
The Ovary–Liver Axis: Molecular Science and Epidemiology



Abstract
1. Introduction
2. Molecular Mechanisms of Action of Ovarian Hormones and Role of the Ovary in Maintaining Metabolic and Liver Health
3. The Liver and PCOS
4. The Liver in Hypogonadal Women
5. The Liver in Menopause and Ovarian Senescence
6. Molecular Pathogenesis of Liver Disease Associated with Ovarian Senescence, Menopause, PCOS, and Female Hypogonadism
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APRI | AST to Platelet Ratio Index |
| AS | Alström syndrome |
| BMI | Body mass index |
| E2 | Estradiol |
| ERα | Estrogen receptor α |
| ERβ | Estrogen receptor β |
| ER(s) | Estrogen receptor(s) |
| ERE(s) | Estrogen response element(s) |
| FIB-4 | Fibrosis 4 |
| FAI | Free androgen index |
| FSH | Follicle-stimulating hormone |
| GGT | Gamma-glutamyl transferase |
| GnRH | Gonadotropin-releasing hormone |
| GPER | G protein-coupled estrogen receptor |
| HA | Hyperandrogenemia |
| HH | Hereditary hemochromastosis |
| HOMA-IR | Homeostasis model of insulin resistance |
| IR | Insulin resistance |
| LH | Luteinizing hormone |
| MAPK | Mitogen-activated protein kinase |
| MAFLD | Metabolic dysfunction-associated fatty liver disease |
| MASH | Metabolic dysfunction-associated steatohepatitis |
| NAFLD | Nonalcoholic fatty liver disease |
| NASH | Nonalcoholic steatohepatitis |
| MASLD | Metabolic dysfunction-associated steatotic liver disease |
| MetS | Meabolic syndrome |
| mTOR | Mammalian target of rapamycin |
| PI3K | Phosphoinositide 3-kinase |
| PCOS | Polycystic ovary syndrome |
| ROS | Reactive oxygen species |
| SHBG | Sex hormone-binding globulin |
| T2DM | Type 2 diabetes mellitus |
| TS | Turner syndrome |
| WC | Waist circumference |
References
- Gustafsson, J.A.; Mode, A.; Norstedt, G.; Skett, P. Sex steroid induced changes in hepatic enzymes. Annu. Rev. Physiol. 1983, 45, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Meda, C.; Dolce, A.; Della Torre, S. Metabolic dysfunction-associated steatotic liver disease across women’s reproductive lifespan and issues. Clin. Mol. Hepatol. 2025, 31, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Won, Y.B.; Seo, S.K.; Yun, B.H.; Cho, S.; Choi, Y.S.; Lee, B.S. Non-alcoholic fatty liver disease in polycystic ovary syndrome women. Sci. Rep. 2021, 11, 7085. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Sung, Y.A.; Hong, Y.S.; Song, D.K.; Jung, H.; Jeong, K.; Chung, H.; Lee, H. Non-alcoholic fatty liver disease is associated with hyperandrogenism in women with polycystic ovary syndrome. Sci. Rep. 2023, 13, 13397. [Google Scholar] [CrossRef]
- Alhermi, A.; Perks, H.; Nigi, V.; Altahoo, N.; Atkin, S.L.; Butler, A.E. The role of the liver in the pathophysiology of PCOS: A literature review. Biomolecules 2025, 15, 51. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The role of estrogens in control of energy balance and glucose homeostasis. Endocr. Rev. 2013, 34, 309–338. [Google Scholar] [CrossRef]
- Gui, Z.; Shi, W.; Zhou, F.; Yan, Y.; Li, Y.; Xu, Y. The role of estrogen receptors in intracellular estrogen signaling pathways, an overview. J. Steroid Biochem. Mol. Biol. 2025, 245, 106632. [Google Scholar] [CrossRef]
- Palmisano, B.T.; Zhu, L.; Stafford, J.M. Role of estrogens in the regulation of liver lipid metabolism. Adv. Exp. Med. Biol. 2017, 1043, 227–256. [Google Scholar] [CrossRef]
- Bushi, A.; Ma, Y.; Adu-Amankwaah, J.; Wang, R.; Cui, F.; Xiao, R.; Zhao, J.; Yuan, J.; Tan, R. G protein-coupled estrogen receptor biased signaling in health and disease. Pharmacol. Ther. 2025, 269, 108822. [Google Scholar] [CrossRef]
- Savova, M.S.; Mihaylova, L.V.; Tews, D.; Wabitsch, M.; Georgiev, M.I. Targeting PI3K/AKT signaling pathway in obesity. Biomed. Pharmacother. 2023, 159, 114244. [Google Scholar] [CrossRef] [PubMed]
- Kasarinaite, A.; Sinton, M.; Saunders, P.T.K.; Hay, D.C. The influence of sex hormones in liver function and disease. Cells 2023, 12, 1604. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, R.; Wang, Y.; Xiao, H.; Lin, W.; Diao, M.; He, S.; Mei, P.; Liao, Y. G protein-coupled estrogen receptor activates PI3K/AKT/mTOR signaling to suppress ferroptosis via SREBP1/SCD1-mediated lipogenesis. Mol. Med. 2024, 30, 28. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Li, X.; Xu, Y.; Cheng, D.; Xia, X.; Lv, X.; Yuan, G.; Peng, C. Estrogen mediates an atherosclerotic-protective action via estrogen receptor alpha/SREBP-1 signaling. Front. Cardiovasc. Med. 2022, 9, 895916. [Google Scholar] [CrossRef]
- Huang, W.Y.; Sun, P.M. Estrogen receptor-associated receptor α and peroxisome proliferator-activated receptor γ in metabolism and disease (Review). Mol. Med. Rep. 2021, 23, 156. [Google Scholar] [CrossRef]
- Antwi, M.B.; Lefere, S.; Clarisse, D.; Koorneef, L.; Heldens, A.; Onghena, L.; Decroix, K.; Fijalkowska, D.; Thommis, J.; Hellemans, M.; et al. PPARα-ERRα crosstalk mitigates metabolic dysfunction-associated steatotic liver disease progression. Metabolism 2025, 164, 156128. [Google Scholar] [CrossRef]
- Ferré, P.; Phan, F.; Foufelle, F. SREBP-1c and lipogenesis in the liver: An update1. Biochem. J. 2021, 478, 3723–3739. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef]
- Ruiz, R.; Jideonwo, V.; Ahn, M.; Surendran, S.; Tagliabracci, V.S.; Hou, Y.; Gamble, A.; Kerner, J.; Irimia-Dominguez, J.M.; Puchowicz, M.A.; et al. Sterol regulatory element-binding protein-1 (SREBP-1) is required to regulate glycogen synthesis and gluconeogenic gene expression in mouse liver. J. Biol. Chem. 2014, 289, 5510–5517. [Google Scholar] [CrossRef]
