
Clinical and Transcriptomic Characterization of Metastatic Hormone-Sensitive Prostate Cancer Patients with Low PTEN Expression
 , , , , , ,
, , , , , ,  , ,
, ,  , , add
Show full author list
, , add
Show full author list
Abstract

1. Introduction
2. Results
2.1. Patients and Samples
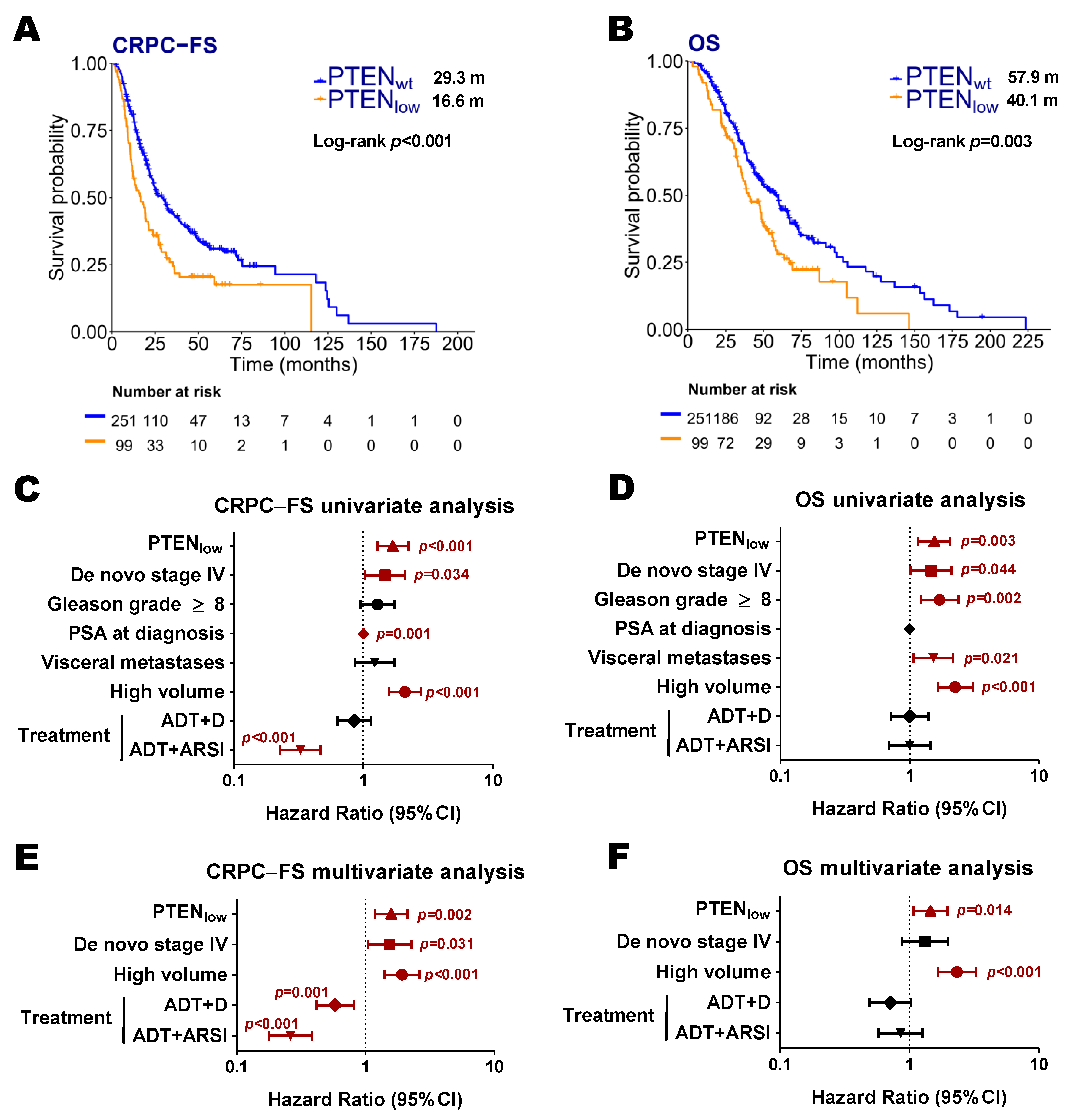
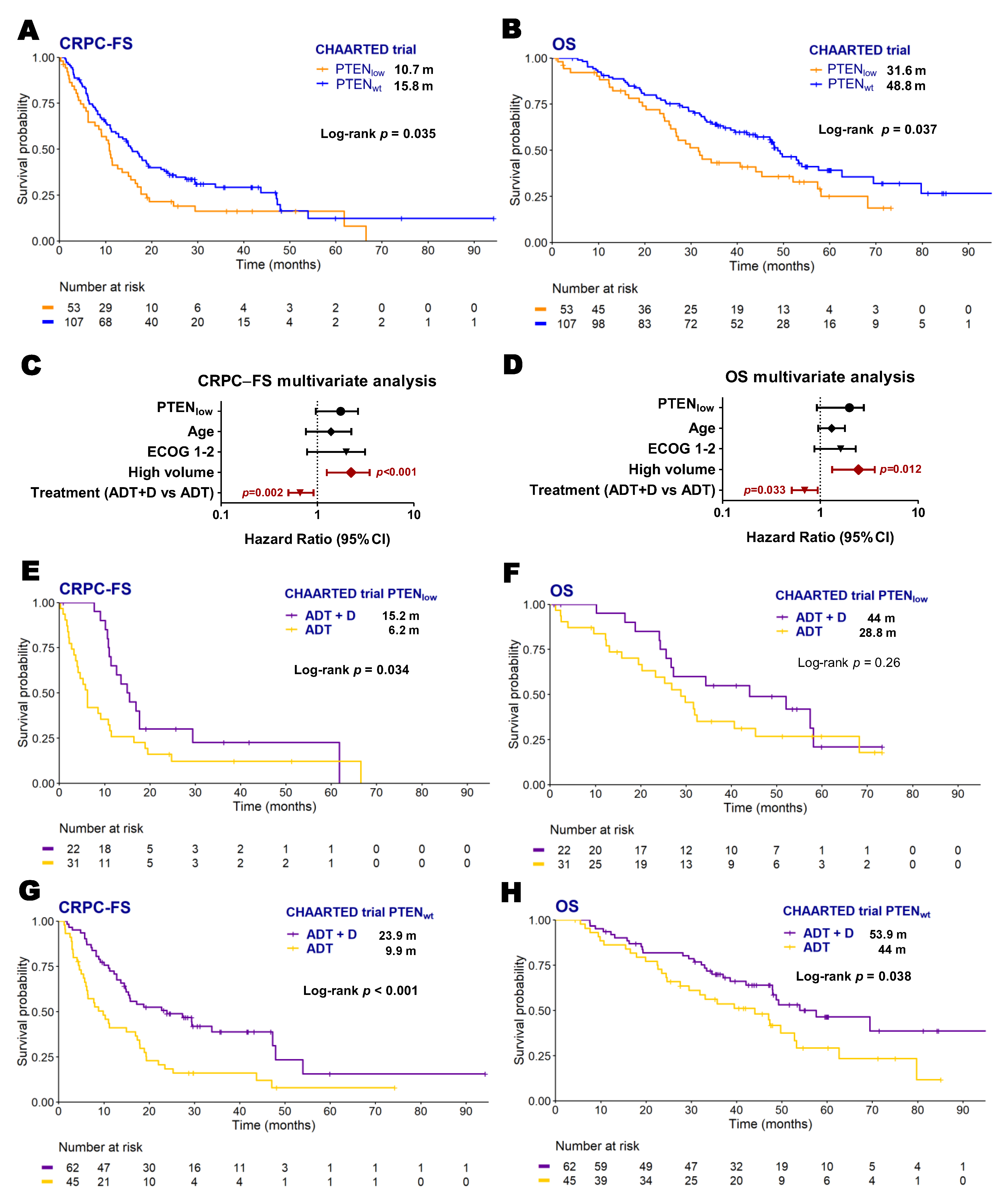
2.2. PTEN Expression and Clinical Evolution
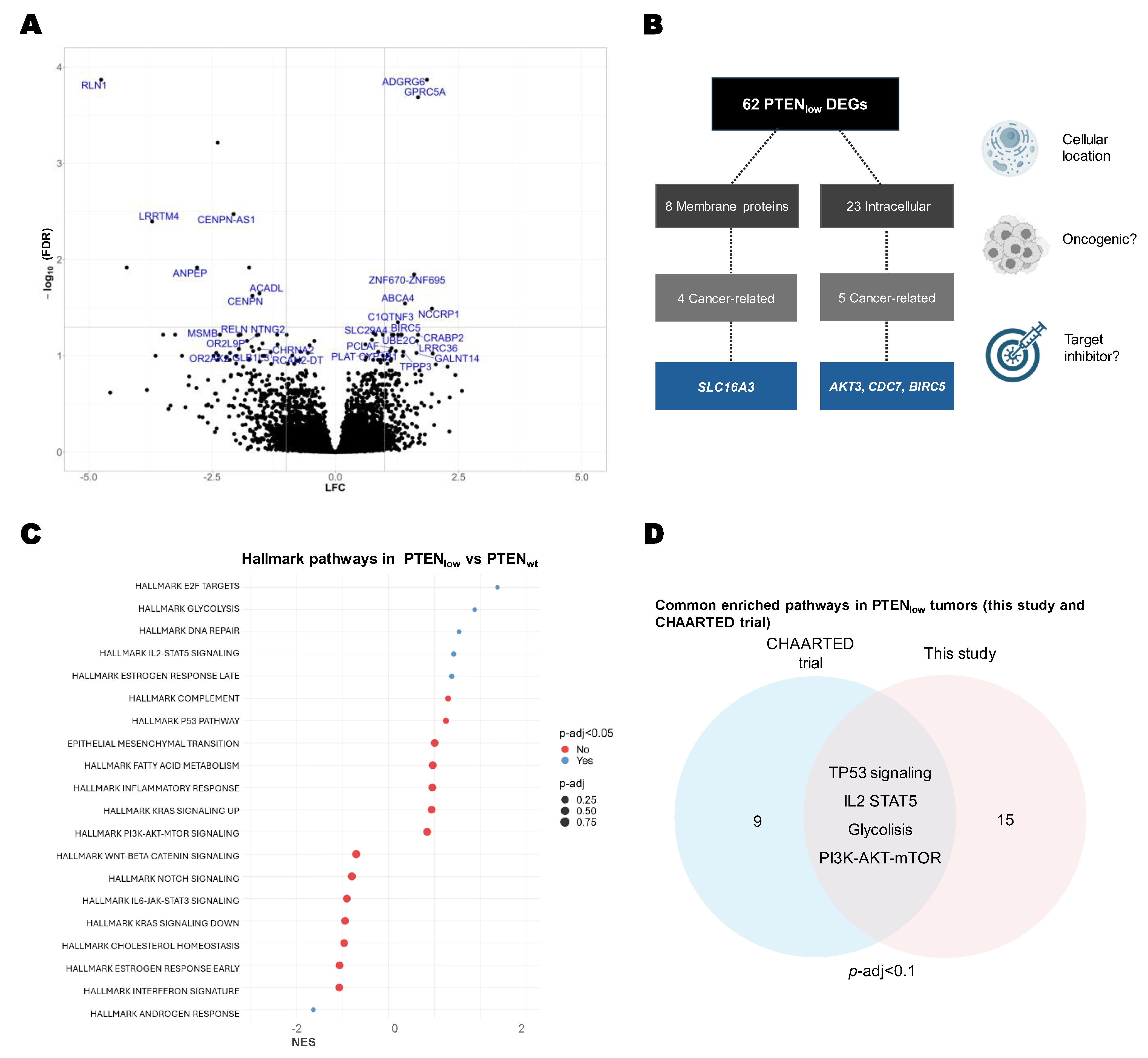
2.3. Transcriptional Characterization of PTENlow Tumors
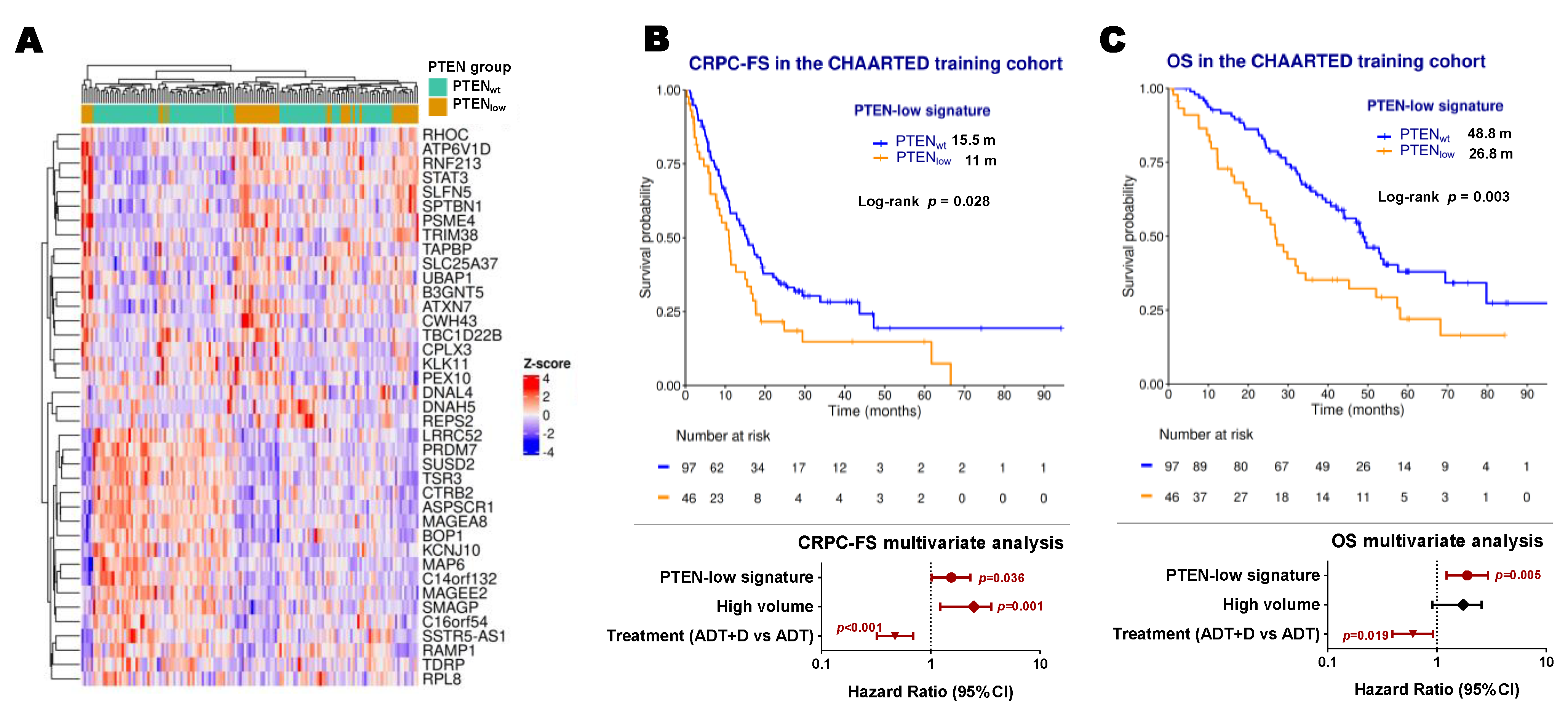
2.4. Development of a PTEN-Low Related Signature
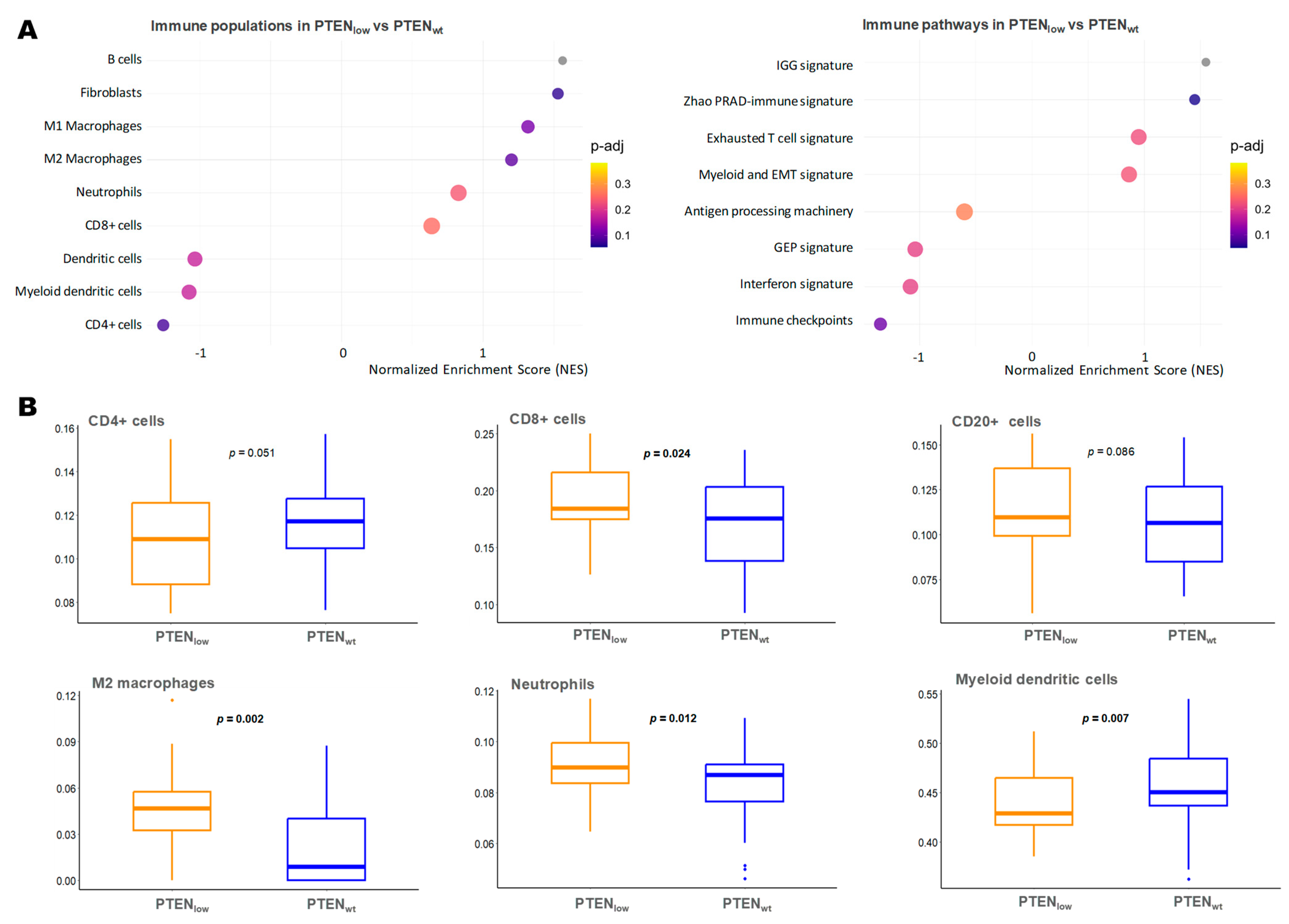
2.5. Immune Microenvironment-Related Gene Expression in PTENlow Tumors
2.6. Neuroendocrine Gene Expression in PTENlow Tumors
3. Discussion
4. Materials and Methods
4.1. Design, Patients, and Samples
4.2. Gene Expression Panel Design
4.3. RNA Extraction
4.4. nCounter Gene Expression Analysis
4.5. RNA Sequencing (RNA-Seq)
4.6. Bioinformatics and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADT | Androgen deprivation therapy |
| AR | Androgen receptor |
| ARSI | Androgen receptor signaling inhibitors |
| AUC | Area under the curve |
| CI | Confidence interval |
| CRPC | Castration-resistance prostate cancer |
| CRPC-FS | CRPC-free survival |
| D | Docetaxel |
| DEGs | Differentially expressed genes |
| ECOG | Eastern Cooperative Oncology Group |
| EMT | Epithelial-mesenchymal transition |
| FDR | False discovery rate |
| GEP | T-cell inflamed gene expression profile |
| IGG | Immunoglobulin signature |
| FFPE | Formalin-fixed paraffin-embedded |
| GSEA | Gene Set Enrichment Analysis |
| HR | Hazard ratio |
| IHC | Immunohistochemistry |
| LDH | Lactate dehydrogenase |
| LFC | Log2 fold change |
| LHRH | Luteinizing hormone-releasing hormone |
| mHSPC | Metastatic hormone-sensitive prostate cancer |
| NE | Neuroendocrine |
| OS | Overall survival |
| PC | Prostate cancer |
| PRAD | Prostate adenocarcinoma |
| PSA | Prostate-specific antigen |
| ssGSEA | Single-sample GSEA |
References
