and educational intervention studies. Neurosci. Biobehav. Rev. 2019, 96, 56–71. [Google Scholar] [CrossRef]
- Cutri-French, C.; Armstrong, D.; Saby, J.; Gorman, C.; Lane, J.; Fu, C.; Peters, S.U.; Percy, A.; Neul, J.L.; Marsh, E.D.; et al. Comparison of Core Features in Four Developmental Encephalopathies in the Rett Natural History Study. Ann. Neurol. 2020, 88, 396–406. [Google Scholar] [CrossRef]
- Pehlivan, D.; Huang, C.; Harris, H.K.; Coquery, C.; Mahat, A.; Maletic-Savatic, M.; Mignon, L.; Aras, S.; Glaze, D.G.; Layne, C.S.; et al. Comprehensive assessment reveals numerous clinical and neurophysiological differences between MECP2-allelic disorders. Ann. Clin. Transl. Neurol. 2025, 12, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Tamaoki, Y.; Kroon, S.L.; Williams, B.M.; Riley, J.R.; Engineer, C.T. Early neural dysfunction reflected in degraded auditory cortex responses in pre-regression heterozygous Mecp2 rats. Neurobiol. Dis. 2025, 210, 106926. [Google Scholar] [CrossRef]
- Sharifi, O.; Haghani, V.; Neier, K.E.; Fraga, K.J.; Korf, I.; Hakam, S.M.; Quon, G.; Johansen, N.; Yasui, D.H.; LaSalle, J.M. Sex-specific single cell-level transcriptomic signatures of Rett syndrome disease progression. Commun. Biol. 2024, 7, 1292. [Google Scholar] [CrossRef]
- Chow, K.; Rezvan, P.H.; Kazerooni, L.; Nguyen, L.; Boyd, N.K.; Vogel, B.N.; Lucas, M.C.; Brown, R.; Quinn, E.A.; Jafarpour, S.; et al. Caregiver burden and familial impact in Down Syndrome Regression Disorder. Orphanet J. Rare Dis. 2025, 20, 126. [Google Scholar] [CrossRef]
- Adang, L.A.; Groeschel, S.; Grzyb, C.; D’Aiello, R.; Gavazzi, F.; Sherbini, O.; Bronner, N.; Patel, A.; Vincent, A.; Sevagamoorthy, A.; et al. Developmental delay can precede neurologic regression in early onset metachromatic leukodystrophy. Mol. Genet. Metab. 2024, 142, 108521. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.; Seo, G.H.; Keum, C.; Heo, S.H.; Kim, T.; Choi, J.; Yum, M.-S.; Lee, B.H. Diagnosis of metachromatic leukodystrophy in a patient with regression and Phelan-McDermid syndrome. Brain Dev. 2020, 42, 414–417. [Google Scholar] [CrossRef]
- Levy, T.; Foss-Feig, J.H.; Betancur, C.; Siper, P.M.; Trelles-Thorne, M.d.P.; Halpern, D.; Frank, Y.; Lozano, R.; Layton, C.; Britvan, B.; et al. Developmental Synaptopathies Consortium. Strong evidence for genotype-phenotype correlations in Phelan-McDermid syndrome: Results from the developmental synaptopathies consortium. Hum. Mol. Genet. 2022, 31, 625–637. [Google Scholar] [CrossRef]
- Levy, T.; Gluckman, J.; Siper, P.M.; Halpern, D.; Zweifach, J.; Filip-Dhima, R.; Holder, J.L.; Trelles, M.P.; Johnson, K.; Bernstein, J.A.; et al. Developmental Synaptopathies Consortium. Clinical, genetic, and cognitive correlates of seizure occurrences in Phelan-McDermid syndrome. J. Neurodev. Disord. 2024, 16, 25. [Google Scholar] [CrossRef]
- Zhang, D.; Bedogni, F.; Boterberg, S.; Camfield, C.; Camfield, P.; Charman, T.; Curfs, L.; Einspieler, C.; Esposito, G.; De Filippis, B.; et al. Towards a consensus on developmental regression. Neurosci. Biobehav. Rev. 2019, 107, 3–5. [Google Scholar] [CrossRef]
- Furley, K.; Mehra, C.; Goin-Kochel, R.P.; Fahey, M.C.; Hunter, M.F.; Williams, K.; Absoud, M. Developmental regression in children: Current and future directions. Cortex 2023, 169, 5–17. [Google Scholar] [CrossRef]
