
Omics-Mediated Treatment for Advanced Prostate Cancer: Moving Towards Precision Oncology
 , , ,
, , ,  , , , , and
, , , , and
Abstract
1. Introduction
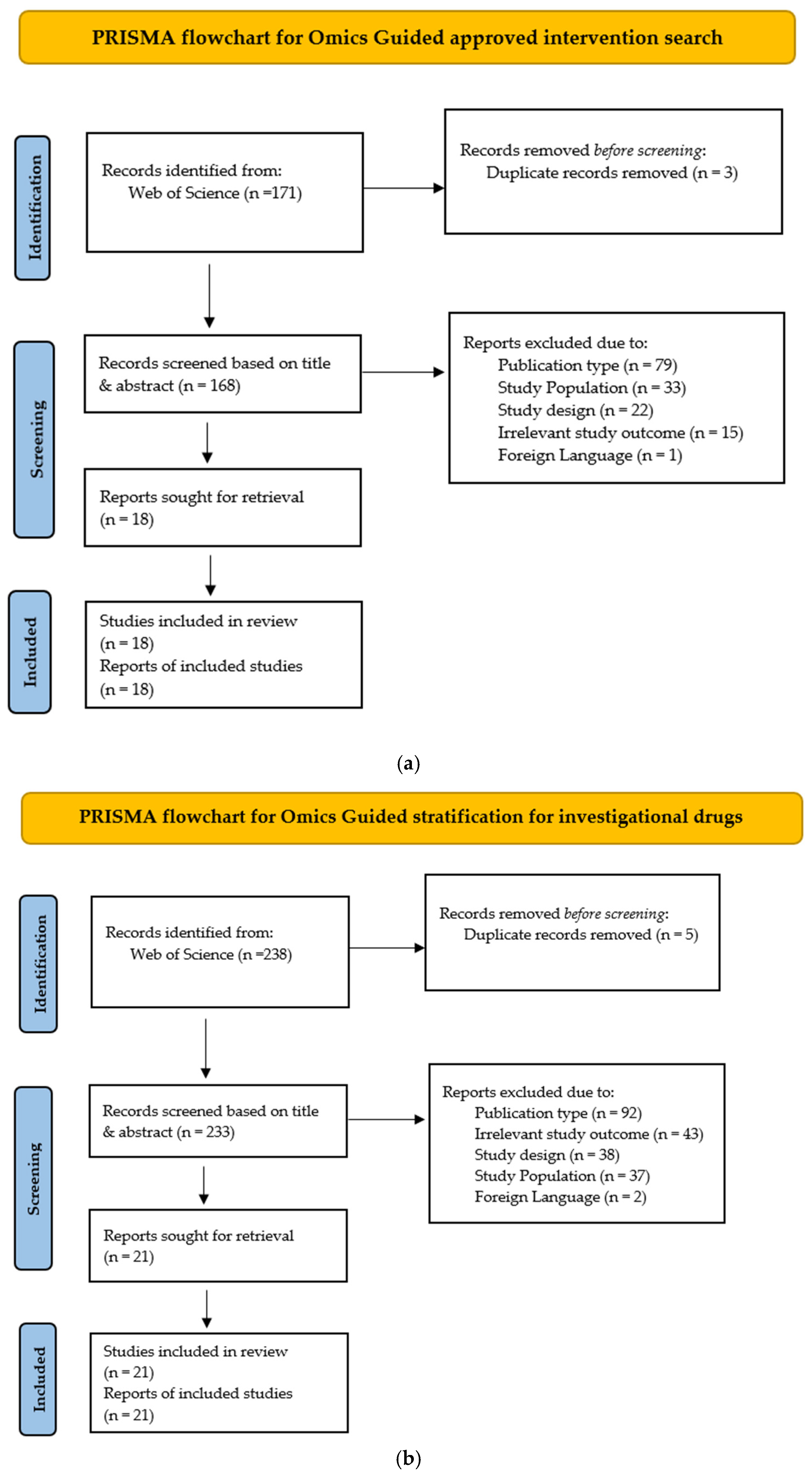
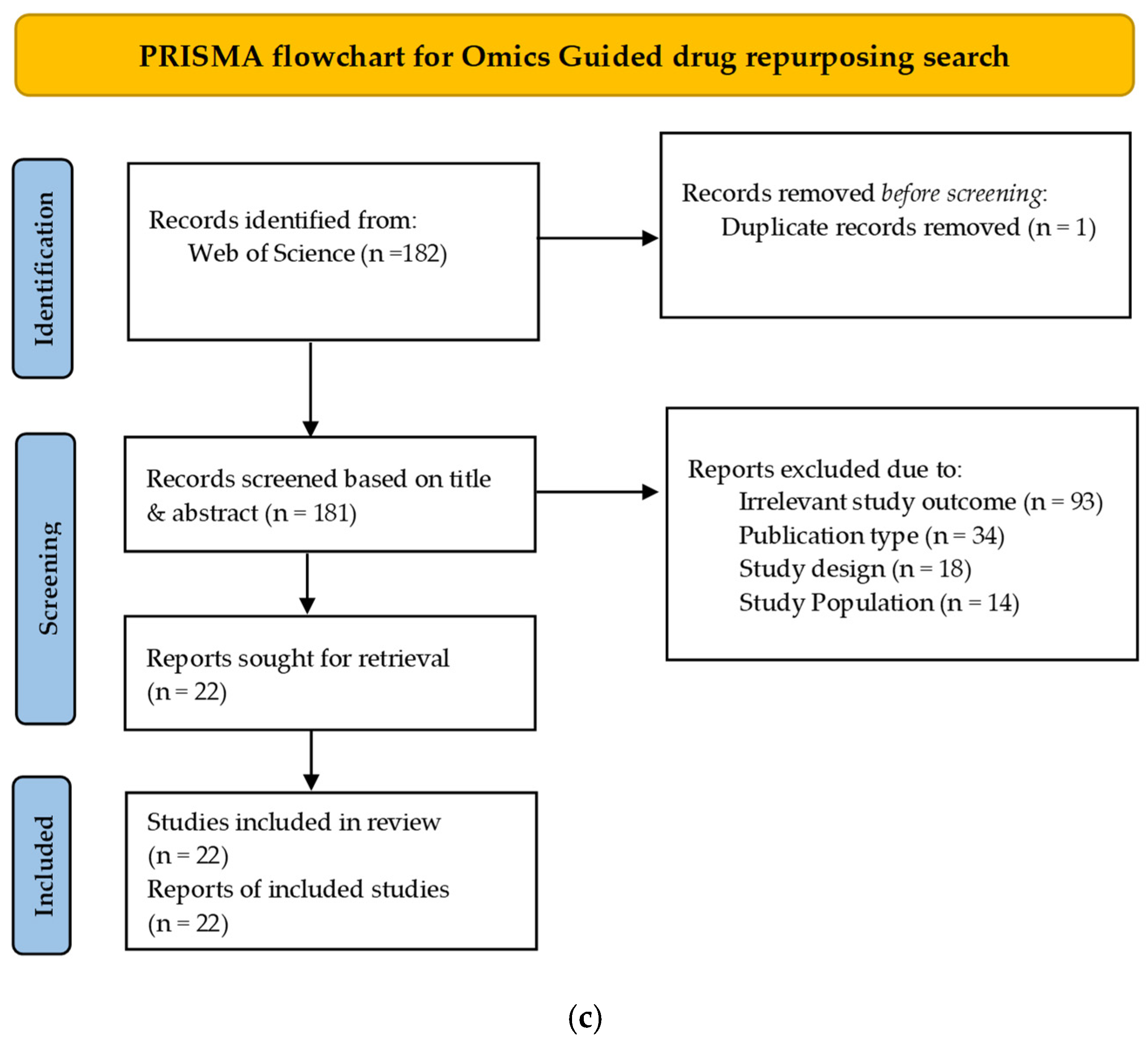
2. Methods
3. Results
3.1. Omics-Based Predictive Markers to Guide Approved Interventions in Advanced PCa
3.1.1. PARP Inhibitors
3.1.2. Combinatorial Therapies
3.1.3. Imaging-Based PSMA (Prostate-Specific Membrane Antigen) Directed Radioligand Therapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Combination/Dose | Molecular Requirement (Companion Diagnostic) | Pivotal Trial (s) | Key Treated Population (n) | Key Efficacy Outcome | p Value (Primary End-Point) | Notable Points |
|---|---|---|---|---|---|---|
| Lutetium-177 vipivotide tetraxetan (Pluvicto™; ^177Lu-PSMA-617) 7.4 GBq i.v. every 6 weeks × 6 cycles and best standard care vs. care alone [71] | PSMA-positive on ^68Ga-PSMA-11 PET/CT (≥1 lesion hotter than liver; no PSMA-negative lesion > 1 cm) | VISION (phase III) | Post-ARPI & taxane mCRPC; PSMA-PET-positive; n = 831 (551 vs. 280) | OS 15.3 vs. 11.3 mo (HR 0.62); rPFS 8.7 vs. 3.4 mo (HR 0.40) | OS p < 0.001; rPFS p < 0.001 | - FDA cleared Pluvicto™ + Locametz™ on 23 Mar 2022. ≥50% PSA fall by week 12 → longer OS & better QoL. - Baseline whole-body TLP outperforms SUVmax for OS prediction DDR-gene alterations not predictive. - Grade ≥ 3 AEs: 52% vs. 38% (xerostomia, nausea, anaemia). |
| Lutetium-177 vipivotide tetraxetan (Pluvicto™; ^177Lu-PSMA-617) 7.4 GBq i.v. every 6 weeks × 6 cycles with best standard care vs. change of ARPI (abiraterone and enzalutamide alternative switching) [72] | PSMA-positive on ^68Ga-PSMA-11 PET/CT (≥1 lesion hotter than liver, no PSMA-negative lesion > 1 cm) | PSMAfore (phase III) | Post-ARPI, taxane-naïve mCRPC; PSMA-PET-positive; n = 468 (234 vs. 234) | Primary rPFS 9.30 vs. 5.55 mo—HR 0.41 (95% CI 0.29–0.56) | p < 0.0001 (two-sided) | - 57% of control pts crossed over to RLT, diluting OS signal. - Updated rPFS at 3rd cut: 11.6 vs. 5.6 mo—HR 0.49. - Interim OS: 23.66 vs. 23.85 mo—HR 0.98, p = 0.44. - Grade ≥ 3 AEs: 36% vs. 48% (lute-177 vs. ARPI). - Improved time to pain/QoL deterioration relative to ARPI switch. |
3.2. Stratification Omics-Based Markers Used in Active Clinical Trials
3.2.1. AKT/PI3K Pathway Trial for PTEN Deficient Disease
3.2.2. Epigenomics Assays to Stratify Patients Before Receiving Chemotherapy
3.2.3. Lipidomics-Based Stratification of mCRPC Patients to Receive a PCSK9 Inhibitor
3.2.4. Stratification Based on Immunogenic Scores and TME Signatures
3.2.5. Transcriptomic Stratification in the CHAARTED Trial
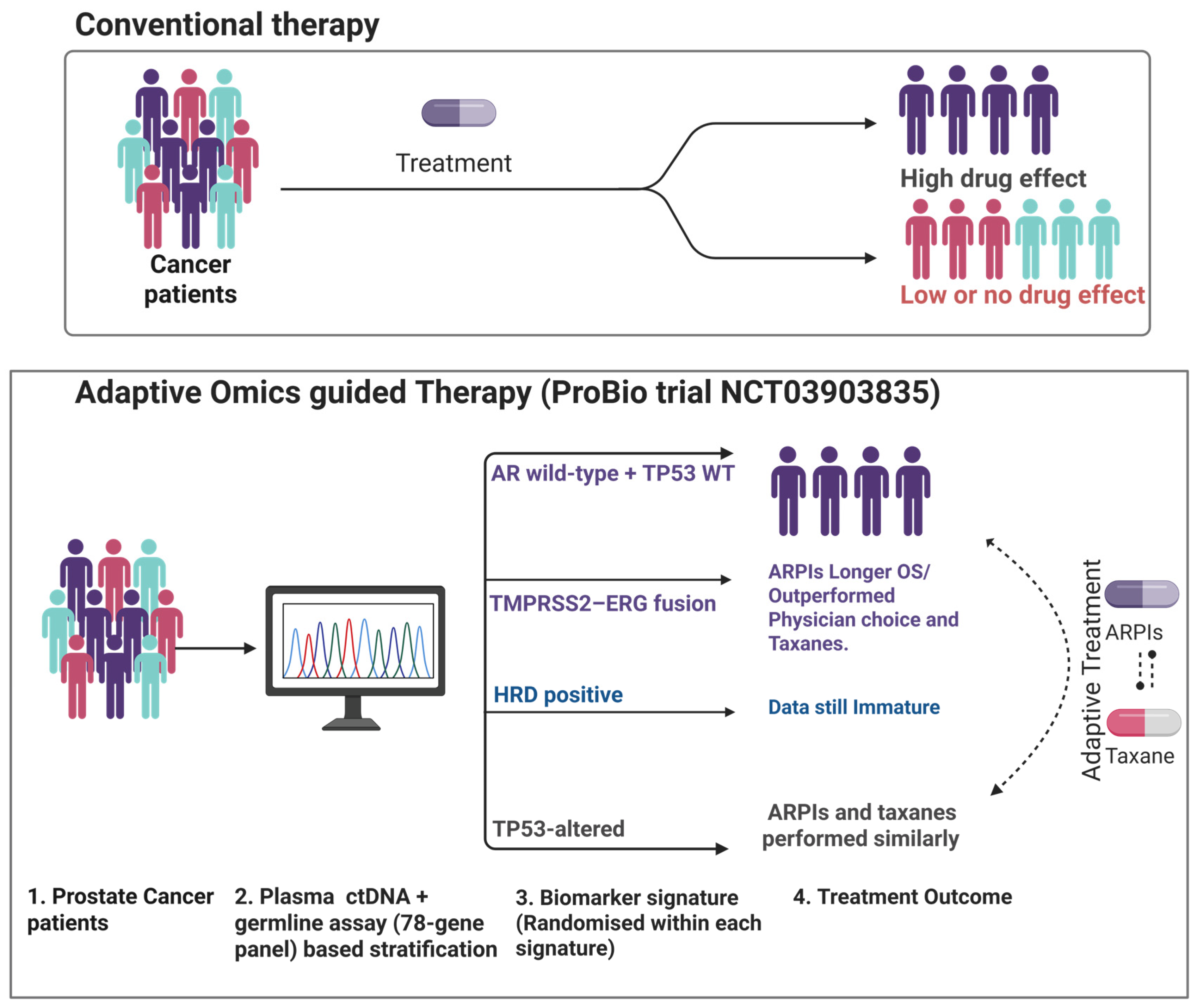
3.2.6. Adaptive Omics-Guided Therapy Based on Multiple Alterations
3.3. Miscellaneous FDA-Approved Agents with Exploratory Omics Markers
3.4. Prospective Trials for Predictive and Prognostic Markers in Advanced PCa
3.5. Omics Driven Drug Development: Drug Repurposing Paradigm
3.5.1. Genomics/Pharmacogenomics-Based Repurposing
3.5.2. Transcriptomic Signature & Network-Based Repurposing
3.5.3. Multi-Omics/AI and Network-Level Drug Prediction
4. Discussion
5. Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AEs | adverse events |
| ARPI | androgen-receptor-pathway inhibitor |
| ASCO-GU | American Society of Clinical Oncology Genitourinary Cancers Symposium |
| ATM | ataxia-telangiectasia mutated |
| BRCA1/2 | BReast CAncer susceptibility genes 1 and 2 |
| CDx | companion diagnostic |
| CT | computed tomography |
| DDR | DNA-damage repair |
| FDA | Food and Drug Administration |
| GBq | gigabecquerel |
| HR | hazard ratio |
| HRR | homologous-recombination repair |
| ITT | intention-to-treat |
| mCRPC | metastatic castration-resistant prostate cancer |
| NE | not estimable |
| NGS | next-generation sequencing |
| N | number of patients |
| OS | overall survival |
| Pred | prednisone/prednisolone |