- Sweeney, C.J.; Chen, Y.-H.; Carducci, M.; Liu, G.; Jarrard, D.F.; Eisenberger, M.; Wong, Y.-N.; Hahn, N.; Kohli, M.; Cooney, M.M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N. Engl. J. Med. 2015, 373, 737–746. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.S.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of Docetaxel, Zoledronic Acid, or Both to First-Line Long-Term Hormone Therapy in Prostate Cancer (STAMPEDE): Survival Results from an Adaptive, Multiarm, Multistage, Platform Randomised Controlled Trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; De Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.S.; Amos, C.L.; Gilson, C.; Jones, R.J.; et al. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N. Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef]
- Davis, I.D.; Martin, A.J.; Stockler, M.R.; Begbie, S.; Chi, K.N.; Chowdhury, S.; Coskinas, X.; Frydenberg, M.; Hague, W.E.; Horvath, L.G.; et al. Enzalutamide with Standard First-Line Therapy in Metastatic Prostate Cancer. N. Engl. J. Med. 2019, 381, 121–131. [Google Scholar] [CrossRef]
- Chi, K.N.; Agarwal, N.; Bjartell, A.; Chung, B.H.; Pereira de Santana Gomes, A.J.; Given, R.; Juárez Soto, Á.; Merseburger, A.S.; Özgüroğlu, M.; Uemura, H.; et al. Apalutamide for Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2019, 381, 13–24. [Google Scholar] [CrossRef]
- Fizazi, K.; Foulon, S.; Carles, J.; Roubaud, G.; McDermott, R.; Fléchon, A.; Tombal, B.; Supiot, S.; Berthold, D.; Ronchin, P.; et al. Abiraterone plus Prednisone Added to Androgen Deprivation Therapy and Docetaxel in de Novo Metastatic Castration-Sensitive Prostate Cancer (PEACE-1): A Multicentre, Open-Label, Randomised, Phase 3 Study with a 2 × 2 Factorial Design. Lancet 2022, 399, 1695–1707. [Google Scholar] [CrossRef]
- Smith, M.R.; Hussain, M.; Saad, F.; Fizazi, K.; Sternberg, C.N.; Crawford, E.D.; Kopyltsov, E.; Park, C.H.; Alekseev, B.; Montesa-Pino, Á.; et al. Darolutamide and Survival in Metastatic, Hormone-Sensitive Prostate Cancer. N. Engl. J. Med. 2022, 386, 1132–1142. [Google Scholar] [CrossRef]
- Aparicio, A.M.; Shen, L.; Tapia, E.L.N.; Lu, J.-F.; Chen, H.-C.; Zhang, J.; Wu, G.; Wang, X.; Troncoso, P.; Corn, P.; et al. Combined Tumor Suppressor Defects Characterize Clinically Defined Aggressive Variant Prostate Cancers. Clin. Cancer Res. 2016, 22, 1520–1530. [Google Scholar] [CrossRef]
- Jiménez, N.; Garcia De Herreros, M.; Reig, Ò.; Marín-Aguilera, M.; Aversa, C.; Ferrer-Mileo, L.; García-Esteve, S.; Rodríguez-Carunchio, L.; Trias, I.; Font, A.; et al. Development and Independent Validation of a Prognostic Gene Expression Signature Based on RB1, PTEN, and TP53 in Metastatic Hormone-Sensitive Prostate Cancer Patients. Eur. Urol. Oncol. 2024, S2588931124000257. [Google Scholar] [CrossRef]