- Santoro, J.D.; Patel, L.; Kammeyer, R.; Filipink, R.A.; Gombolay, G.Y.; Cardinale, K.M.; de Asua, D.R.; Zaman, S.; Santoro, S.L.; Marzouk, S.M.; et al. Assessment and Diagnosis of Down Syndrome Regression Disorder: International Expert Consensus. Front. Neurol. 2022, 13, 940175. [Google Scholar] [CrossRef]
- Fernandez, A.; Drozd, M.M.; Thümmler, S.; Dor, E.; Capovilla, M.; Askenazy, F.; Bardoni, B. Childhood-Onset Schizophrenia: A Systematic Overview of Its Genetic Heterogeneity From Classical Studies to the Genomic Era. Front. Genet. 2019, 10, 1137. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.E.; Atmaram, S.K.; Khadanga, P.; Mukherjee, N.; Madegowda, R.K.; Manohar, H. The diagnostic conundrum of late-onset developmental regression in child psychiatry: Case series. BJPsych Open 2025, 11, e25. [Google Scholar] [CrossRef]
- Mehra, C.; Sil, A.; Hedderly, T.; Kyriakopoulos, M.; Lim, M.; Turnbull, J.; Happe, F.; Baird, G.; Absoud, M. Childhood disintegrative disorder and autism spectrum disorder: A systematic review. Dev. Med. Child Neurol. 2019, 61, 523–534. [Google Scholar] [CrossRef]
- Reierson, G.; Bernstein, J.; Froehlich-Santino, W.; Urban, A.; Purmann, C.; Berquist, S.; Jordan, J.; O’Hara, R.; Hallmayer, J. Characterizing regression in Phelan McDermid Syndrome (22q13 deletion syndrome). J. Psychiatr. Res. 2017, 91, 139–144. [Google Scholar] [CrossRef]
- Dille, Y.; Lagae, L.; Swillen, A.; Buggenhout, G.V. Neurodevelopmental profile and stages of regression in Phelan-McDermid syndrome. Dev. Med. Child Neurol. 2023, 65, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.J.; Larsen, K.; Havighurst, S.S. Childhood Disintegrative Disorder (CDD): Symptomatology of the Norwegian Patient Population and Parents’ Experiences of Patient Regression. J. Autism Dev. Disord. 2022, 52, 1495–1506. [Google Scholar] [CrossRef]
- Barger, B.D.; Campbell, J.M.; McDonough, J.D. Prevalence and onset of regression within autism spectrum disorders: A meta-analytic review. J. Autism Dev. Disord. 2013, 43, 817–828. [Google Scholar] [CrossRef]
- Tan, C.; Frewer, V.; Cox, G.; Williams, K.; Ure, A. Prevalence and Age of Onset of Regression in Children with Autism Spectrum Disorder: A Systematic Review and Meta-analytical Update. Autism Res. 2021, 14, 582–598. [Google Scholar] [CrossRef]
- Malhotra, S.; Gupta, N. Childhood disintegrative disorder. Eur. Child Adolesc. Psychiatry 2002, 11, 108. [Google Scholar] [CrossRef]
- Rosman, N.P.; Bergia, B.M. Childhood disintegrative disorder: Distinction from autistic disorder and predictors of outcome. J. Child Neurol. 2013, 28, 1587–1598. [Google Scholar] [CrossRef] [PubMed]
- Volkmar, F.R.; Cohen, D.J. Disintegrative disorder or ‘late onset’ autism. J. Child Psychol. Psychiatry 1989, 30, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Bonne, S.; Iftimovici, A.; Mircher, C.; Conte, M.; Louveau, C.; Legrand, A.; Danset-Alexandre, C.; Cannarsa, C.; Debril, A.; Consoli, A.; et al. Down syndrome regression disorder, a case series: Clinical characterization and therapeutic approaches. Front. Neurosci. 2023, 17, 1126973. [Google Scholar] [CrossRef] [PubMed]