- Zhao, Q.; Lin, X.; Wang, G. Targeting SREBP-1-mediated lipogenesis as potential strategies for cancer. Front. Oncol. 2022, 12, 952371. [Google Scholar] [CrossRef]
- Yoon, M. PPARα in obesity: Sex difference and estrogen involvement. PPAR Res. 2010, 2010, 584296. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Nakajima, M.; Yokoi, T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005, 227, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Bosch, E.; Alviggi, C.; Lispi, M.; Conforti, A.; Hanyaloglu, A.C.; Chuderland, D.; Simoni, M.; Raine-Fenning, N.; Crépieux, P.; Kol, S.; et al. Reduced FSH and LH action: Implications for medically assisted reproduction. Hum. Reprod. 2021, 36, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- De Paoli, M.; Zakharia, A.; Werstuck, G.H. The role of estrogen in insulin resistance: A review of clinical and preclinical data. Am. J. Pathol. 2021, 191, 1490–1498. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Goulis, D.G. Menopause and metabolic dysfunction-associated steatotic liver disease. Maturitas 2024, 186, 108024. [Google Scholar] [CrossRef]
- Ko, S.H.; Jung, Y. Energy metabolism changes and dysregulated lipid metabolism in postmenopausal women. Nutrients 2021, 13, 4556. [Google Scholar] [CrossRef]
- Emanuel, R.H.K.; Roberts, J.; Docherty, P.D.; Lunt, H.; Campbell, R.E.; Möller, K. A review of the hormones involved in the endocrine dysfunctions of polycystic ovary syndrome and their interactions. Front. Endocrinol. 2022, 13, 1017468. [Google Scholar] [CrossRef]
- Unluhizarci, K.; Karaca, Z.; Kelestimur, F. Role of insulin and insulin resistance in androgen excess disorders. World J. Diabetes 2021, 12, 616–629. [Google Scholar] [CrossRef]
- Arvanitakis, K.; Chatzikalil, E.; Kalopitas, G.; Patoulias, D.; Popovic, D.S.; Metallidis, S.; Kotsa, K.; Germanidis, G.; Koufakis, T. Metabolic dysfunction-associated steatotic liver disease and polycystic ovary syndrome: A complex interplay. J. Clin. Med. 2024, 13, 4243. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, J.; Lu, Y.; Wu, L. Association of metabolic-dysfunction associated steatotic liver disease with polycystic ovary syndrome. iScience 2024, 27, 108783. [Google Scholar] [CrossRef]
- Ko, S.H.; Kim, H.S. Menopause-associated lipid metabolic disorders and foods beneficial for postmenopausal women. Nutrients 2020, 12, 202. [Google Scholar] [CrossRef] [PubMed]
- Kodoth, V.; Scaccia, S.; Aggarwal, B. Adverse changes in body composition during the menopausal transition and relation to cardiovascular risk: A contemporary review. Womens Health Rep. 2022, 3, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Ramezani-Binabaj, M.; Motalebi, M.; Karimi-Sari, H.; Rezaee-Zavareh, M.S.; Alavian, S.M. Are women with polycystic ovarian syndrome at a high risk of non-alcoholic Fatty liver disease; a meta-analysis. Hepat. Mon. 2014, 14, e23235. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.L.L.; Faria, L.C.; Guimarães, T.C.M.; Moreira, G.V.; Cândido, A.L.; Couto, C.A.; Reis, F.M. Non-alcoholic fatty liver disease in women with polycystic ovary syndrome: Systematic review and meta-analysis. J. Endocrinol. Investig. 2017, 40, 1279–1288. [Google Scholar] [CrossRef]
- Wu, J.; Yao, X.Y.; Shi, R.X.; Liu, S.F.; Wang, X.Y. A potential link between polycystic ovary syndrome and non-alcoholic fatty liver disease: An update meta-analysis. Reprod. Health 2018, 15, 77. [Google Scholar] [CrossRef]
- Shengir, M.; Chen, T.; Guadagno, E.; Ramanakumar, A.V.; Ghali, P.; Deschenes, M.; Wong, P.; Krishnamurthy, S.; Sebastiani, G. Non-alcoholic fatty liver disease in premenopausal women with polycystic ovary syndrome: A systematic review and meta-analysis. JGH Open 2021, 5, 434–445. [Google Scholar] [CrossRef]
- Manzano-Nunez, R.; Santana-Dominguez, M.; Rivera-Esteban, J.; Sabiote, C.; Sena, E.; Bañares, J.; Tacke, F.; Pericàs, J.M. Non-alcoholic fatty liver disease in patients with polycystic ovary syndrome: A systematic review, meta-analysis, and meta-regression. J. Clin. Med. 2023, 12, 856. [Google Scholar] [CrossRef]
- Liu, D.; Gao, X.; Pan, X.F.; Zhou, T.; Zhu, C.; Li, F.; Fan, J.G.; Targher, G.; Zhao, J. The hepato-ovarian axis: Genetic evidence for a causal association between non-alcoholic fatty liver disease and polycystic ovary syndrome. BMC Med. 2023, 21, 62. [Google Scholar] [CrossRef]
- Yao, K.; Zheng, H.; Peng, H. Association between polycystic ovary syndrome and risk of non-alcoholic fatty liver disease: A meta-analysis. Endokrynol. Pol. 2023, 74, 520–527. [Google Scholar] [CrossRef]
- Wu, Z.; Liang, G.; Zhang, Y.; Li, R. Risk factors for metabolic dysfunction-associated steatotic liver disease in patients with polycystic ovary syndrome in East Asia: A review and meta-analysis. Endocr. Pract. 2025, 31, 668–676. [Google Scholar] [CrossRef]
- Loria, P.; Adinolfi, L.E.; Bellentani, S.; Bugianesi, E.; Grieco, A.; Fargion, S.; Gasbarrini, A.; Loguercio, C.; Lonardo, A.; Marchesini, G.; et al. Practice guidelines for the diagnosis and management of nonalcoholic fatty liver disease. A decalogue from the Italian Association for the Study of the Liver (AISF) Expert. Committee. Dig. Liver Dis. 2010, 42, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Temple, J.L.; Cordero, P.; Li, J.; Nguyen, V.; Oben, J.A. A guide to non-alcoholic fatty liver disease in childhood and adolescence. Int. J. Mol. Sci. 2016, 17, 947. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Rasquin, L.I.; Anastasopoulou, C. Polycystic Ovarian Syndrome. [Updated 2025 May 4]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK459251/ (accessed on 30 May 2025).