- Truqap Combination in PTEN-Deficient Metastatic Hormone-Sensitive Prostate Cancer Demonstrated Statistically Significant and Clinically Meaningful Improvement in Radiographic Progression-Free Survival in CAPItello-281 Phase III Trial; News Release; AstraZeneca: Cambridge, UK, 2024.
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-Kinase Pathway in Cancer. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-R.; Chen, M.; Pandolfi, P.P. The Functions and Regulation of the PTEN Tumour Suppressor: New Modes and Prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical Implications of PTEN Loss in Prostate Cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef]
- Tortorella, E.; Giantulli, S.; Sciarra, A.; Silvestri, I. AR and PI3K/AKT in Prostate Cancer: A Tale of Two Interconnected Pathways. Int. J. Mol. Sci. 2023, 24, 2046. [Google Scholar] [CrossRef]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef]
- Velez, M.G.; Kosiorek, H.E.; Egan, J.B.; McNatty, A.L.; Riaz, I.B.; Hwang, S.R.; Stewart, G.A.; Ho, T.H.; Moore, C.N.; Singh, P.; et al. Differential Impact of Tumor Suppressor Gene (TP53, PTEN, RB1) Alterations and Treatment Outcomes in Metastatic, Hormone-Sensitive Prostate Cancer. Prostate Cancer Prostatic Dis. 2022, 25, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Ferraldeschi, R.; Nava Rodrigues, D.; Riisnaes, R.; Miranda, S.; Figueiredo, I.; Rescigno, P.; Ravi, P.; Pezaro, C.; Omlin, A.; Lorente, D.; et al. PTEN Protein Loss and Clinical Outcome from Castration-Resistant Prostate Cancer Treated with Abiraterone Acetate. Eur. Urol. 2015, 67, 795–802. [Google Scholar] [CrossRef]
- Hamid, A.A.; Huang, H.-C.; Wang, V.; Chen, Y.-H.; Feng, F.; Den, R.; Attard, G.; Van Allen, E.M.; Tran, P.T.; Spratt, D.E.; et al. Transcriptional Profiling of Primary Prostate Tumor in Metastatic Hormone-Sensitive Prostate Cancer and Association with Clinical Outcomes: Correlative Analysis of the E3805 CHAARTED Trial. Ann. Oncol. 2021, 32, 1157–1166. [Google Scholar] [CrossRef]