| PSMA | prostate-specific membrane antigen |
| QoL | quality of life |
| RECIST | Response Evaluation Criteria in Solid Tumors |
| rPFS | radiographic progression-free survival |
| STR | survival-time ratio |
| SUVmax | maximum standardised uptake value |
| TLP | total-lesion PSMA |
| ^177Lu | lutetium-177 |
| ^68Ga | gallium-68 |
| bid | bis in die (twice daily) |
| i.v. | intravenous |
| AAP | abiraterone acetate + prednisone/prednisolone |
| ACTRN12622001003763 | Australian New Zealand Clinical Trials Registry identifier |
| ADT | androgen-deprivation therapy |
| APC | Advanced Prostate Cancer (context: transcriptomic signature study) |
| APCCC | Advanced Prostate Cancer Consensus Conference |
| AR | androgen receptor |
| AR-A | androgen-receptor-activity (9-gene score) |
| ARSI | androgen receptor signalling inhibitor |
| CMLHMS | Comprehensive Machine-Learning Histone-Modification Score |
| CNA | copy-number alteration |
| CRPC | castration-resistant prostate cancer |
| CSPC | castration-sensitive prostate cancer |
| ctDNA | circulating tumor DNA |
| CTC | circulating tumor cell |
| ENZA | enzalutamide |
| ECOG | Eastern Co-operative Oncology Group (performance status) |
| GC | Decipher genomic classifier (22-gene) |
| GMT | genetically matched therapy |
| GR | glucocorticoid receptor |
| HRD | homologous-recombination deficiency |
| ICI | immune-checkpoint inhibitor |
| IFN | interferon |
| ImS+ | immunogenic-signature–positive |
| IPI | ipilimumab |
| IPATential150 | phase III ipatasertib ± abiraterone trial |
| mCSPC | metastatic castration-sensitive prostate cancer |
| mHSPC | metastatic hormone-sensitive prostate cancer |
| MRI | magnetic-resonance imaging |
| NIVO | nivolumab |
| PARP | poly (ADP-ribose) polymerase |
| PCa | prostate cancer |
| PCPro | Prostate Cancer Prognostic (lipidomic) score |
| PFS | progression-free survival |
| PET | positron-emission tomography |
| PDOs | patient-derived organoids |
| Q4 | quartile 4 (highest Decipher risk group) |
| RRM2 | ribonucleotide-diphosphate reductase subunit M2 |
| RSS | replication-stress signature (47-gene) |
| TALA | talazoparib |
| TALAPRO-2 | phase III talazoparib + enzalutamide study |
| TIL | tumor-infiltrating lymphocyte |
| TME | tumor micro-environment |
| UCO | University of Cordoba |
| VISION | phase III ^177Lu-PSMA-617 trial |
| WT | wild type |
| (NCT numbers) | ClinicalTrials.gov study identifiers |
Appendix A
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA. Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van Der Kwast, T.; Bristow, R.G. Prostate Cancer. Nat. Rev. Dis. Primer 2021, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Gillessen, S.; Bossi, A.; Davis, I.D.; De Bono, J.; Fizazi, K.; James, N.D.; Mottet, N.; Shore, N.; Small, E.; Smith, M.; et al. Management of Patients with Advanced Prostate Cancer—Metastatic and/or Castration-Resistant Prostate Cancer: Report of the Advanced Prostate Cancer Consensus Conference (APCCC) 2022. Eur. J. Cancer 2023, 185, 178–215. [Google Scholar] [CrossRef]
- Tiwari, R.; Manzar, N.; Ateeq, B. Dynamics of Cellular Plasticity in Prostate Cancer Progression. Front. Mol. Biosci. 2020, 7, 130. [Google Scholar] [CrossRef]
- Varga, J.; Greten, F.R. Cell Plasticity in Epithelial Homeostasis and Tumorigenesis. Nat. Cell Biol. 2017, 19, 1133–1141. [Google Scholar] [CrossRef]
- Wasim, S.; Lee, S.-Y.; Kim, J. Complexities of Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 14257. [Google Scholar] [CrossRef]
- Varaprasad, G.L.; Gupta, V.K.; Prasad, K.; Kim, E.; Tej, M.B.; Mohanty, P.; Verma, H.K.; Raju, G.S.R.; Bhaskar, L.; Huh, Y.S. Recent Advances and Future Perspectives in the Therapeutics of Prostate Cancer. Exp. Hematol. Oncol. 2023, 12, 80. [Google Scholar] [CrossRef]