- Rosso, M.; Fremion, E.; Santoro, S.L.; Oreskovic, N.M.; Chitnis, T.; Skotko, B.G.; Santoro, J.D. Down Syndrome Disintegrative Disorder: A Clinical Regression Syndrome of Increasing Importance. Pediatrics 2020, 145, e20192939. [Google Scholar] [CrossRef]
- Kadam, S.D.; Sullivan, B.J.; Goyal, A.; Blue, M.E.; Smith-Hicks, C. Rett Syndrome and CDKL5 Deficiency Disorder: From Bench to Clinic. Int. J. Mol. Sci. 2019, 20, 5098. [Google Scholar] [CrossRef]
- Ta, D.; Downs, J.; Baynam, G.; Wilson, A.; Richmond, P.; Leonard, H. A brief history of MECP2 duplication syndrome: 20-years of clinical understanding. Orphanet J. Rare Dis. 2022, 17, 131. [Google Scholar] [CrossRef]
- Miguet, M.; Faivre, L.; Amiel, J.; Nizon, M.; Touraine, R.; Prieur, F.; Pasquier, L.; Lefebvre, M.; Thevenon, J.; Dubourg, C.; et al. Further delineation of the MECP2 duplication syndrome phenotype in 59 French male patients, with a particular focus on morphological and neurological features. J. Med. Genet. 2018, 55, 359–371. [Google Scholar] [CrossRef]
- Marafi, D.; Suter, B.; Schultz, R.; Glaze, D.; Pavlik, V.N.; Goldman, A.M. Spectrum and time course of epilepsy and the associated cognitive decline in MECP2 duplication syndrome. Neurology 2019, 92, e108–e114. [Google Scholar] [CrossRef]
- Petriti, U.; Dudman, D.C.; Scosyrev, E.; Lopez-Leon, S. Global prevalence of Rett syndrome: Systematic review and meta-analysis. Syst. Rev. 2023, 12, 5. [Google Scholar] [CrossRef]
- Neul, J.L.; Benke, T.A.; Marsh, E.D.; Skinner, S.A.; Merritt, J.; Lieberman, D.N.; Standridge, S.; Feyma, T.; Heydemann, P.; Peters, S.; et al. The array of clinical phenotypes of males with mutations in Methyl-CpG binding protein 2. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2019, 180, 55–67. [Google Scholar] [CrossRef]
- Young, J.I.; Hong, E.P.; Castle, J.C.; Crespo-Barreto, J.; Bowman, A.B. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. USA 2005, 102, 17551–17558. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Flamier, A.; Bell, G.W.; Diao, A.J.; Whitfield, T.W.; Wang, H.-C.; Wu, Y.; Schulte, F.; Friesen, M.; Guo, R.; et al. MECP2 directly interacts with RNA polymerase II to modulate transcription in human neurons. Neuron 2024, 112, 1943–1958.e10. [Google Scholar] [CrossRef] [PubMed]
- Mottis, A.; Mouchiroud, L.; Auwerx, J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev. 2013, 27, 819–835. [Google Scholar] [CrossRef]
- Lyst, M.J.; Ekiert, R.; Ebert, D.H.; Merusi, C.; Nowak, J.; Selfridge, J.; Guy, J.; Kastan, N.R.; Robinson, N.D.; de Lima Alves, F.; et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 2013, 16, 898–902. [Google Scholar] [CrossRef]
- Zylka, M.J.; Simon, J.M.; Philpot, B.D. Gene length matters in neurons. Neuron 2015, 86, 353–355. [Google Scholar] [CrossRef]
- King, I.F.; Yandava, C.N.; Mabb, A.M.; Hsiao, J.S.; Huang, H.S.; Pearson, B.L.; Calabrese, J.M.; Starmer, J.; Parker, J.S.; Magnuson, T.; et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature 2013, 501, 58–62. [Google Scholar] [CrossRef]
- Gabel, H.W.; Kinde, B.; Stroud, H.; Gilbert, C.S.; Harmin, D.A.; Kastan, N.R.; Hemberg, M.; Ebert, D.H.; Greenberg, M.E. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 2015, 522, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Sugino, K.; Hempel, C.M.; Okaty, B.W.; Arnson, H.A.; Kato, S.; Dani, V.S.; Nelson, S.B. Cell-type-specific repression by methyl-CpG-binding protein 2 is biased toward long genes. J. Neurosci. 2014, 34, 12877–12883. [Google Scholar] [CrossRef]