- Soundararajan, R.; Viscuse, P.; Pilie, P.; Liu, J.; Logotheti, S.; Laberiano Fernández, C.; Lorenzini, D.; Hoang, A.; Lu, W.; Soto, L.M.S.; et al. Genotype-to-Phenotype Associations in the Aggressive Variant Prostate Cancer Molecular Profile (AVPC-m) Components. Cancers 2022, 14, 3233. [Google Scholar] [CrossRef]
- Parry, M.A.; Grist, E.; Mendes, L.; Dutey-Magni, P.; Sachdeva, A.; Brawley, C.; Murphy, L.; Proudfoot, J.; Lall, S.; Liu, Y.; et al. Clinical Testing of Transcriptome-Wide Expression Profiles in High-Risk Localized and Metastatic Prostate Cancer Starting Androgen Deprivation Therapy: An Ancillary Study of the STAMPEDE Abiraterone Phase 3 Trial. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Cetintas, V.B.; Batada, N.N. Is There a Causal Link between PTEN Deficient Tumors and Immunosuppressive Tumor Microenvironment? J. Transl. Med. 2020, 18, 45. [Google Scholar] [CrossRef] [PubMed]
- Imada, E.L.; Sanchez, D.F.; Dinalankara, W.; Vidotto, T.; Ebot, E.M.; Tyekucheva, S.; Franco, G.R.; Mucci, L.A.; Loda, M.; Schaeffer, E.M.; et al. Transcriptional Landscape of PTEN Loss in Primary Prostate Cancer. BMC Cancer 2021, 21, 856. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Prat, A.; Parker, J.S.; Liu, Y.; Carey, L.A.; Troester, M.A.; Perou, C.M. Building Prognostic Models for Breast Cancer Patients Using Clinical Variables and Hundreds of Gene Expression Signatures. BMC Med. Genom. 2011, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zeng, Y.; Han, Z.; Zhuo, Y.; Liang, Y.; Hon, C.T.; Wan, S.; Wu, S.; Dahl, D.; Zhong, W.; et al. Novel Immune-Related Signature for Risk Stratification and Prognosis in Prostatic Adenocarcinoma. Cancer Sci. 2021, 112, 4365–4376. [Google Scholar] [CrossRef]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.S.K.; Varambally, S.; et al. Divergent Clonal Evolution of Castration-Resistant Neuroendocrine Prostate Cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef]
- Yang, R.; Wang, M.; Zhang, G.; Bao, Y.; Wu, Y.; Li, X.; Yang, W.; Cui, H. E2F7−EZH2 Axis Regulates PTEN/AKT/mTOR Signalling and Glioblastoma Progression. Br. J. Cancer 2020, 123, 1445–1455. [Google Scholar] [CrossRef]
- Saal, L.H.; Johansson, P.; Holm, K.; Gruvberger-Saal, S.K.; She, Q.-B.; Maurer, M.; Koujak, S.; Ferrando, A.A.; Malmström, P.; Memeo, L.; et al. Poor Prognosis in Carcinoma Is Associated with a Gene Expression Signature of Aberrant PTEN Tumor Suppressor Pathway Activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7564–7569. [Google Scholar] [CrossRef]