- Cooperberg, M.R.; Broering, J.M.; Carroll, P.R. Risk Assessment for Prostate Cancer Metastasis and Mortality at the Time of Diagnosis. JNCI J. Natl. Cancer Inst. 2009, 101, 878–887. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The Landscape of Somatic Copy-Number Alteration across Human Cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Frantzi, M.; Hupe, M.C.; Merseburger, A.S.; Schanstra, J.P.; Mischak, H.; Latosinska, A. Omics Derived Biomarkers and Novel Drug Targets for Improved Intervention in Advanced Prostate Cancer. Diagnostics 2020, 10, 658. [Google Scholar] [CrossRef]
- Mateo, J.; Seed, G.; Bertan, C.; Rescigno, P.; Dolling, D.; Figueiredo, I.; Miranda, S.; Nava Rodrigues, D.; Gurel, B.; Clarke, M.; et al. Genomics of Lethal Prostate Cancer at Diagnosis and Castration Resistance. J. Clin. Investig. 2020, 130, 1743–1751. [Google Scholar] [CrossRef]
- Mena, E.; Lindenberg, L.; Choyke, P. The Impact of PSMA PET/CT Imaging in Prostate Cancer Radiation Treatment. Semin. Nucl. Med. 2022, 52, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Gillessen, S.; Attard, G.; Beer, T.M.; Beltran, H.; Bjartell, A.; Bossi, A.; Briganti, A.; Bristow, R.G.; Chi, K.N.; Clarke, N.; et al. Management of Patients with Advanced Prostate Cancer: Report of the Advanced Prostate Cancer Consensus Conference 2019. Eur. Urol. 2020, 77, 508–547. [Google Scholar] [CrossRef] [PubMed]
- Lawhn-Heath, C.; Flavell, R.R.; Behr, S.C.; Yohannan, T.; Greene, K.L.; Feng, F.; Carroll, P.R.; Hope, T.A. Single-Center Prospective Evaluation of 68Ga-PSMA-11 PET in Biochemical Recurrence of Prostate Cancer. Am. J. Roentgenol. 2019, 213, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.; Cochlin, D.; Delahunt, B.; Kynaston, H.; Rees, J.; Rous, B.; Narahari, K. TNM Clinical Staging of Prostate Cancer: Issues and Solutions. BJU Int. 2019, 123, 382–384. [Google Scholar] [CrossRef]
- Horgan, R.P.; Kenny, L.C. ‘Omic’ Technologies: Genomics, Transcriptomics, Proteomics and Metabolomics. Obstet. Gynaecol. 2011, 13, 189–195. [Google Scholar] [CrossRef]
- Chen, C.; Cao, F.-H.; Li, Z.-G.; Zhang, L.-G.; Liu, J.; Chen, N.; Yao, A.-L.; Kang, S.-S.; Gao, W.-X.; Han, H.; et al. Bioinformatics Analysis of Differentially Expressed Proteins in Prostate Cancer Based on Proteomics Data. OncoTargets Ther. 2016, 9, 1545–1557. [Google Scholar] [CrossRef]
- Zhang, N.; Kandalai, S.; Zhou, X.; Hossain, F.; Zheng, Q. Applying Multi-omics toward Tumor Microbiome Research. iMeta 2023, 2, e73. [Google Scholar] [CrossRef]
- Cai, Z.; Jiang, Z.; Li, S.; Mo, S.; Wang, S.; Liang, M.; Tan, X.; Zhong, W.; Zhang, L.; Deng, J.; et al. RNA Modification Regulators’ Co-Expression Score (RMRCoeS) Predicts Biochemical Recurrence and Therapy Response in Prostate Cancer: A Multi-Omics and Experimental Validation Study. Int. Immunopharmacol. 2024, 139, 112723. [Google Scholar] [CrossRef]
- Torkamani, A.; Andersen, K.G.; Steinhubl, S.R.; Topol, E.J. High-Definition Medicine. Cell 2017, 170, 828–843. [Google Scholar] [CrossRef] [PubMed]
- Erho, N.; Crisan, A.; Vergara, I.A.; Mitra, A.P.; Ghadessi, M.; Buerki, C.; Bergstralh, E.J.; Kollmeyer, T.; Fink, S.; Haddad, Z.; et al. Discovery and Validation of a Prostate Cancer Genomic Classifier That Predicts Early Metastasis Following Radical Prostatectomy. PLoS ONE 2013, 8, e66855. [Google Scholar] [CrossRef] [PubMed]
- Cullen, J.; Rosner, I.L.; Brand, T.C.; Zhang, N.; Tsiatis, A.C.; Moncur, J.; Ali, A.; Chen, Y.; Knezevic, D.; Maddala, T.; et al. A Biopsy-Based 17-Gene Genomic Prostate Score Predicts Recurrence After Radical Prostatectomy and Adverse Surgical Pathology in a Racially Diverse Population of Men with Clinically Low- and Intermediate-Risk Prostate Cancer. Eur. Urol. 2015, 68, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J.; Swanson, G.P.; Fisher, G.; Brothman, A.R.; Berney, D.M.; Reid, J.E.; Mesher, D.; Speights, V.; Stankiewicz, E.; Foster, C.S.; et al. Prognostic Value of an RNA Expression Signature Derived from Cell Cycle Proliferation Genes in Patients with Prostate Cancer: A Retrospective Study. Lancet Oncol. 2011, 12, 245–255. [Google Scholar] [CrossRef]