- Skene, P.J.; Illingworth, R.S.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Andrews, R.; Bird, A.P. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 2010, 37, 457–468. [Google Scholar] [CrossRef]
- Suter, B.; Treadwell-Deering, D.; Zoghbi, H.Y.; Glaze, D.G.; Neul, J.L. Brief report: MECP2 mutations in people without Rett syndrome. J. Autism Dev. Disord. 2014, 44, 703–711. [Google Scholar] [CrossRef]
- Canton, A.P.M.; Tinano, F.R.; Guasti, L.; Montenegro, L.R.; Ryan, F.; Shears, D.; de Melo, M.E.; Gomes, L.G.; Piana, M.P.; Brauner, R.; et al. Rare variants in the MECP2 gene in girls with central precocious puberty: A translational cohort study. Lancet Diabetes Endocrinol. 2023, 11, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Fang, P.; Barrish, J.; Lane, J.; Caeg, E.B.; Smith, E.O.; Zoghbi, H.; Percy, A.; Glaze, D.G. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 2008, 70, 1313–1321. [Google Scholar] [CrossRef]
- Fu, C.; Armstrong, D.; Marsh, E.; Lieberman, D.; Motil, K.; Witt, R.; Standridge, S.; Nues, P.; Lane, J.; Dinkel, T.; et al. Consensus guidelines on managing Rett syndrome across the lifespan. BMJ Paediatr. Open 2020, 4, e000717. [Google Scholar] [CrossRef]
- Neul, J.L.; Lane, J.B.; Lee, H.S.; Geerts, S.; Barrish, J.O.; Annese, F.; Baggett, L.M.; Barnes, K.; Skinner, S.A.; Motil, K.J.; et al. Developmental delay in Rett syndrome: Data from the natural history study. J. Neurodev. Disord. 2014, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Lyst, M.J.; Bird, A. Rett syndrome: A complex disorder with simple roots. Nat. Rev. Genet. 2015, 16, 261–275. [Google Scholar] [CrossRef]
- Chao, H.T.; Chen, H.; Samaco, R.C.; Xue, M.; Chahrour, M.; Yoo, J.; Gong, S.; Lu, H.-C.; Heintz, N.; Ekker, M.; et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 2010, 468, 263–269. [Google Scholar] [CrossRef]
- Haase, F.; Singh, R.; Gloss, B.; Tam, P.; Gold, W. Meta-Analysis Identifies BDNF and Novel Common Genes Differently Altered in Cross-Species Models of Rett Syndrome. Int. J. Mol. Sci. 2022, 23, 11125. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Pozzo-Miller, L. BDNF deregulation in Rett syndrome. Neuropharmacology 2014, 76, 737–746. [Google Scholar] [CrossRef]
- Itoh, M.; Ide, S.; Takashima, S.; Kudo, S.; Nomura, Y.; Segawa, M.; Kubota, T.; Mori, H.; Tanaka, S.; Horie, H.; et al. Methyl CpG-binding protein 2 (a mutation of which causes Rett syndrome) directly regulates insulin-like growth factor binding protein 3 in mouse and human brains. J. Neuropathol. Exp. Neurol. 2007, 66, 117–123. [Google Scholar] [CrossRef]
- Tillotson, R.; Bird, A. The Molecular Basis of MeCP2 Function in the Brain. J. Mol. Biol. 2020, 432, 1602–1623. [Google Scholar] [CrossRef]
- Agarwal, N.; Becker, A.; Jost, K.L.; Haase, S.; Thakur, B.K.; Brero, A.; Hardt, T.; Kudo, S.; Leonhardt, H.; Cardoso, M.C. MeCP2 Rett mutations affect large scale chromatin organization. Hum. Mol. Genet. 2011, 20, 4187–4195. [Google Scholar] [CrossRef] [PubMed]