- Liu, D.; Augello, M.A.; Grbesa, I.; Prandi, D.; Liu, Y.; Shoag, J.E.; Karnes, R.J.; Trock, B.J.; Klein, E.A.; Den, R.B.; et al. Tumor Subtype Defines Distinct Pathways of Molecular and Clinical Progression in Primary Prostate Cancer. J. Clin. Investig. 2021, 131, e147878. [Google Scholar] [CrossRef]
- Liao, C.-H.; Wang, Y.-H.; Chang, W.-W.; Yang, B.-C.; Wu, T.-J.; Liu, W.-L.; Yu, A.L.; Yu, J. Leucine-Rich Repeat Neuronal Protein 1 Regulates Differentiation of Embryonic Stem Cells by Post-Translational Modifications of Pluripotency Factors. Stem Cells 2018, 36, 1514–1524. [Google Scholar] [CrossRef]
- Quintanal-Villalonga, A.; Kawasaki, K.; Redin, E.; Uddin, F.; Rakhade, S.; Durani, V.; Sabet, A.; Shafer, M.; Karthaus, W.R.; Zaidi, S.; et al. CDC7 Inhibition Impairs Neuroendocrine Transformation in Lung and Prostate Tumors through MYC Degradation. Signal Transduct. Target. Ther. 2024, 9, 1–14. [Google Scholar] [CrossRef]
- Frazzi, R. BIRC3 and BIRC5: Multi-faceted Inhibitors in Cancer. Cell Biosci. 2021, 11, 8. [Google Scholar] [CrossRef]
- Yu, T.; Liu, Z.; Tao, Q.; Xu, X.; Li, X.; Li, Y.; Chen, M.; Liu, R.; Chen, D.; Wu, M.; et al. Targeting Tumor-Intrinsic SLC16A3 to Enhance Anti-PD-1 Efficacy via Tumor Immune Microenvironment Reprogramming. Cancer Lett. 2024, 589, 216824. [Google Scholar] [CrossRef] [PubMed]
- Coleman, N.; Moyers, J.T.; Harbery, A.; Vivanco, I.; Yap, T.A. Clinical Development of AKT Inhibitors and Associated Predictive Biomarkers to Guide Patient Treatment in Cancer Medicine. Pharmacogenom. Pers. Med. 2021, 14, 1517–1535. [Google Scholar] [CrossRef]
- Carceles-Cordon, M.; Orme, J.J.; Domingo-Domenech, J.; Rodriguez-Bravo, V. The Yin and Yang of Chromosomal Instability in Prostate Cancer. Nat. Rev. Urol. 2024, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Al-Subhi, N.; Ali, R.; Abdel-Fatah, T.; Moseley, P.M.; Chan, S.Y.T.; Green, A.R.; Ellis, I.O.; Rakha, E.A.; Madhusudan, S. Targeting Ataxia Telangiectasia-Mutated- and Rad3-Related Kinase (ATR) in PTEN-Deficient Breast Cancers for Personalized Therapy. Breast Cancer Res. Treat. 2018, 169, 277–286. [Google Scholar] [CrossRef]