- Lonardo, A.; Suzuki, A. Sexual dimorphism of NAFLD in adults. Focus on clinical aspects and implications for practice and translational research. J. Clin. Med. 2020, 9, 1278. [Google Scholar] [CrossRef]
- Sarkar, M.; Terrault, N.; Chan, W.; Cedars, M.I.; Huddleston, H.G.; Duwaerts, C.C.; Balitzer, D.; Gill, R.M. Polycystic ovary syndrome (PCOS) is associated with NASH severity and advanced fibrosis. Liver Int. 2020, 40, 355–359. [Google Scholar] [CrossRef]
- Stefan, N.; Lonardo, A.; Targher, G. Role of steatotic liver disease in prediction and prevention of cardiometabolic diseases. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 136–137. [Google Scholar] [CrossRef]
- Lonardo, A.; Stefan, N.; Mantovani, A. Widening research horizons on metabolic dysfunction-associated steatotic liver disease and cancer. Trends Endocrinol. Metab. 2025. online ahead of print. [Google Scholar] [CrossRef]
- Kumarendran, B.; O’Reilly, M.W.; Manolopoulos, K.N.; Toulis, K.A.; Gokhale, K.M.; Sitch, A.J.; Wijeyaratne, C.N.; Coomarasamy, A.; Arlt, W.; Nirantharakumar, K. Polycystic ovary syndrome, androgen excess, and the risk of nonalcoholic fatty liver disease in women: A longitudinal study based on a United Kingdom primary care database. PLoS Med. 2018, 15, e1002542. [Google Scholar] [CrossRef]
- Ballestri, S.; Mantovani, A.; Byrne, C.D.; Lonardo, A.; Targher, G. Diagnostic accuracy of ultrasonography for the detection of hepatic steatosis: An updated meta-analysis of observational studies. Metab. Target Organ Damage 2021, 1, 7. [Google Scholar] [CrossRef]
- Diamond, T.; Stiel, D.; Lunzer, M.; Wilkinson, M.; Roche, J.; Posen, S. Osteoporosis and skeletal fractures in chronic liver disease. Gut 1990, 31, 82–87. [Google Scholar] [CrossRef]
- Calanchini, M.; Moolla, A.; Tomlinson, J.W.; Cobbold, J.F.; Grossman, A.; Fabbri, A.; Turner, H.E. Liver biochemical abnormalities in Turner syndrome: A comprehensive characterization of an adult population. Clin. Endocrinol. 2018, 89, 667–676. [Google Scholar] [CrossRef]
- Han, J.C.; Reyes-Capo, D.P.; Liu, C.Y.; Reynolds, J.C.; Turkbey, E.; Turkbey, I.B.; Bryant, J.; Marshall, J.D.; Naggert, J.K.; Gahl, W.A.; et al. Comprehensive endocrine-metabolic evaluation of patients with Alström syndrome compared with BMI-matched controls. J. Clin. Endocrinol. Metab. 2018, 103, 2707–2719. [Google Scholar] [CrossRef] [PubMed]
- Viuff, M.H.; Stochholm, K.; Grønbaek, H.; Berglund, A.; Juul, S.; Gravholt, C.H. Increased occurrence of liver and gastrointestinal diseases and anaemia in women with Turner syndrome-a nationwide cohort study. Aliment. Pharmacol. Ther. 2021, 53, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Bourcigaux, N.; Dubost, E.; Buzzi, J.C.; Donadille, B.; Corpechot, C.; Poujol-Robert, A.; Christin-Maitre, S. Focus on liver function abnormalities in patients with Turner syndrome: Risk factors and evaluation of fibrosis risk. J. Clin. Endocrinol. Metab. 2023, 108, 2255–2261. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.A.; Lee, H.W.; Ahn, S.H.; Lee, E.J.; Ku, C.R.; Kim, S.U. Positive association between nonalcoholic fatty liver disease and growth hormone deficiency in patients with nonfunctioning pituitary adenoma. Front. Endocrinol. 2023, 13, 1057769. [Google Scholar] [CrossRef]
- Lam, J.; Stoppa-Vaucher, S.; Antoniou, M.C.; Bouthors, T.; Ruiz, I.; Sekarski, N.; Rutz, T.; Fries, S.; Binz, P.A.; Bütschi, F.N.; et al. Turner syndrome: Skin, liver, eyes, dental and ENT evaluation should be improved. Front. Endocrinol. 2023, 14, 1190670. [Google Scholar] [CrossRef]
- Twohig, P.; Li, L.; Danford, D.; Craft, M.; Yetman, A.T. Prevalence of hepatic steatosis and fibrosis in Turner syndrome: A prospective case-control study. Liver Int. 2024, 44, 1309–1315. [Google Scholar] [CrossRef]
- Zaegel, N.; Brahimaj, R.; Battaglia-Hsu, S.; Lamiral, Z.; Feigerlova, E. Systemic inflammatory indices and liver dysfunction in Turner syndrome patients: A retrospective case-control study. J. Endocr. Soc. 2024, 8, bvae099. [Google Scholar] [CrossRef]
- Robeva, R.; Elenkova, A.; Kirilov, G.; Zacharieva, S. Metabolic risk in patients with a diminished ovarian reserve and premature ovarian insufficiency. J. Clin. Med. 2024, 13, 5105. [Google Scholar] [CrossRef]
- Ridder, L.O.R.; Just, J.; Hvas, C.L.; Nielsen, M.M.; Møller, H.J.; Grønbæk, H.; Gravholt, C.H. Elevated liver enzymes in Turner syndrome: The role of low-grade inflammation and hormonal imbalances. J. Endocr. Soc. 2025, 9, bvaf059. [Google Scholar] [CrossRef]
- Shankar Kikkeri, N.; Nagalli, S. Turner Syndrome. [Updated 2023 Aug 8]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK554621/ (accessed on 30 May 2025).
- Khan, N.; Farooqui, A.; Ishrat, R. Turner Syndrome where are we? Orphanet J. Rare Dis. 2024, 19, 314. [Google Scholar] [CrossRef]
- Lonardo, A.; Weiskirchen, R. From hypothalamic obesity to metabolic dysfunction-associated steatotic liver disease: Physiology meets the clinics via metabolomics. Metabolites 2024, 14, 408. [Google Scholar] [CrossRef] [PubMed]
- McDermott, J.H.; Walsh, C.H. Hypogonadism in hereditary hemochromatosis. J. Clin. Endocrinol. Metab. 2005, 90, 2451–2455. [Google Scholar] [CrossRef] [PubMed]
- Roemhild, K.; von Maltzahn, F.; Weiskirchen, R.; Knüchel, R.; von Stillfried, S.; Lammers, T. Iron metabolism: Pathophysiology and pharmacology. Trends Pharmacol. Sci. 2021, 42, 640–656. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.L.; Rawla, P. Hemochromatosis. [Updated 2024 Oct 6]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK430862/ (accessed on 30 May 2025).
- World Health Organization. Menopause. Available online: https://www.who.int/news-room/fact-sheets/detail/menopause (accessed on 30 May 2025).
- Jersild, M. Infectious hepatitis with subacute atrophy of the liver; an epidemic in women after the menopause. N. Engl. J. Med. 1947, 237, 8–11. [Google Scholar] [CrossRef]