- Cuzick, J.; Stone, S.; Fisher, G.; Yang, Z.H.; North, B.V.; Berney, D.M.; Beltran, L.; Greenberg, D.; Møller, H.; Reid, J.E.; et al. Validation of an RNA Cell Cycle Progression Score for Predicting Death from Prostate Cancer in a Conservatively Managed Needle Biopsy Cohort. Br. J. Cancer 2015, 113, 382–389. [Google Scholar] [CrossRef]
- Kreuz, M.; Otto, D.J.; Fuessel, S.; Blumert, C.; Bertram, C.; Bartsch, S.; Loeffler, D.; Puppel, S.-H.; Rade, M.; Buschmann, T.; et al. ProstaTrend—A Multivariable Prognostic RNA Expression Score for Aggressive Prostate Cancer. Eur. Urol. 2020, 78, 452–459. [Google Scholar] [CrossRef]
- Medori, M.; Micheletti, C.; Gadler, M. Omics Sciences and Precision Medicine in Prostate Cancer. Clin. Ter. 2023, 174, 95–103. [Google Scholar] [CrossRef]
- Pamarthy, S.; Sabaawy, H.E. Patient Derived Organoids in Prostate Cancer: Improving Therapeutic Efficacy in Precision Medicine. Mol. Cancer 2021, 20, 125. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Snyder, M.P. Integrative Omics for Health and Disease. Nat. Rev. Genet. 2018, 19, 299–310. [Google Scholar] [CrossRef]
- Olmos, D.; Lorente, D.; Jambrina, A.; Tello-Velasco, D.; Ovejero-Sánchez, M.; Gonzalez-Ginel, I.; Romero-Laorden, N.; Nunes-Carneiro, D.; Balongo, M.; Gutierrez-Pecharromán, A.M.; et al. BRCA1/2 and Homologous Recombination Repair Alterations in High- and Low-Volume Metastatic Hormone-Sensitive Prostate Cancer: Prevalence and Impact on Outcomes. Ann. Oncol. 2025, S0923753425007392. [Google Scholar] [CrossRef]
- Carneiro, B.A.; Lotan, T.L.; De Souza, A.; Aggarwal, R. Emerging Subtypes and New Treatments for Castration-Resistant Prostate Cancer. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, e319–e332. [Google Scholar] [CrossRef]
- Seed, G.; Beije, N.; Yuan, W.; Bertan, C.; Goodall, J.; Lundberg, A.; Tyler, M.; Figueiredo, I.; Pereira, R.; Baker, C.; et al. Elucidating Acquired PARP Inhibitor Resistance in Advanced Prostate Cancer. Cancer Cell 2024, 42, 2113–2123.e4. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.K.; Kosoff, D.; Emamekhoo, H.; Lang, J.M.; Kyriakopoulos, C.E. PARP Inhibitors in Metastatic Prostate Cancer. Front. Oncol. 2023, 13, 1159557. [Google Scholar] [CrossRef] [PubMed]
- Kwon, W.-A. PARP Inhibitors in the Treatment of Prostate Cancer: From Scientific Rationale to Clinical Development. World J. Men's Health 2024, 42, 290. [Google Scholar] [CrossRef] [PubMed]
- Cornford, P.; Bellmunt, J.; Bolla, M.; Briers, E.; De Santis, M.; Gross, T.; Henry, A.M.; Joniau, S.; Lam, T.B.; Mason, M.D.; et al. EAU-ESTRO-SIOG Guidelines on Prostate Cancer. Part II: Treatment of Relapsing, Metastatic, and Castration-Resistant Prostate Cancer. Eur. Urol. 2017, 71, 630–642. [Google Scholar] [CrossRef]
- Kwon, W.-A.; Joung, J.Y. Precision Targeting in Metastatic Prostate Cancer: Molecular Insights to Therapeutic Frontiers. Biomolecules 2025, 15, 625. [Google Scholar] [CrossRef]
- Sokolova, A.O.; Cheng, H.H. Genetic Testing in Prostate Cancer. Curr. Oncol. Rep. 2020, 22, 5. [Google Scholar] [CrossRef]
- Yan, L.; Su, P.; Sun, X. Role of Multi-omics in Advancing the Understanding and Treatment of Prostate Cancer (Review). Mol. Med. Rep. 2025, 31, 1–17. [Google Scholar] [CrossRef]
- Iacobas, S.; Iacobas, D.A. A Personalized Genomics Approach of the Prostate Cancer. Cells 2021, 10, 1644. [Google Scholar] [CrossRef]
- Lakshmanan, V.-K.; Ojha, S.; Jung, Y.D. A Modern Era of Personalized Medicine in the Diagnosis, Prognosis, and Treatment of Prostate Cancer. Comput. Biol. Med. 2020, 126, 104020. [Google Scholar] [CrossRef]
- Rayyan—A Web and Mobile App for Systematic Reviews | Systematic Reviews | Full Text. Available online: https://systematicreviewsjournal.biomedcentral.com/articles/10.1186/s13643-016-0384-4 (accessed on 19 June 2025).