- Kim, O.; Butler, M.; Sergi, Z.; Robey, R.W.; Zhang, M.; Chari, R.; Pang, Y.; Yu, G.; Zhang, W.; Song, H.; et al. Combined Inhibition of Topoisomerase I and Poly(ADP-Ribose) Polymerase: A Synergistic Therapeutic Strategy for Glioblastoma with Phosphatase and Tensin Homolog Deficiency. Neurooncol. Adv. 2023, 5, vdad102. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, C.; Bracarda, S.; Sternberg, C.N.; Chi, K.N.; Olmos, D.; Sandhu, S.; Massard, C.; Matsubara, N.; Alekseev, B.; Parnis, F.; et al. Ipatasertib plus Abiraterone and Prednisolone in Metastatic Castration-Resistant Prostate Cancer (IPATential150): A Multicentre, Randomised, Double-Blind, Phase 3 Trial. Lancet 2021, 398, 131–142. [Google Scholar] [CrossRef]
- Fizazi, K.; George, D.J.; De Santis, M.; Clarke, N.; Fay, A.P.; Uemura, H.; Grinsted, L.; Rooney, C.; Verheijen, R.B.; Anjum, R.; et al. A Phase III Trial of Capivasertib and Abiraterone versus Placebo and Abiraterone in Patients with de Novo Metastatic Hormone-Sensitive Prostate Cancer Characterized by PTEN Deficiency (CAPItello-281). J. Clin. Oncol. 2021, 39, TPS178. [Google Scholar] [CrossRef]
- Chen, J.; Shi, M.; Chuen Choi, S.Y.; Wang, Y.; Lin, D.; Zeng, H.; Wang, Y. Genomic Alterations in Neuroendocrine Prostate Cancer: A Systematic Review and Meta-analysis. BJUI Compass 2023, 4, 256–265. [Google Scholar] [CrossRef]
- Duan, R.; Du, W.; Guo, W. EZH2: A Novel Target for Cancer Treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef]
- Calvo, M.; Penkov, K.; Spira, A.I.; Moreno Candilejo, I.; Shore, N.D.; Zhang, T.; Mellado-Gonzalez, B.; Alonso Gordoa, T.; Paz-Ares Rodriguez, L.; Tarantolo, S.R.; et al. A Multi-Center, Open-Label, Randomized Dose Expansion Study of PF-06821497, a Potent and Selective Inhibitor of Enhancer of Zeste Homolog 2 (EZH2), in Patients with Metastatic Castration-Resistant Prostate Cancer (mCRPC). J. Clin. Oncol. 2023, 41, TPS282. [Google Scholar] [CrossRef]
- Schweizer, M.T.; Penkov, K.; Choudhury, A.D.; Calvo, E.; Frank, R.C.; Liu, L.; Mittapalli, R.K.; Tougias, J.; Andreu-Vieyra, C.; Bowler, T.G.; et al. Phase 1 Trial of Mevrometostat (PF-06821497), a Potent and Selective Inhibitor of Enhancer of Zeste Homolog 2 (EZH2), in Castration-Resistant Prostate Cancer (CRPC). J. Clin. Oncol. 2024, 42, 5061. [Google Scholar] [CrossRef]