- Codes, L.; Asselah, T.; Cazals-Hatem, D.; Tubach, F.; Vidaud, D.; Paraná, R.; Bedossa, P.; Valla, D.; Marcellin, P. Liver fibrosis in women with chronic hepatitis C: Evidence for the negative role of the menopause and steatosis and the potential benefit of hormone replacement therapy. Gut 2007, 56, 390–395. [Google Scholar] [CrossRef]
- Yang, J.D.; Abdelmalek, M.F.; Pang, H.; Guy, C.D.; Smith, A.D.; Diehl, A.M.; Suzuki, A. Gender and menopause impact severity of fibrosis among patients with nonalcoholic steatohepatitis. Hepatology 2014, 59, 1406–1414. [Google Scholar] [CrossRef]
- Matsuo, K.; Gualtieri, M.R.; Cahoon, S.S.; Jung, C.E.; Paulson, R.J.; Shoupe, D.; Muderspach, L.I.; Wakatsuki, A.; Wright, J.D.; Roman, L.D. Surgical menopause and increased risk of nonalcoholic fatty liver disease in endometrial cancer. Menopause 2016, 23, 189–196. [Google Scholar] [CrossRef]
- Veronese, N.; Notarnicola, M.; Osella, A.R.; Cisternino, A.M.; Reddavide, R.; Inguaggiato, R.; Guerra, V.; Rotolo, O.; Zinzi, I.; Chiloiro, M.; et al. Menopause does not affect fatty liver severity in women: A population study in a Mediterranean area. Endocr. Metab. Immune Disord. Drug Targets 2018, 18, 513–521. [Google Scholar] [CrossRef]
- Park, S.H.; Park, Y.E.; Lee, J.; Choi, J.H.; Heo, N.Y.; Park, J.; Kim, T.O.; Moon, Y.S.; Kim, H.K.; Jang, H.J.; et al. Lack of association between early menopause and non-alcoholic fatty liver disease in postmenopausal women. Climacteric 2020, 23, 173–177. [Google Scholar] [CrossRef]
- Jaroenlapnopparat, A.; Charoenngam, N.; Ponvilawan, B.; Mariano, M.; Thongpiya, J.; Yingchoncharoen, P. Menopause is associated with increased prevalence of nonalcoholic fatty liver disease: A systematic review and meta-analysis. Menopause 2023, 30, 48–354. [Google Scholar] [CrossRef]
- Raverdy, V.; Chatelain, E.; Lasailly, G.; Caiazzo, R.; Vandel, J.; Verkindt, H. Combining diabetes, sex, and menopause as meaningful clinical features associated with NASH and liver fibrosis in individuals with class II and III obesity: A retrospective cohort study. Obesity 2023, 31, 3066–3076. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Lee, Y.J.; Kwon, Y.J.; Lee, J.W. Age at menopause and risk of metabolic dysfunction-associated fatty liver disease: A 14-year cohort study. Dig. Liver Dis. 2024, 56, 1880–1886. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, S.; Feng, B.; Lu, Y.; Wang, Y.; Liao, W.; Wu, S.; Wang, L. Association between menopause, body composition, and nonalcoholic fatty liver disease: A prospective cohort in northern China. Maturitas 2025, 192, 108148. [Google Scholar] [CrossRef] [PubMed]
- Bagheri Lankarani, K.; Jamalinia, M.; Zare, F.; Heydari, S.T.; Ardekani, A.; Lonardo, A. Liver-kidney-metabolic health, sex, and menopause impact total scores and monovessel vs. multivessel coronary artery calcification. Adv. Ther. 2025, 42, 1729–1744. [Google Scholar] [CrossRef]
- Bruno, S.; Maisonneuve, P.; Castellana, P.; Rotmensz, N.; Rossi, S.; Maggioni, M.; Persico, M.; Colombo, A.; Monasterolo, F.; Casadei-Giunchi, D.; et al. Incidence and risk factors for non-alcoholic steatohepatitis: Prospective study of 5408 women enrolled in Italian tamoxifen chemoprevention trial. BMJ 2005, 330, 932. [Google Scholar] [CrossRef]
- Maldonado, S.S.; Cedars, M.I.; Yates, K.P.; Wilson, L.A.; Gill, R.; Terrault, N.A.; Suzuki, A.; Sarkar, M.A. Antimullerian hormone, a marker of ovarian reserve, is protective against presence and severity of NASH in premenopausal women. Clin. Gastroenterol. Hepatol. 2024, 22, 339–346.e5. [Google Scholar] [CrossRef]
- Hense, J.D.; Isola, J.V.V.; Garcia, D.N.; Magalhães, L.S.; Masternak, M.M.; Stout, M.B.; Schneider, A. The role of cellular senescence in ovarian aging. NPJ Aging 2024, 10, 35. [Google Scholar] [CrossRef]
- Verdiesen, R.M.G.; von Berg, J.; Said, M.A.; van der Harst, P.; Mahajan, A.; van Gils, C.H.; van der Schouw, Y.T.; Onland-Moret, N.C. Anti-Müllerian hormone and cardiometabolic disease in women: A two-sample Mendelian randomization study. Rev. Cardiovasc. Med. 2022, 23, 269. [Google Scholar] [CrossRef]
- Kim, S.E.; Min, J.S.; Lee, S.; Lee, D.Y.; Choi, D. Different effects of menopausal hormone therapy on non-alcoholic fatty liver disease based on the route of estrogen administration. Sci. Rep. 2023, 13, 15461. [Google Scholar] [CrossRef]
- Xu, X.L.; Huang, Z.Y.; Yu, K.; Li, J.; Fu, X.W.; Deng, S.L. Estrogen biosynthesis and signal transduction in ovarian disease. Front. Endocrinol. 2022, 13, 827032. [Google Scholar] [CrossRef]
- Ma, Y.; Fan, X.; Han, J.; Cheng, Y.; Zhao, J.; Fang, W.; Gao, L. Critical illness and sex hormones: Response and impact of the hypothalamic-pituitary-gonadal axis. Ther. Adv. Endocrinol. Metab. 2025, 16, 20420188251328192. [Google Scholar] [CrossRef] [PubMed]
- McCartney, C.R.; Campbell, R.E. Abnormal GnRH pulsatility in polycystic ovary syndrome: Recent insights. Curr. Opin. Endocr. Metab. Res. 2020, 12, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Szybiak-Skora, W.; Cyna, W.; Lacka, K. New insights in the diagnostic potential of sex hormone-binding globulin (SHBG)-Clinical approach. Biomedicines 2025, 13, 1207. [Google Scholar] [CrossRef]
- Wallace, I.R.; McKinley, M.C.; Bell, P.M.; Hunter, S.J. Sex hormone binding globulin and insulin resistance. Clin. Endocrinol. 2013, 78, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Li, M.; Pan, F.; Xiao, Y.; Cui, W.; Hu, Y. Non-alcoholic fatty liver disease is an influencing factor for the association of SHBG with metabolic syndrome in diabetes patients. Sci. Rep. 2017, 7, 14532. [Google Scholar] [CrossRef]
- Wang, X.; Xie, J.; Pang, J.; Zhang, H.; Chen, X.; Lin, J.; Li, Q.; Chen, Q.; Ma, J.; Xu, X.; et al. Serum SHBG is associated with the development and regression of nonalcoholic fatty liver disease: A prospective study. J. Clin. Endocrinol. Metab. 2020, 105, dgz244. [Google Scholar] [CrossRef]