- Tilki, D.; Van Den Bergh, R.C.N.; Briers, E.; Van Den Broeck, T.; Brunckhorst, O.; Darraugh, J.; Eberli, D.; De Meerleer, G.; De Santis, M.; Farolfi, A.; et al. EAU-EANM-ESTRO-ESUR-ISUP-SIOG Guidelines on Prostate Cancer. Part II—2024 Update: Treatment of Relapsing and Metastatic Prostate Cancer. Eur. Urol. 2024, 86, 164–182. [Google Scholar] [CrossRef]
- Companion Diagnostics | FDA. Available online: https://www.fda.gov/medical-devices/in-vitro-diagnostics/companion-diagnostics?utm_source (accessed on 30 May 2025).
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. FDA Approves Olaparib for HRR Gene-Mutated Metastatic Castration-Resistant Prostate Cancer. 19 May 2020. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-hrr-gene-mutated-metastatic-castration-resistant-prostate-cancer (accessed on 30 July 2025).
- Taza, F.; Holler, A.E.; Fu, W.; Wang, H.; Adra, N.; Albany, C.; Ashkar, R.; Cheng, H.H.; Sokolova, A.O.; Agarwal, N.; et al. Differential Activity of PARP Inhibitors in BRCA1- Versus BRCA2-Altered Metastatic Castration-Resistant Prostate Cancer. JCO Precis. Oncol. 2021, 5, 1200–1220. [Google Scholar] [CrossRef] [PubMed]
- Zurita, A.J.; Graf, R.P.; Villacampa, G.; Raskina, K.; Sokol, E.; Jin, D.; Antonarakis, E.S.; Li, G.; Huang, R.S.P.; Casanova-Salas, I.; et al. Genomic Biomarkers and Genome-Wide Loss-of-Heterozygosity Scores in Metastatic Prostate Cancer Following Progression on Androgen-Targeting Therapies. JCO Precis. Oncol. 2022, 6, e2200195. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef]
- Hussain, M.; Corcoran, C.; Sibilla, C.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Mateo, J.; Olmos, D.; Mehra, N.; et al. Tumor Genomic Testing for >4,000 Men with Metastatic Castration-Resistant Prostate Cancer in the Phase III Trial PROfound (Olaparib). Clin. Cancer Res. 2022, 28, 1518–1530. [Google Scholar] [CrossRef]
- Matsubara, N.; De Bono, J.; Olmos, D.; Procopio, G.; Kawakami, S.; Ürün, Y.; Van Alphen, R.; Flechon, A.; Carducci, M.A.; Choi, Y.D.; et al. Olaparib Efficacy in Patients with Metastatic Castration-Resistant Prostate Cancer and BRCA1, BRCA2, or ATM Alterations Identified by Testing Circulating Tumor DNA. Clin. Cancer Res. 2023, 29, 92–99. [Google Scholar] [CrossRef]
- Mateo, J.; De Bono, J.S.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Agarwal, N.; Olmos, D.; Thiery-Vuillemin, A.; et al. Olaparib for the Treatment of Patients With Metastatic Castration-Resistant Prostate Cancer and Alterations in BRCA1 and/or BRCA2 in the PROfound Trial. J. Clin. Oncol. 2024, 42, 571–583. [Google Scholar] [CrossRef]
- Triner, D.; Graf, R.P.; Madison, R.W.; Gjoerup, O.; Tukachinsky, H.; Ross, J.S.; Quintanilha, J.C.F.; Li, G.; Cheng, H.H.; Pritchard, C.C.; et al. Durable Benefit from Poly(ADP-Ribose) Polymerase Inhibitors in Metastatic Prostate Cancer in Routine Practice: Biomarker Associations and Implications for Optimal Clinical next-Generation Sequencing Testing. ESMO Open 2024, 9, 103684. [Google Scholar] [CrossRef]
- Loehr, A.; Hussain, A.; Patnaik, A.; Bryce, A.H.; Castellano, D.; Font, A.; Shapiro, J.; Zhang, J.; Sautois, B.; Vogelzang, N.J.; et al. Emergence of BRCA Reversion Mutations in Patients with Metastatic Castration-Resistant Prostate Cancer After Treatment with Rucaparib. Eur. Urol. 2023, 83, 200–209. [Google Scholar] [CrossRef]
- Loehr, A.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Response to Rucaparib in BRCA-Mutant Metastatic Castration-Resistant Prostate Cancer Identified by Genomic Testing in the TRITON2 Study. Clin. Cancer Res. 2021, 27, 6677–6686. [Google Scholar] [CrossRef] [PubMed]
- Anscher, M.S.; Chang, E.; Gao, X.; Gong, Y.; Weinstock, C.; Bloomquist, E.; Adeniyi, O.; Charlab, R.; Zimmerman, S.; Serlemitsos-Day, M.; et al. FDA Approval Summary: Rucaparib for the Treatment of Patients with Deleterious BRCA-Mutated Metastatic Castrate-Resistant Prostate Cancer. Oncologist 2021, 26, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Piulats, J.M.; Reaume, M.N.; Ostler, P.; McDermott, R.; Gingerich, J.R.; Pintus, E.; Sridhar, S.S.; Bambury, R.M.; Emmenegger, U.; et al. Rucaparib or Physician’s Choice in Metastatic Prostate Cancer. N. Engl. J. Med. 2023, 388, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhou, L.; Zhou, X.; Wei, Q.; Ouyang, N.; Shao, J.; Huang, J.; Liang, Z. Challenges in next Generation Sequencing of Homology Recombination Repair Genomic Variants in Prostate Cancer: A Nationwide Survey and Calibration Project in China. Prostate Int. 2022, 10, 181–187. [Google Scholar] [CrossRef]
- Schweizer, M.T.; Sivakumar, S.; Tukachinsky, H.; Coleman, I.; De Sarkar, N.; Yu, E.Y.; Konnick, E.Q.; Nelson, P.S.; Pritchard, C.C.; Montgomery, B. Concordance of DNA Repair Gene Mutations in Paired Primary Prostate Cancer Samples and Metastatic Tissue or Cell-Free DNA. JAMA Oncol. 2021, 7, 1378. [Google Scholar] [CrossRef]
- Fallah, J.; Xu, J.; Weinstock, C.; Brave, M.H.; Bloomquist, E.; Fiero, M.H.; Schaefer, T.; Pathak, A.; Abukhdeir, A.; Bhatnagar, V.; et al. FDA Approval Summary: Olaparib in Combination With Abiraterone for Treatment of Patients With BRCA-Mutated Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2024, 42, 605–613. [Google Scholar] [CrossRef]