- Vidotto, T.; Melo, C.M.; Lautert-Dutra, W.; Chaves, L.P.; Reis, R.B.; Squire, J.A. Pan-Cancer Genomic Analysis Shows Hemizygous PTEN Loss Tumors Are Associated with Immune Evasion and Poor Outcome. Sci. Rep. 2023, 13, 5049. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell–Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef]
- Li, S.; Zhu, M.; Pan, R.; Fang, T.; Cao, Y.-Y.; Chen, S.; Zhao, X.; Lei, C.-Q.; Guo, L.; Chen, Y.; et al. The Tumor Suppressor PTEN Has a Critical Role in Antiviral Innate Immunity. Nat. Immunol. 2016, 17, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Trigunaite, A.; Dimo, J.; Jørgensen, T.N. Suppressive Effects of Androgens on the Immune System. Cell. Immunol. 2015, 294, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, N.; Reig, Ò.; Marín-Aguilera, M.; Aversa, C.; Ferrer-Mileo, L.; Font, A.; Rodriguez-Vida, A.; Climent, M.Á.; Cros, S.; Chirivella, I.; et al. Transcriptional Profile Associated with Clinical Outcomes in Metastatic Hormone-Sensitive Prostate Cancer Treated with Androgen Deprivation and Docetaxel. Cancers 2022, 14, 4757. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Larionova, I.; Tashireva, L. Immune Gene Signatures as Prognostic Criteria for Cancer Patients. Ther. Adv. Med. Oncol. 2023, 15, 17588359231189436. [Google Scholar] [CrossRef]
- Scher, H.I.; Halabi, S.; Tannock, I.; Morris, M.; Sternberg, C.N.; Carducci, M.A.; Eisenberger, M.A.; Higano, C.; Bubley, G.J.; Dreicer, R.; et al. Design and End Points of Clinical Trials for Patients with Progressive Prostate Cancer and Castrate Levels of Testosterone: Recommendations of the Prostate Cancer Clinical Trials Working Group. J. Clin. Oncol. 2008, 26, 1148–1159. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Patients | PTENlow | PTENwt | p-Value | |
|---|---|---|---|---|
| Patients, n (%) | 355 | 100 (28.2) | 255 (71.8) * | |
| Age (years) | ||||
| Median (range) | 68.1 (46.3–92.8) | 69.7 (47.7–86.5) | 67.8 (46.3–92.8) | 0.456 |
| PSA at diagnosis (ng/mL) | ||||
| Median (range) | 40.3 (0.02–7448) | 32.5 (1.8–4860) | 42 (0.02–7448) | 0.452 |
| ECOG performance status score, n (%) | ||||
| 0 | 135 (38) | 35 (35) | 100 (39.2) | 0.542 |
| 1 or 2 | 196 (55.2) | 61 (61) | 135 (52.9) | |
| NA | 24 (6.8) | 4 (4) | 20 (7.8) | |
| Stage at diagnosis, n (%) | ||||
| <IV | 70 (19.7) | 16 (16) | 54 (21.2) | 0.3 |
| IV | 271 (76.3) | 80 (80) | 191 (74.9) | |
| NA | 14 (3.9) | 4 (4) | 10 (3.9) | |
| Gleason sum at diagnosis, n (%) | ||||