- Ohlsson, C.; Hellberg, N.; Parini, P.; Vidal, O.; Bohlooly, M.; Rudling, M.; Lindberg, M.K.; Warner, M.; Angelin, B.; Gustafsson, J.A. Obesity and disturbed lipoprotein profile in estrogen receptor-alpha-deficient male mice. Biochem. Biophys. Res. Commun. 2000, 278, 640–645, Erratum in: Biochem. Biophys. Res. Commun. 2006, 348, 326. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, Z.; Geng, Y.; Li, F.; Hu, R.; Song, Y.; Zhang, M.; Song, K. The risk factors, pathogenesis and treatment of premature ovarian insufficiency. J. Ovarian Res. 2025, 18, 134. [Google Scholar] [CrossRef]
- Khan, M.S.; Kim, H.S.; Kim, R.; Yoon, S.H.; Kim, S.G. Dysregulated liver metabolism and polycystic ovarian syndrome. Int. J. Mol. Sci. 2023, 24, 7454. [Google Scholar] [CrossRef]
- Gupta, M.; Babic, A.; Beck, A.H.; Terry, K. TNF-α expression, risk factors, and inflammatory exposures in ovarian cancer: Evidence for an inflammatory pathway of ovarian carcinogenesis? Hum. Pathol. 2016, 54, 82–91. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Papavassiliou, A.G. Molecular mechanisms of insulin resistance in polycystic ovary syndrome. Trends Mol. Med. 2006, 12, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, P.; Weiskirchen, R. The signaling pathways in obesity-related complications. J. Cell Commun. Signal. 2024, 18, e12039. [Google Scholar] [CrossRef] [PubMed]
- Dinakaran, A.; Ar, S.; Rajagambeeram, R.; Nanda, S.K.; Daniel, M. SHBG and Insulin resistance—Nexus revisited. Bioinformation 2024, 20, 816–821. [Google Scholar] [CrossRef]
- Xing, C.; Zhang, J.; Zhao, H.; He, B. Effect of sex hormone-binding globulin on polycystic ovary syndrome: Mechanisms, manifestations, genetics, and treatment. Int. J. Womens Health 2022, 14, 91–105. [Google Scholar] [CrossRef]
- Boots, C.E.; Jungheim, E.S. Inflammation and human ovarian follicular dynamics. Semin. Reprod. Med. 2015, 33, 270–275. [Google Scholar] [CrossRef]
- Stanciu, S.M.; Jinga, M.; Miricescu, D.; Stefani, C.; Nica, R.I.; Stanescu-Spinu, I.I.; Vacaroiu, I.A.; Greabu, M.; Nica, S. mTOR dysregulation, insulin resistance, and hypertension. Biomedicines 2024, 12, 1802. [Google Scholar] [CrossRef]
- Parazzini, F.; Gerli, S.; Favilli, A.; Vignali, M.; Ricci, E.; Cipriani, S.; Chiaffarino, F.; Dell’acqua, A.; Harari, S.; Bianchi, S. mTOR inhibitors and risk of ovarian cysts: A systematic review and meta-analysis. BMJ Open 2021, 11, e048190. [Google Scholar] [CrossRef]
- Ghafari, A.; Maftoohi, M.; Eslami Samrin, M.; Barani, S.; Banimohammad, M.; Samie, R. The last update on polycystic ovary syndrome (PCOS), diagnosis criteria, and novel treatment. Endocr. Metab. Sci. 2025, 17, 100228. [Google Scholar] [CrossRef]
- Iqbal, J.; Zaidi, M. Understanding estrogen action during menopause. Endocrinology 2009, 150, 3443–3445. [Google Scholar] [CrossRef]
- Kumariya, S.; Ubba, V.; Jha, R.K.; Gayen, J.R. Autophagy in ovary and polycystic ovary syndrome: Role, dispute and future perspective. Autophagy 2021, 17, 2706–2733. [Google Scholar] [CrossRef]
- Nash, Z.; Al-Wattar, B.H.; Davies, M. Bone and heart health in menopause. Best Pract. Res. Clin. Obstet. Gynaecol. 2022, 81, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Jayasena, C.N.; Devine, K.; Barber, K.; Comninos, A.N.; Conway, G.S.; Crown, A.; Davies, M.C.; Ewart, A.; Seal, L.J.; Smyth, A.; et al. Society for endocrinology guideline for understanding, diagnosing and treating female hypogonadism. Clin. Endocrinol. 2024, 101, 409–442. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Wang, L.; Sun, D.; Wu, Y.; Yu, C.; Huang, Y.; Chan, S.O.; Ling, W.; Lv, J.; Li, L.; et al. Associations of alcohol consumption and genetic predisposition to hepatic steatosis with liver-related events: Results from large population-based cohort studies. Gastroenterology 2025. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Perrett, R.M.; McArdle, C.A. Molecular mechanisms of gonadotropin-releasing hormone signaling: Integrating cyclic nucleotides into the network. Front. Endocrinol. 2013, 4, 180. [Google Scholar] [CrossRef]
- Lala, V.; Zubair, M.; Minter, D.A. Liver Function Tests. [Updated 2023 Jul 30]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK482489/ (accessed on 30 May 2025).
- García, O.P.; Long, K.Z.; Rosado, J.L. Impact of micronutrient deficiencies on obesity. Nutr. Rev. 2009, 67, 559–572. [Google Scholar] [CrossRef]
- Mu, L.; Wang, G.; Yang, X.; Liang, J.; Tong, H.; Li, L.; Geng, K.; Bo, Y.; Hu, X.; Yang, R.; et al. Physiological premature aging of ovarian blood vessels leads to decline in fertility in middle-aged mice. Nat. Commun. 2025, 16, 72. [Google Scholar] [CrossRef]
- Gonçalves, C.R.; Vasconcellos, A.S.; Rodrigues, T.R.; Comin, F.V.; Reis, F.M. Hormone therapy in women with premature ovarian insufficiency: A systematic review and meta-analysis. Reprod. Biomed. Online 2022, 44, 1143–1157. [Google Scholar] [CrossRef]
- Lan, Y.; Jin, B.; Fan, Y.; Huang, Y.; Zhou, J. The circadian rhythm regulates the hepato-ovarian axis linking polycystic ovary syndrome and non-alcoholic fatty liver disease. Biochem. Genet. 2025. online ahead of print. [Google Scholar] [CrossRef]
- Vassilatou, E. Nonalcoholic fatty liver disease and polycystic ovary syndrome. World J. Gastroenterol. 2014, 20, 8351–8363. [Google Scholar] [CrossRef]
- Riemann, A.; Blaschke, M.; Jauho-Ghadimi, A.; Siggelkow, H.; Gollisch, K.S.C. Metformin improves the hepatic steatosis index in non-obese patients with polycystic ovary syndrome. J. Clin. Med. 2022, 11, 4294. [Google Scholar] [CrossRef]