- Singleton, M.K.; Gonzales, M.L.; Leung, K.N.; Yasui, D.H.; Schroeder, D.I.; Dunaway, K.; LaSalle, J.M. MeCP2 is required for global heterochromatic and nucleolar changes during activity-dependent neuronal maturation. Neurobiol. Dis. 2011, 43, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.; Banerjee, A.; Sur, M. Developmental Dynamics of Rett Syndrome. Neural Plast. 2016, 2016, 6154080. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, B.; Hanefeld, F.; Percy, A.; Skjeldal, O. An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur. J. Paediatr. Neurol. 2002, 6, 293–297. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Sun, Y.; Zoghbi, H.Y. Balanced X chromosome inactivation patterns in the Rett syndrome brain. Am. J. Med. Genet. 2002, 111, 164–168. [Google Scholar] [CrossRef]
- Fang, X.; Butler, K.M.; Abidi, F.; Gass, J.; Beisang, A.; Feyma, T.; Ryther, R.C.; Standridge, S.; Heydemann, P.; Jones, M.; et al. Analysis of X-inactivation status in a Rett syndrome natural history study cohort. Mol. Genet. Genom. Med. 2022, 10, e1917. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Antalffy, B.; Armstrong, D.L.; Zoghbi, H.Y. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 2002, 11, 115–124. [Google Scholar] [CrossRef]
- Ladd-Acosta, C.; Pevsner, J.; Sabunciyan, S.; Yolken, R.H.; Webster, M.J.; Dinkins, T.; Callinan, P.A.; Fan, J.-B.; Potash, J.B.; Feinberg, A.P. DNA methylation signatures within the human brain. Am. J. Hum. Genet. 2007, 81, 1304–1315. [Google Scholar] [CrossRef]
- Liu, H.; Zhou, J.; Tian, W.; Luo, C.; Bartlett, A.; Aldridge, A.; Lucero, J.; Osteen, J.K.; Nery, J.R.; Chen, H.; et al. DNA methylation atlas of the mouse brain at single-cell resolution. Nature 2021, 598, 120–128. [Google Scholar] [CrossRef]
- Alessio, N.; Riccitiello, F.; Squillaro, T.; Capasso, S.; Del Gaudio, S.; Di Bernardo, G.; Cipollaro, M.; Melone, M.A.B.; Peluso, G.; Galderisi, U. Neural stem cells from a mouse model of Rett syndrome are prone to senescence, show reduced capacity to cope with genotoxic stress, and are impaired in the differentiation process. Exp. Mol. Med. 2018, 50, 1. [Google Scholar] [CrossRef]
- Achilly, N.P.; He, L.J.; Kim, O.A.; Ohmae, S.; Wojaczynski, G.J.; Lin, T.; Sillitoe, R.V.; Medina, J.F.; Zoghbi, H.Y. Deleting Mecp2 from the cerebellum rather than its neuronal subtypes causes a delay in motor learning in mice. eLife 2021, 10, e64833. [Google Scholar] [CrossRef] [PubMed]
- Bajikar, S.S.; Zhou, J.; O’Hara, R.; Tirumala, H.P.; Durham, M.A.; Trostle, A.J.; Dias, M.; Shao, Y.; Chen, H.; Wang, W.; et al. Acute MeCP2 loss in adult mice reveals transcriptional and chromatin changes that precede neurological dysfunction and inform pathogenesis. Neuron 2024, 113, 380–395.e8. [Google Scholar] [CrossRef]
- Bedogni, F.; Cobolli Gigli, C.; Pozzi, D.; Rossi, R.L.; Scaramuzza, L.; Rossetti, G.; Pagani, M.; Kilstrup-Nielsen, C.; Matteoli, M.; Landsberger, N. Defects During Mecp2 Null Embryonic Cortex Development Precede the Onset of Overt Neurological Symptoms. Cereb. Cortex 2016, 26, 2517–2529. [Google Scholar] [CrossRef]
- Alonso, M.G.D.P.; Marzal, C.C.; González MDSGomez, E.G. Adolescent with neurocognitive regression as onset of atypical Rett syndrome. Neurol. Perspect. 2024, 4, 100150. [Google Scholar] [CrossRef]
- Venkateswaran, S.; McMillan, H.J.; Doja, A.; Humphreys, P. Adolescent onset cognitive regression and neuropsychiatric symptoms associated with the A140V MECP2 mutation. Dev. Med. Child Neurol. 2014, 56, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Rolando, S. Rett syndrome: Report of eight cases. Brain Dev. 1985, 7, 290–296. [Google Scholar] [CrossRef]