- Clarke, N.W.; Armstrong, A.J.; Thiery-Vuillemin, A.; Oya, M.; Shore, N.; Loredo, E.; Procopio, G.; de Menezes, J.; Girotto, G.; Arslan, C.; et al. Abiraterone and Olaparib for Metastatic Castration-Resistant Prostate Cancer. NEJM Evid. 2022, 1, EVIDoa2200043. [Google Scholar] [CrossRef]
- Saad, F.; Armstrong, A.J.; Oya, M.; Vianna, K.; Özgüroğlu, M.; Gedye, C.; Buchschacher, G.L.; Lee, J.Y.; Emmenegger, U.; Navratil, J.; et al. Tolerability of Olaparib Combined with Abiraterone in Patients with Metastatic Castration-Resistant Prostate Cancer: Further Results from the Phase 3 PROpel Trial. Eur. Urol. Oncol. 2024, 7, 1394–1402. [Google Scholar] [CrossRef]
- Chi, K.N.; Sandhu, S.; Smith, M.R.; Attard, G.; Saad, M.; Olmos, D.; Castro, E.; Roubaud, G.; Pereira De Santana Gomes, A.J.; Small, E.J.; et al. Niraparib plus Abiraterone Acetate with Prednisone in Patients with Metastatic Castration-Resistant Prostate Cancer and Homologous Recombination Repair Gene Alterations: Second Interim Analysis of the Randomized Phase III MAGNITUDE Trial. Ann. Oncol. 2023, 34, 772–782. [Google Scholar] [CrossRef]
- Agarwal, N.; Azad, A.A.; Carles, J.; Fay, A.P.; Matsubara, N.; Heinrich, D.; Szczylik, C.; De Giorgi, U.; Young Joung, J.; Fong, P.C.C.; et al. Talazoparib plus Enzalutamide in Men with First-Line Metastatic Castration-Resistant Prostate Cancer (TALAPRO-2): A Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2023, 402, 291–303. [Google Scholar] [CrossRef]
- Agarwal, N.; Azad, A.; Carles, J.; Fay, A.P.; Matsubara, N.; Szczylik, C.; De Giorgi, U.; Joung, J.Y.; Fong, P.C.C.; Voog, E.; et al. Final overall survival (OS) with talazoparib (TALA) + enzalutamide (ENZA) as first-line treatment in unselected patients with metastatic castration-resistant prostate cancer (mCRPC) in the phase 3 TALAPRO-2 trial. J. Clin. Oncol. 2025, 43, LBA18. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network—Home. Available online: https://www.nccn.org (accessed on 18 July 2025).
- Food and Drug Administration. List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools). 5 March 2025. Available online: https://www.fda.gov/medical-devices/in-vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-in-vitro-and-imaging-tools (accessed on 30 July 2025).
- Serritella, A.V.; Taylor, A.; Haffner, M.C.; Abida, W.; Bryce, A.; Karsh, L.I.; Tagawa, S.T.; Twardowski, P.; Armstrong, A.J.; Lang, J.M. Therapeutic Implications of Homologous Repair Deficiency Testing in Patients with Prostate Cancer (Part 2 of 2). Prostate Cancer Prostatic Dis. 2024, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Madan, R.A.; Karzai, F.; VanderWeele, D.J.; Cheng, H.H.; de Bono, J.S. Poly(ADP-Ribose) Polymerase Inhibitor Combinations in First-Line Metastatic Castration-Resistant Prostate Cancer: Increasing Toxicity with Unclear Benefits. J. Clin. Oncol. 2023, 41, 5501–5504. [Google Scholar] [CrossRef] [PubMed]
- Bakht, M.K.; Beltran, H. Biological Determinants of PSMA Expression, Regulation and Heterogeneity in Prostate Cancer. Nat. Rev. Urol. 2025, 22, 26–45. [Google Scholar] [CrossRef]
- Bakht, M.K.; Yamada, Y.; Ku, S.-Y.; Venkadakrishnan, V.B.; Korsen, J.A.; Kalidindi, T.M.; Mizuno, K.; Ahn, S.H.; Seo, J.-H.; Garcia, M.M.; et al. Landscape of Prostate-Specific Membrane Antigen Heterogeneity and Regulation in AR-Positive and AR-Negative Metastatic Prostate Cancer. Nat. Cancer 2023, 4, 699–715. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Sartor, O.; De Bono, J.; Chi, K.; Fizazi, K.; Krause, B.J.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Saad, F.; et al. Association of Declining Prostate-Specific Antigen Levels with Clinical Outcomes in Patients with Metastatic Castration-Resistant Prostate Cancer Receiving [177Lu]Lu-PSMA-617 in the Phase 3 VISION Trial. Eur. Urol. 2024, 86, 552–562. [Google Scholar] [CrossRef]
- Morris, M.J.; Castellano, D.; Herrmann, K.; de Bono, J.S.; Shore, N.D.; Chi, K.N.; Crosby, M.; Piulats, J.M.; Fléchon, A.; Wei, X.X.; et al. 177Lu-PSMA-617 versus a change of androgen receptor pathway inhibitor therapy for taxane-naive patients with progressive metastatic castration-resistant prostate cancer (PSMAfore): A phase 3, randomised, controlled trial. Lancet 2024, 404, 1227–1239. [Google Scholar] [CrossRef]
- Rami, A.; Rashid, N.S.; Zhong, C.; Xie, W.; Stoltenberg, H.; Wheeler, E.J.; Wolanski, A.; Ritzer, J.; Choudhury, A.D.; Taplin, M.-E.; et al. Association between DNA damage repair alterations and outcomes to 177Lu-PSMA-617 in advanced prostate cancer. ESMO Open 2025, 10, 104131. [Google Scholar] [CrossRef]
- Hein, C.; Burgard, C.; Blickle, A.; Bastian, M.B.; Maus, S.; Schaefer-Schuler, A.; Hoffmann, M.A.; Schreckenberger, M.; Ezziddin, S.; Rosar, F. Analysis of Molecular Imaging and Laboratory Baseline Biomarkers in PSMA-RLT: Whole-Body Total Lesion PSMA (TLP) Predicts Overall Survival. Cancers 2024, 16, 2670. [Google Scholar] [CrossRef]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef]
- Beltran, H.; Hruszkewycz, A.; Scher, H.I.; Hildesheim, J.; Isaacs, J.; Yu, E.Y.; Kelly, K.; Lin, D.; Dicker, A.; Arnold, J.; et al. The Role of Lineage Plasticity in Prostate Cancer Therapy Resistance. Clin. Cancer Res. 2019, 25, 6916–6924. [Google Scholar] [CrossRef]