- Wang, D.; He, B. Current perspectives on nonalcoholic fatty liver disease in women with polycystic ovary syndrome. Diabetes Metab. Syndr. Obes. 2022, 15, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- El Sobky, S.A.; Aboud, N.K.; El Assaly, N.M.; Fawzy, I.O.; El-Ekiaby, N.; Abdelaziz, A.I. Regulation of lipid droplet (LD) formation in hepatocytes via regulation of SREBP1c by non-coding RNAs. Front. Med. 2022, 9, 903856. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.; Brady, C.W.; Fleckenstein, J.; Forde, K.A.; Khungar, V.; Molleston, J.P.; Afshar, Y.; Terrault, N.A. Reproductive health and liver disease: Practice guidance by the American Association for the Study of Liver Diseases. Hepatology 2021, 73, 318–335. [Google Scholar] [CrossRef] [PubMed]
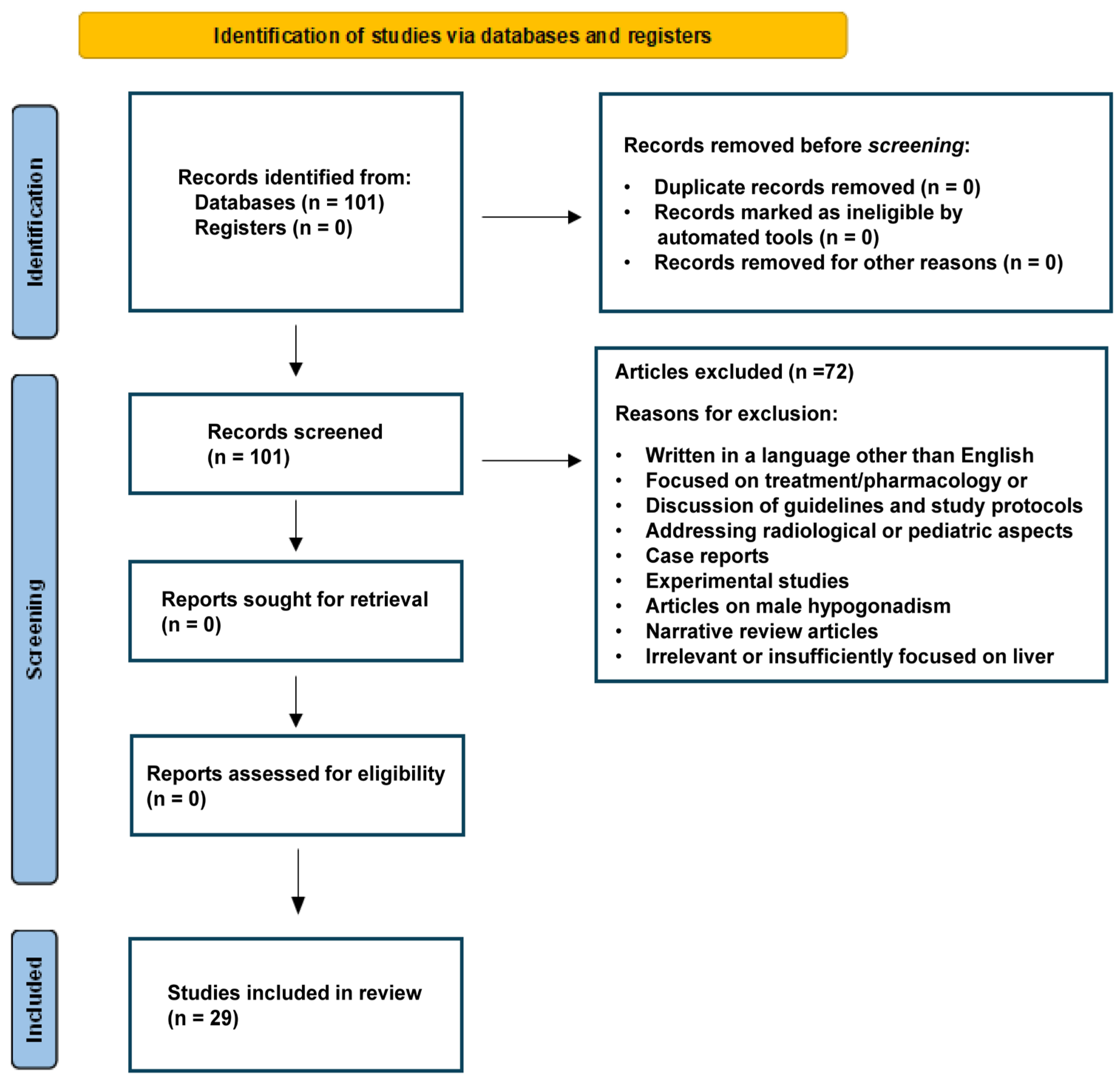

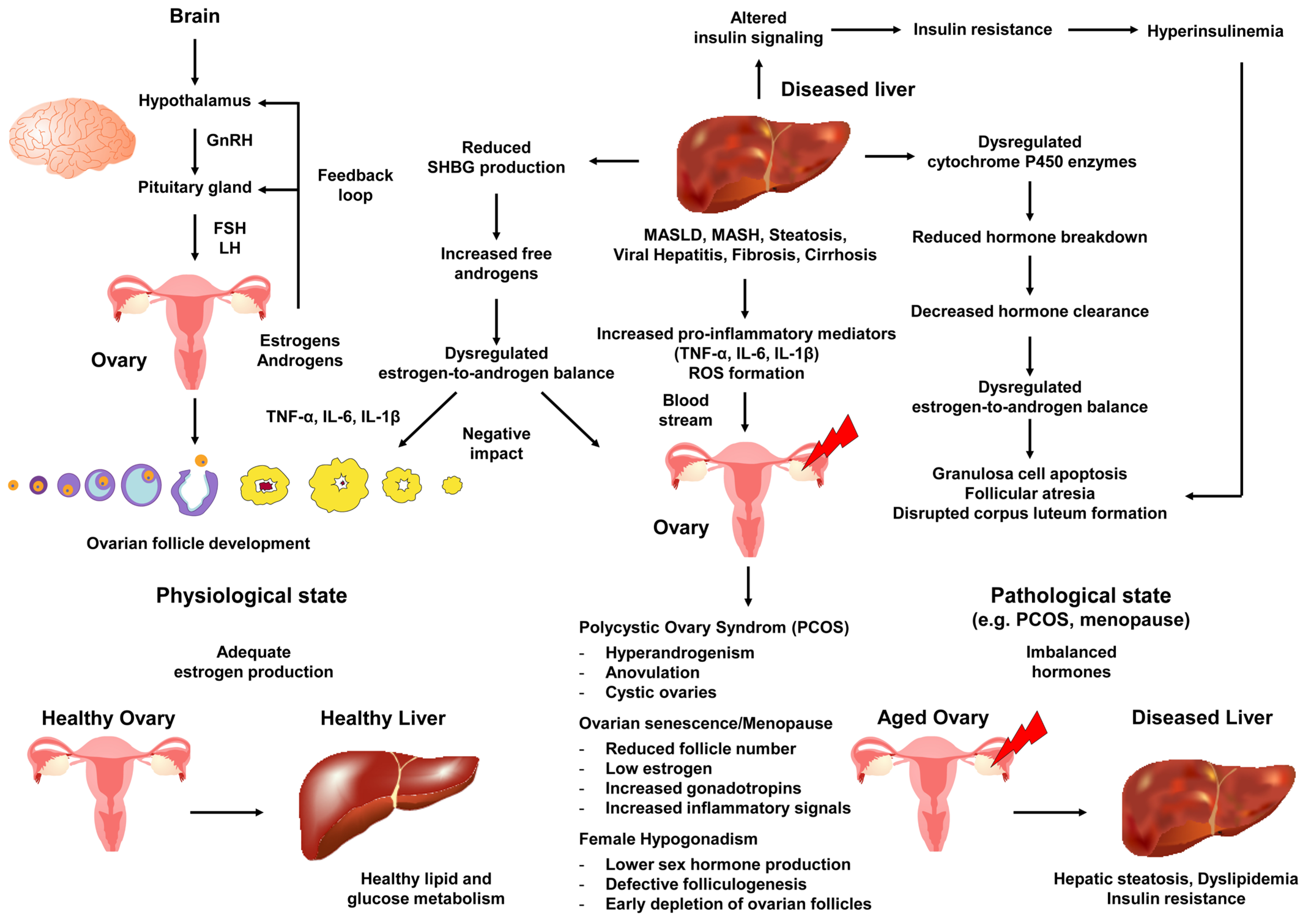

{kind=link}
{kind=link}
{kind=link}
| Author(s), Year [Ref] | Method | Findings | Conclusion |
|---|---|---|---|
| Ramezani-Binabaj et al., 2014 [33] | In total, 7 eligible studies were included, totaling 616 PCOS cases (defined according to the Rotterdam criteria) and 569 healthy controls. NAFLD was assessed with USG. | NAFLD is more prevalent among PCOS subjects than in healthy controls (OR 3.93, 95% CI: 2.17, 7.11). | Women with PCOS have an increased risk of NAFLD. |
| Rocha et al., 2017 [34] | In total, 17 eligible studies were included, totaling 2734 PCOS individuals (diagnosed based on Rotterdam criteria except for two studies using androgen excess and PCOD society, one study using the NIH criteria, and one study with missing information was NA) and 2561 controls of similar age and BMI. The criteria for diagnosing NAFLD were NA. | PCOS subjects exhibit an increased prevalence of NAFLD (OR 2.54, 95%, CI 2.19–2.95). | Women with PCOS have an increased prevalence of NAFLD. This is linked to high serum total testosterone, FAI, obesity, and IR. |
| Wu et al., 2018 [35] | In total, 17 eligible studies were included, totaling 2715 PCOS subjects (diagnosed based on Rotterdam criteria except for two studies using androgen excess and PCOD society, one study using the NIH criteria, and one study with missing information was NA) and 2619 controls. The criteria for diagnosing NAFLD were NA. | PCOS individuals have a higher prevalence of NAFLD than controls (OR 2.25, 95%, CI = 1.95–2.60). HA is a risk factor for NALD among those with PCOS (OR 3.31, 95% CI 2.58–4.24). | PCOS is associated with a higher risk of NAFLD, likely due to HA. |
| Shengir et al., 2021 [36] | In total, 23 eligible studies were included, totaling 4162 PCOS women and 2983 controls. The diagnosis of PCOS was based on one of the following definitions: Rotterdam criteria, NIH criteria, or AES criteria. NAFLD was diagnosed with imaging studies or noninvasive biomarkers. | PCOS women have a higher risk of NAFLD than controls (OR 2.49, 95%, CI 2.20–2.82). South American/Middle East PCOS patients exhibited a greater risk of NAFLD (OR 3.55, 95% CI 2.27–5.55) compared to their European and Asian counterparts (OR 2.22, 95% CI 1.85–2.67 and OR 2.63, 95% CI 2.20–3.15, respectively). IR (OR 1.97, 95% CI 1.44–2.71 and MetS (OR 3.39, 95% CI 2.42–4.76) were more frequent among PCOS women than among controls. | Premenopausal women with PCOS have a 2.5-fold increased risk of NAFLD, with BMI playing a significant role in this association. |
| Manzano-Nunez et al., 2023 [37] | In total, 36 eligible studies were included, totaling 5021 individuals with PCOS (diagnosed by NIH, Rotterdam, or AES criteria) and 2156 controls. NAFLD was identified either histologically or non-invasively. | MA of proportions found a pooled NAFLD prevalence of 43%, 95% CI 35–52%, with high heterogeneity. BMI, WC, ALT, HOMA-IR, FAI, HA, and TG were associated with significantly higher risk of NAFLD among women with PCOS. | NAFLD is common in young women with PCOS, influenced by both metabolic and PCOS-specific endocrine factors. |
| Liu et al., 2023 [38] | Bidirectional two-sample MR analysis was conducted using glycemic-related traits in up to 200,622 individuals and sex hormones in 189,473 women from a large-scale biopsy-confirmed NAFLD UKB GWAS database. | Subjects with a higher genetic propensity to NAFLD were at increased risk to develop PCOS (OR per one-unit log odds increase in NAFLD: 1.10, 95% CI: 1.02–1.18; p = 0.013). MR mediation analysis showed indirect causal effects of NAFLD on PCOS via fasting insulin alone (OR 1.02, 95% CI 1.01–1.03; p = 0.004) and in concert with androgen concentrations. | While genetically predicted NAFLD is linked to a higher likelihood of PCOS, the idea that PCOS may lead to NAFLD is not well-documented, suggesting the need for additional studies. |
| Yao et al., 2023 [39] | In total, 32 studies were included, totaling 145,131 PCOS patients and 50,832,503 controls. NAFLD was assessed with USG | PCOS was associated with a high risk of NAFLD (OR 2.93, p < 0.001, 95% CI 2.38–3.62). Age and BMI did not explain heterogeneity across the studies while a significant risk of publication bias was found. | PCOS is associated with a high risk of NAFLD. |
| Wu et al., 2025 [40] | In total, 26 eligible studies (all from China except one from Korea) were included, totaling 4510 participants. The diagnosis of PCOS was based on Rotterdam criteria in all studies except for five using the PCOS-PRCHIS criteria. MASLD was identified with USG in all studies except one where the criterion was NA. | At the pooled MVA, WHR (p < 0.001), testosterone (p = 0.034), and HOMA-IR (p = 0.02) were significantly greater in PCOS women with MASLD. | In East Asia, MASLD is associated with obesity, IR, and HA in women with PCOS. |
| Author(s), Year [Ref] | Method | Findings | Conclusion |
|---|---|---|---|