- Sahafnejad, Z.; Ramazi, S.; Allahverdi, A. An Update of Epigenetic Drugs for the Treatment of Cancers and Brain Diseases: A Comprehensive Review. Genes 2023, 14, 873. [Google Scholar] [CrossRef]
- Brendel, C.; Klahold, E.; Gärtner, J.; Huppke, P. Suppression of nonsense mutations in Rett syndrome by aminoglycoside antibiotics. Pediatr. Res. 2009, 65, 520–523. [Google Scholar] [CrossRef]
- Wong, K.M.; Wegener, E.; Baradaran-Heravi, A.; Huppke, B.; Gärtner, J.; Huppke, P. Evaluation of Novel Enhancer Compounds in Gentamicin-Mediated Readthrough of Nonsense Mutations in Rett Syndrome. Int. J. Mol. Sci. 2023, 24, 11665. [Google Scholar] [CrossRef]
- Percy, A.K.; Ananth, A.; Neul, J.L. Rett Syndrome: The Emerging Landscape of Treatment Strategies. CNS Drugs 2024, 38, 851–867. [Google Scholar] [CrossRef]
- Neul, J.L.; Percy, A.K.; Benke, T.A.; Berry-Kravis, E.M.; Glaze, D.G.; Marsh, E.D.; Lin, T.; Stankovic, S.; Bishop, K.M.; Youakim, J.M. Trofinetide for the treatment of Rett syndrome: A randomized phase 3 study. Nat. Med. 2023, 29, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Parent, H.; Ferranti, A.; Niswender, C. Trofinetide: A pioneering treatment for Rett syndrome. Trends Pharmacol. Sci. 2023, 44, 740–741. [Google Scholar] [CrossRef] [PubMed]
- Santoro, J.D.; Partridge, R.; Tanna, R.; Pagarkar, D.; Khoshnood, M.; Rehmani, M.; Kammeyer, R.M.; Gombolay, G.Y.; Fisher, K.; Conravey, A.; et al. Evidence of neuroinflammation and immunotherapy responsiveness in individuals with down syndrome regression disorder. J. Neurodev. Disord. 2022, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.L.; Paraíso-Luna, J.; Bustos-Martínez, I.; Barco, Á. Targeting epigenetic dysregulation in autism spectrum disorders. Trends Mol. Med. 2024, 30, 1028–1046. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Comments | Reference |
|---|---|---|
| ||
Autism Spectrum Disorder (ASD)
|
| [54,58,59] |
Childhood Disintegrative Disorder (CDD)
|
| [50,54,57] |
Down Syndrome Regression Disorder (DSRD)
|
| [51,63,64] |
CDKL5-deficiency disorder
|
| [40,65] |
FOXG1 disorder
|
| [40] |
Landa–Kleffner syndrome
|
| [50] |
MECP2 Duplication syndrome
|
| [41,67,68] |
Phelan–McDermid syndrome
|
| [48,50,55,56] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, J.; Santosh, P. Molecular Insights into Neurological Regression with a Focus on Rett Syndrome—A Narrative Review. Int. J. Mol. Sci. 2025, 26, 5361. https://doi.org/10.3390/ijms26115361
Singh J, Santosh P. Molecular Insights into Neurological Regression with a Focus on Rett Syndrome—A Narrative Review. International Journal of Molecular Sciences. 2025; 26(11):5361. https://doi.org/10.3390/ijms26115361
Chicago/Turabian StyleSingh, Jatinder, and Paramala Santosh. 2025. "Molecular Insights into Neurological Regression with a Focus on Rett Syndrome—A Narrative Review" International Journal of Molecular Sciences 26, no. 11: 5361. https://doi.org/10.3390/ijms26115361
APA StyleSingh, J., & Santosh, P. (2025). Molecular Insights into Neurological Regression with a Focus on Rett Syndrome—A Narrative Review. International Journal of Molecular Sciences, 26(11), 5361. https://doi.org/10.3390/ijms26115361