- Emmett, L.; Yin, C.; Crumbaker, M.; Hruby, G.; Kneebone, A.; Epstein, R.; Nguyen, Q.; Hickey, A.; Ihsheish, N.; O’Neill, G.; et al. Rapid Modulation of PSMA Expression by Androgen Deprivation: Serial 68Ga-PSMA-11 PET in Men with Hormone-Sensitive and Castrate-Resistant Prostate Cancer Commencing Androgen Blockade. J. Nucl. Med. 2019, 60, 950–954. [Google Scholar] [CrossRef]
- Li, E.V.; Schaeffer, E.M.; Ramesh Kumar, S.K.S.; Zhou, R.; Yang, X.J.; Mana-Ay, M.; Vescovo, M.; Ho, A.; Keeter, M.K.; Carr, J.; et al. Utility of 18F-DCFPyL PET for Local Staging for High or Very High Risk Prostate Cancer for Patients Undergoing Radical Prostatectomy. Eur. J. Nucl. Med. Mol. Imaging 2025, 52, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Bakht, M.K.; Lovnicki, J.M.; Tubman, J.; Stringer, K.F.; Chiaramonte, J.; Reynolds, M.R.; Derecichei, I.; Ferraiuolo, R.-M.; Fifield, B.-A.; Lubanska, D.; et al. Differential Expression of Glucose Transporters and Hexokinases in Prostate Cancer with a Neuroendocrine Gene Signature: A Mechanistic Perspective for 18F-FDG Imaging of PSMA-Suppressed Tumors. J. Nucl. Med. 2020, 61, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Kichloo, A.; Amir, R.; Aljadah, M.; Wani, F.; Solanki, S.; Singh, J.; Chugh, S.S. FDG-PET Versus PSMA-PET: A Patient With Prostate Cancer. J. Investig. Med. High Impact Case Rep. 2020, 8, 2324709620941313. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.R.; Rottey, S.; Bernard-Tessier, A.; Mellado-Gonzalez, B.; Kosaka, T.; Stadler, W.M.; Sandhu, S.; Yu, B.; Shaw, C.; Ju, C.-H.; et al. Phase 1b Study of Tarlatamab in de Novo or Treatment-Emergent Neuroendocrine Prostate Cancer (NEPC). J. Clin. Oncol. 2024, 42, 5012. [Google Scholar] [CrossRef]
- Verhoeven, M.; Ruigrok, E.A.M.; Van Leenders, G.J.L.H.; Van Den Brink, L.; Balcioglu, H.E.; Van Weerden, W.M.; Dalm, S.U. GRPR versus PSMA: Expression Profiles during Prostate Cancer Progression Demonstrate the Added Value of GRPR-Targeting Theranostic Approaches. Front. Oncol. 2023, 13, 1199432. [Google Scholar] [CrossRef]
- Jadvar, H. The VISION Forward: Recognition and Implication of PSMA−/18F-FDG + mCRPC. J. Nucl. Med. 2022, 63, 812–815. [Google Scholar] [CrossRef]
- Sweeney, C.; Bracarda, S.; Sternberg, C.N.; Chi, K.N.; Olmos, D.; Sandhu, S.; Massard, C.; Matsubara, N.; Alekseev, B.; Parnis, F.; et al. Ipatasertib plus Abiraterone and Prednisolone in Metastatic Castration-Resistant Prostate Cancer (IPATential150): A Multicentre, Randomised, Double-Blind, Phase 3 Trial. Lancet 2021, 398, 131–142. [Google Scholar] [CrossRef]
- Conduit, C.; Mak, B.; Qu, W.; Lulio, J.D.; Burder, R.; Bressel, M.; Cusick, T.; Dhillon, H.M.; Lourenço, R.D.A.; Underhill, C.; et al. GUIDE: A Randomised Non-Comparative Phase II Trial of Biomarker-Driven Intermittent Docetaxel versus Standard-of-Care Docetaxel in Metastatic Castration-Resistant Prostate Cancer (Clinical Trial Protocol). Ther. Adv. Med. Oncol. 2022, 14, 17588359221092486. [Google Scholar] [CrossRef]
- Australian and New Zealand Urogenital and Prostate Cancer Trials Group. A Phase II Trial of Biomarker-Driven Intermittent Docetaxel in Metastatic Castration-Resistant Prostate Cancer (mCRPC); Clinicaltrials.gov: Bethesda, MD, USA, 2024.
- Scheinberg, T.; Lin, H.-M.; Fitzpatrick, M.; Azad, A.A.; Bonnitcha, P.; Davies, A.; Heller, G.; Huynh, K.; Mak, B.; Mahon, K.; et al. PCPro: A Clinically Accessible, Circulating Lipid Biomarker Signature for Poor-Prognosis Metastatic Prostate Cancer. Prostate Cancer Prostatic Dis. 2024, 27, 136–143. [Google Scholar] [CrossRef]
- Mellor, R.; Ardolino, L.; Scheinberg, T.; Fitzpatrick, M.; Lin, H.-M.; Bonnitcha, P.; Sullivan, D.; Meikle, P.J.; Stockler, M.R.; Moujaber, T.; et al. Evolocumab in Metastatic Castration-Resistant Prostate Cancer: Study Protocol for a Single-Arm, Phase II Trial, and Initial Experience with Use of a Validated Lipid Biomarker to Direct Therapy. Ther. Adv. Med. Oncol. 2024, 16, 17588359241307814. [Google Scholar] [CrossRef]
- Linch, M.D.; Leone, G.; Wong, Y.N.S.; Jones, R.J.; Sankey, P.; Josephs, D.H.; Crabb, S.J.; Harris, L.; Tasnim, A.; Rashid, M.; et al. Nivolumab and Ipilimumab for Metastatic Prostate Cancer with an Immunogenic Signature: The NEPTUNES Multi-Centre Two-Cohort, Biomarker-Selected Phase 2 Trial. J. Clin. Oncol. 2024, 42, 5013. [Google Scholar] [CrossRef]
- Lim, E.A.; Bendell, J.C.; Falchook, G.S.; Bauer, T.M.; Drake, C.G.; Choe, J.H.; George, D.J.; Karlix, J.L.; Ulahannan, S.; Sachsenmeier, K.F.; et al. Phase Ia/b, Open-Label, Multicenter Study of AZD4635 (an Adenosine A2A Receptor Antagonist) as Monotherapy or Combined with Durvalumab, in Patients with Solid Tumors. Clin. Cancer Res. 2022, 28, 4871–4884. [Google Scholar] [CrossRef] [PubMed]
- Hamid, A.A.; Huang, H.-C.; Wang, V.; Chen, Y.-H.; Feng, F.; Den, R.; Attard, G.; Van Allen, E.M.; Tran, P.T.; Spratt, D.E.; et al. Transcriptional Profiling of Primary Prostate Tumor in Metastatic Hormone-Sensitive Prostate Cancer and Association with Clinical Outcomes: Correlative Analysis of the E3805 CHAARTED Trial. Ann. Oncol. 2021, 32, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- De Laere, B.; Crippa, A.; Discacciati, A.; Larsson, B.; Persson, M.; Johansson, S.; D’hondt, S.; Bergström, R.; Chellappa, V.; Mayrhofer, M.; et al. Androgen Receptor Pathway Inhibitors and Taxanes in Metastatic Prostate Cancer: An Outcome-Adaptive Randomized Platform Trial. Nat. Med. 2024, 30, 3291–3302. [Google Scholar] [CrossRef]