| Diamond et al., 1990 [50] | A comparative study was conducted on 115 individuals with biopsy-proven CLD and 113 age- and sex-matched controls. | LC and hypogonadism were risk factors for both spinal (β coef = 0.190 and 0.176; SE = 0.079 and 0.086, respectively) and forearm osteoporosis (β coef = 0.20 and 0.29; SE = 0.073 and 0.80, respectively). SBD was the predominant determinant of spinal fractures (β coef = −0.007; SE = 0.001), while hypogonadism (β coef = 0.363; SE = 0.075) and LC (β coef = 0.185; SE = 0.068) predicted peripheral fractures. | Hypogonadism is strongly linked to osteoporosis and peripheral fractures in individuals with biopsy-proven CLD. |
| Calanchini et al., 2018 [51] | A retrospective assessment was performed on 125 TS women. | RLE (most often altered GGT values) were identified in 49.6% of cases, and individuals exhibiting RLE had greater diameters of the sinuses and ascending aorta. A higher prevalence of RLE was found among women with isochromosome of the X long arm whereas patients with 45,X/46,XX, 45,X/47,XXX or 45,X/46,XX/47,XXX exhibited a lower prevalence of abnormal GGT values. FIB-4 > 1.3 was found in 11.8%. Fibroscan suggested significant LF in 38.1% of cases and histological changes (including 2 with LC) were found in 45.4% of 11 liver biopsies. | RLE are frequently seen in TS women, especially in connection with specific karyotype abnormalities and aortic dilatation, and may indicate fibrosing liver disease. |
| Han et al., 2018 [52] | A total of 38 patients with AS (ranging in age from 2 to 38 years) were compared to 76 controls matched for age, sex, race, and BMI. | AS individuals exhibited significantly higher fasting and MMT IR indices, higher MMT glucose, insulin, and C-peptide values, higher HbA1c, and TG, higher prevalence of T2DM (p < 0.001) lower HDL-Chol, and a 10-fold higher prevalence of MetS (p < 0.001), a significantly greater steatosis extent and higher transaminase values (p < 0.001). | Severe IR, T2DM, and SLD are the hallmarks of AS. |
| Viuff et al., 2021 [53] | A total of 1156 individuals with TS identified with the DCCR were compared to 577 age-matched female controls. | Women with TS have a 13-fold (IRR 12.9 (95% CI 5.8–28.8)) increased risk of liver disease owing to TLD (IRR 8.0 (95% CI 1.8–35.4)), liver insufficiency (IRR 6.7 (95% CI 1.7–26.9)), LF/LC (IRR 16.5 (95% CI 2.2–122.1)) and unspecified liver disease (IRR 10.6 (95% CI 4.4–25.3)). Furthermore, the presence of RLE was increased 12-fold (IRR 12.4 (95% CI 4.2–36.6)). | TS individuals have a high risk of liver disease, with a potential beneficial effect of HRT on liver diseases. |
| Bourcigaux et al., 2023 [54] | A single-center, retrospective cross-sectional analysis of 264 TS individuals. | In total, 42.8% of these individuals had raised liver enzymes. However, fewer than 10% were at risk of developing LF and LC was observed in 2 out of 19 liver biopsies. | Approximately 10% of TS subjects are at risk of LF, and the FIB-4 score should be included in the screening strategy. |
| Hwang, et al., 2023 [55] | A total of 278 individuals with NFPA who underwent transsphenoidal adenectomy were enrolled. | Gonadal function was unassociated with the prevalence of NAFLD among NFPA subjects (29.3% eugonadic vs. 47.8% hypogonadic, p = 0.14). | Hypogonadism is not linked to NAFLD prevalence in NFPA subjects. |
| Lam et al., 2023 [56] | In total, 68 TS patients were recruited. | Liver disease was found in 4.4% of cases, more often among individuals with Y structural rearrangement than among those with complete X monosomy (8.3% vs. 4.2% p = 0.771). | Steatohepatitis and inflammatory liver disease can be present in TS individuals. |
| Twohig et al., 2023 [57] | In total, 55 TS and 50 controls were enrolled. | Compared to controls, women with TS more often had steatosis (65% vs. 12%, stage 1 vs. 0, p < 0.0001) and fibrosis (39% vs. 2%, average Metavir F2 vs. F0, p < 0.00001) irrespective of BMI) (p < 0.01). GGT is more sensitive than AST or ALT in identifying liver changes. | Compared to healthy controls, TS is associated with a higher risk of SLD which can be identified by GGT serum values and SWE. |
| Zaegel et al., 2024 [58] | In total, 66 patients with TS were compared to 66 healthy controls using matched-pair analyses. | At LRA, TS was significantly associated with ALRI, APRI, GPR and liver dysfunction. | ALRI, APRI, and GPR show promise as biomarkers of liver disease in women with TS. |
| Robeva et al., 2024 [59] | A retrospective analysis of 28 healthy women, 77 individuals with DOR; and 121 patients with POI (of whom 36 had TS and 85 had non-TS POI). | Compared to controls, women with DOR, non-TS POI, and TS had RLE, pronounced IR, and worse lipidemic asset (p < 0.008 for all). Moreover, TS subjects had significantly higher AAT, GGT, and TSH concentrations compared to non-TS POI and DOR individuals. | Regardless of the severity of endocrinological issues and karyotypic abnormalities, decreased ovarian function is connected to IR, dyslipidemia, and RLE. |
| Ridder, et al., 2025 [60] | In total, 82 women with TS and 59 female controls were studied. | TS subjects showed higher values of GGT, AST, ALT (which were correlated with the inflammatory biomarkers CRP and sCD163 and 11β-hydroxytestosterone concentrations) and FIB-4 than controls (p < 0.001, all). Additionally, the neutrophil activation marker MPO was elevated in TS and correlated with liver parameters and sCD163. | In TS, RLE are associated with low-grade chronic inflammation and hormonal imbalances. |
| Author(s), Year [PMID] | Method | Findings | Conclusion |
|---|---|---|---|
| Codes et al., 2007 [69] | In total, 251 women were enrolled, of which were 22 menopausal and 65 received HRT. | Women with F2-F4 Metavir scores had a known infection duration of >15 years, higher BMI and more frequent steatosis compared to thosie with F0-F1 LF. They were more likely to be menopausal and less likely to receive HRT. Steatosis was more common and severe in menopausal women. | Factors associated with the severity of LF include a duration of infection of more than 15 years, a higher BMI, severe steatosis, and menopause. Additionally, HRT in menopausal woman is linked to a lower LF stage in CHC. |