| ≤7 | 84 (23.7) | 20 (20) | 64 (25.1) | 0.402 |
| ≥8 | 261 (73.5) | 76 (76) | 185 (72.5) | |
| NA | 10 (2.8) | 4 (4) | 6 (2.4) | |
| Presence of visceral metastases, n (%) | ||||
| Yes | 55 (15.5) | 20 (20) | 35 (13.7) | 0.142 |
| No | 296 (83.4) | 78 (78) | 218 (85.5) | |
| NA | 4 (1.1) | 2 (2) | 2 (0.8) | |
| Disease volume, n (%) | ||||
| High | 221 (62.3) | 64 (64) | 157 (61.6) | 0.537 |
| Low | 129 (36.6) | 33 (33) | 96 (37.6) | |
| NA | 5 (1.4) | 3 (3) | 2 (0.8) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia de Herreros, M.; Jiménez, N.; Padrosa, J.; Aversa, C.; Ferrer-Mileo, L.; García-Esteve, S.; Rodríguez-Carunchio, L.; Trias, I.; Fernández-Mañas, L.; Marín-Aguilera, M.; et al. Clinical and Transcriptomic Characterization of Metastatic Hormone-Sensitive Prostate Cancer Patients with Low PTEN Expression. Int. J. Mol. Sci. 2025, 26, 6244. https://doi.org/10.3390/ijms26136244
Garcia de Herreros M, Jiménez N, Padrosa J, Aversa C, Ferrer-Mileo L, García-Esteve S, Rodríguez-Carunchio L, Trias I, Fernández-Mañas L, Marín-Aguilera M, et al. Clinical and Transcriptomic Characterization of Metastatic Hormone-Sensitive Prostate Cancer Patients with Low PTEN Expression. International Journal of Molecular Sciences. 2025; 26(13):6244. https://doi.org/10.3390/ijms26136244
Chicago/Turabian StyleGarcia de Herreros, Marta, Natalia Jiménez, Joan Padrosa, Caterina Aversa, Laura Ferrer-Mileo, Samuel García-Esteve, Leonardo Rodríguez-Carunchio, Isabel Trias, Laia Fernández-Mañas, Mercedes Marín-Aguilera, and et al. 2025. "Clinical and Transcriptomic Characterization of Metastatic Hormone-Sensitive Prostate Cancer Patients with Low PTEN Expression" International Journal of Molecular Sciences 26, no. 13: 6244. https://doi.org/10.3390/ijms26136244
APA StyleGarcia de Herreros, M., Jiménez, N., Padrosa, J., Aversa, C., Ferrer-Mileo, L., García-Esteve, S., Rodríguez-Carunchio, L., Trias, I., Fernández-Mañas, L., Marín-Aguilera, M., Altamirano, M., Mazariegos, M., Font, A., Rodriguez-Vida, A., Climent, M. Á., Cros, S., Chirivella, I., Figols, M., Sala-González, N., ... Mellado, B. (2025). Clinical and Transcriptomic Characterization of Metastatic Hormone-Sensitive Prostate Cancer Patients with Low PTEN Expression. International Journal of Molecular Sciences, 26(13), 6244. https://doi.org/10.3390/ijms26136244