| Yang et al., 2014 [70] | In total, 541 adults with biopsy-proven NASH were enrolled in the study. | After multiple adjustments (ACOR) and 95% CI, the risk for more severe LF was 1.4 (0.9, 2.1) for postmenopausal women (p = 0.17) and 1.6 (1.0, 2.5) for men (p = 0.03, compared to premenopausal women. The risk of greater fibrosis severity in men compared to women was 1.8 (1.1, 2.9) for patients <50 years (p = 0.02) and 1.2 (0.7, 2.1) for those ≥50 years (p = 0.59). | Compared to pre-menopausal women, men are at a higher risk of developing more severe LF, while postmenopausal women have a similar risk of LF severity compared to men. |
| Matsuo et al., 2016 [71] | A retrospective study was conducted on 666 endometrial cancer cases that underwent surgical staging, along with 209 endometrial hyperplasia cases that underwent hysterectomy-based treatment. This study also included 712 oophorectomy cases and 163 nonoophorectomy cases. | Oophorectomy was strongly associated with NAFLD risk (HR, 1.70; 95% CI, 1.01–2.86; p = 0.047) and NAFLD was significantly associated with postoperative T2DM (HR, 2.26; 95% CI, 1.52–3.35; p < 0.0001) and hypercholesterolemia (HR, 1.71; 95% CI, 1.12–2.63; p = 0.014). | In young women with endometrial cancer, oophorectomy is significantly associated with an increased NAFLD risk due to post-operative T2DM and hypercholesterolemia. |
| Veronese et al., 2018 [72] | An analysis was performed on 752 women in menopause and 535 in pre-menopause. | The years from menopause were not associated with the severity of NAFLD (p for trend = 0.74; Spearman correlation = 0.04; 95% CI: −0.09 to 0.17), whereas all the indexes of adiposity and the number of MetS components were associated with a higher liver steatosis score. | The higher prevalence of NAFLD observed post-menopausally may be attributed to abdominal adiposity and MetS, rather than menopause itself. |
| Park et al., 2020 [73] | A study was conducted on 4354 postmenopausal women who participated in the 2010–2012 KNHANES. | The OR for NAFLD per 1-year increase in age at menopause was 1.01 (95% CI, 0.99–1.03; p = 0.329). The prevalence of advanced fibrosis was 2.1% (95% CI, 0.7–6.4%), 2.2% (95% CI, 1.3–3.8%), and 3.9% (95% CI, 1.2–12.2%) in early (<45 years), normal (45–54 years), and late (≥55 years) menopausal women, respectively. | While there is no evidence linking early menopause to the risk of NAFLD, this study demonstrates that advanced fibrosis related to NAFLD is highly prevalent post-menopause. |
| Jaroenlapnopparat et al., 2023 [74] | A metanalysis was performed of 12 published studies. | Menopause and NAFLD are significantly associated (pooled OR 2.37, 95% CI, 1.99–2.82) and this association remained significant in a sensitivity meta-analysis of six studies with adjustment for age and metabolic factors (pooled OR 2.19, 95% CI, 1.73–2.78), without any evidence of publication bias. | Menopause doubles the risk of NAFLD. |
| Raverdy et al., 2023 [75] | In total, 1446 participants with obesity were enrolled in this study. | NASH and F ≥ 2 prevalence was 15.4% (33/215) and 15.5% (32/206) among premenopausal women with T2DM vs. 29.5% (33/112) and 30.3% (N = 36/119) in postmenopausal women with T2DM (p < 0.01). The distinct contribution of menopause was proven by the interaction between sex and age with respect to NASH among T2DM patients (p = 0.048). | A notably high prevalence of advanced SLD occurs after menopause among women with T2DM. |
| Kim et al., 2024 [76] | In total, 1888 participants were enrolled and followed for a median of 12.3 years. | After adjusting for confounding factors, the HR for new-onset MAFLD was 1.40 (CI 1.00–1.95) in women with menopause at <40 years compared to those in whom menopause occurred at the age of ≥50 years. | The risk of MAFLD risk is higher in women with premature menopause (under 40 years) compared to those who experienced menopause at 50 years or older. |
| Yang et al., 2025 [77] | In total, 1316 postmenopausal and 3049 premenopausal women were enrolled between 2006 and 2017 and followed up till 2021. | Compared to premenopausal subjects, women who experienced menopause exhibited higher chances of NAFLD (9-year aHR = 1.219, 95%, CI 1.088–1.365). BWL of ≥3% or WC reduction by ≥5% was associated with a 31.1% reduction (95% CI, 20.8–40.0%) or a 14.2% reduction (95% CI, 1.1–25.6%) in the risk of NAFLD among premenopausal women. | Menopause is associated with an increased risk of NAFLD, partly due to the accumulation of visceral fat. |
| Bagheri et al., 2025 [78] | In total, 446 patients undergoing CT coronary scanning were included in this study. | MASLD and CKD increased CAC risk in male but not female patients, with menopause significantly modifying LKMH’s effect. | The impact of LKMH on CAC burden is significantly influenced by liver fat content and menopause, |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiskirchen, R.; Lonardo, A. The Ovary–Liver Axis: Molecular Science and Epidemiology. Int. J. Mol. Sci. 2025, 26, 6382. https://doi.org/10.3390/ijms26136382
Weiskirchen R, Lonardo A. The Ovary–Liver Axis: Molecular Science and Epidemiology. International Journal of Molecular Sciences. 2025; 26(13):6382. https://doi.org/10.3390/ijms26136382
Chicago/Turabian StyleWeiskirchen, Ralf, and Amedeo Lonardo. 2025. "The Ovary–Liver Axis: Molecular Science and Epidemiology" International Journal of Molecular Sciences 26, no. 13: 6382. https://doi.org/10.3390/ijms26136382
APA StyleWeiskirchen, R., & Lonardo, A. (2025). The Ovary–Liver Axis: Molecular Science and Epidemiology. International Journal of Molecular Sciences, 26(13), 6382. https://doi.org/10.3390/ijms26136382