- Tolmeijer, S.H.; Boerrigter, E.; Sumiyoshi, T.; Kwan, E.M.; Ng, S.W.S.; Annala, M.; Donnellan, G.; Herberts, C.; Benoist, G.E.; Hamberg, P.; et al. Early On-Treatment Changes in Circulating Tumor DNA Fraction and Response to Enzalutamide or Abiraterone in Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2023, 29, 2835–2844. [Google Scholar] [CrossRef]
- Swami, U.; Graf, R.P.; Nussenzveig, R.H.; Fisher, V.; Tukachinsky, H.; Schrock, A.B.; Li, G.; Ross, J.S.; Sayegh, N.; Tripathi, N.; et al. SPOP Mutations as a Predictive Biomarker for Androgen Receptor Axis–Targeted Therapy in De Novo Metastatic Castration-Sensitive Prostate Cancer. Clin. Cancer Res. 2022, 28, 4917–4925. [Google Scholar] [CrossRef]
- Vellky, J.E.; Kirkpatrick, B.J.; Gutgesell, L.C.; Morales, M.; Brown, R.M.; Wu, Y.; Maienschein-Cline, M.; Notardonato, L.D.; Weinfeld, M.S.; Nguyen, R.H.; et al. ERBB3 Overexpression Is Enriched in Diverse Patient Populations with Castration-Sensitive Prostate Cancer and Is Associated with a Unique AR Activity Signature. Clin. Cancer Res. 2024, 30, 1530–1543. [Google Scholar] [CrossRef]
- Brown, M.C.; D’Anniballe, V.M.; Boczkowski, D.; Kandadi, H.; Sheikh, N.; Kornahrens, W.; Heath, E.I.; Thakur, A.; Chen, W.; Lum, L.; et al. Peripheral Blood IFN Responses to Toll-Like Receptor 1/2 Signaling Associate with Longer Survival in Men with Metastatic Prostate Cancer Treated with Sipuleucel-T. Cancer Res. Commun. 2024, 4, 2724–2733. [Google Scholar] [CrossRef]
- Liontos, M.; Goussia, A.; Korfiatis, N.; Papadopoulou, K.; Kanellis, G.; Visvikis, A.; Petrakis, G.; Tsiatas, M.; Fountzilas, E.; Samantas, E.; et al. The Role of Cabazitaxel in Patients With Castration-Resistant and Osseous Metastases Prostate Cancer. A Hellenic Cooperative Oncology Group Phase II Study. Clin. Genitourin. Cancer 2025, 23, 102253. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.Y.; Thomas, S.; Saad, F.; Gormley, M.; Yu, M.K.; Ricci, D.S.; Rooney, B.; Brookman-May, S.; McCarthy, S.; Olmos, D.; et al. Association of Molecular Subtypes With Differential Outcome to Apalutamide Treatment in Nonmetastatic Castration-Resistant Prostate Cancer. JAMA Oncol. 2021, 7, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- McKay, R.R.; Kwak, L.; Crowdis, J.P.; Sperger, J.M.; Zhao, S.G.; Xie, W.; Werner, L.; Lis, R.T.; Zhang, Z.; Wei, X.X.; et al. Phase II Multicenter Study of Enzalutamide in Metastatic Castration-Resistant Prostate Cancer to Identify Mechanisms Driving Resistance. Clin. Cancer Res. 2021, 27, 3610–3619. [Google Scholar] [CrossRef] [PubMed]
- Slootbeek, P.H.J.; Kloots, I.S.H.; Smits, M.; Van Oort, I.M.; Gerritsen, W.R.; Schalken, J.A.; Ligtenberg, M.J.L.; Grünberg, K.; Kroeze, L.I.; Bloemendal, H.J.; et al. Impact of Molecular Tumour Board Discussion on Targeted Therapy Allocation in Advanced Prostate Cancer. Br. J. Cancer 2022, 126, 907–916. [Google Scholar] [CrossRef]
- Parry, M.; Grist, E.; Brawley, C.; Proudfoot, J.A.; Mendes, L.; Lall, S.; Hoyle, A.P.; Sachdeva, A.; Liu, Y.; Amos, C.; et al. 1358O Clinical Qualification of Transcriptome Signatures for Advanced Prostate Cancer (APC) Starting Androgen Deprivation Therapy (ADT) with or without Abiraterone Acetate and Prednisolone (AAP): An Ancillary Study of the STAMPEDE AAP Trial. Ann. Oncol. 2022, 33, S1161. [Google Scholar] [CrossRef]
- Powles, T.; Yuen, K.C.; Gillessen, S.; Kadel, E.E.; Rathkopf, D.; Matsubara, N.; Drake, C.G.; Fizazi, K.; Piulats, J.M.; Wysocki, P.J.; et al. Atezolizumab with Enzalutamide versus Enzalutamide Alone in Metastatic Castration-Resistant Prostate Cancer: A Randomized Phase 3 Trial. Nat. Med. 2022, 28, 144–153. [Google Scholar] [CrossRef]
- Huang, J.; Du, M.; Soupir, A.; Wang, L.; Tan, W.; Kalari, K.R.; Kilari, D.; Park, J.; Huang, C.-C.; Kohli, M.; et al. Plasma Copy Number Alteration-Based Prognostic and Predictive Multi-Gene Risk Score in Metastatic Castration-Resistant Prostate Cancer. Cancers 2022, 14, 4714. [Google Scholar] [CrossRef]
- Shaya, J.; Nonato, T.; Cabal, A.; Randall, J.M.; Millard, F.; Stewart, T.; McKay, R.R. Analysis of the Prognostic Significance of Circulating Tumor DNA in Metastatic Castrate Resistant Prostate Cancer. Clin. Genitourin. Cancer 2021, 19, 564.e1–564.e10. [Google Scholar] [CrossRef]
- Jayaram, A.; Wingate, A.; Wetterskog, D.; Wheeler, G.; Sternberg, C.N.; Jones, R.; Berruti, A.; Lefresne, F.; Lahaye, M.; Thomas, S.; et al. Plasma Tumor Gene Conversions after One Cycle Abiraterone Acetate for Metastatic Castration-Resistant Prostate Cancer: A Biomarker Analysis of a Multicenter International Trial. Ann. Oncol. 2021, 32, 726–735. [Google Scholar] [CrossRef]
- Pan, J.; Zhao, J.; Ni, X.; Gan, H.; Wei, Y.; Wu, J.; Zhang, T.; Wang, Q.; Freedland, S.J.; Wang, B.; et al. The Prevalence and Prognosis of Next-generation Therapeutic Targets in Metastatic Castration-resistant Prostate Cancer. Mol. Oncol. 2022, 16, 4011–4022. [Google Scholar] [CrossRef]
- Zhu, X.; Farsh, T.; Vis, D.; Yu, I.; Li, H.; Liu, T.; Sjöström, M.; Shrestha, R.; Kneppers, J.; Severson, T.; et al. Genomic and Transcriptomic Features of Androgen Receptor Signaling Inhibitor Resistance in Metastatic Castration-Resistant Prostate Cancer. J. Clin. Investig. 2024, 134, e178604. [Google Scholar] [CrossRef]
- Raut, R.; Srivastava, D.; Nayak, V.; Saini, T.; Gupta, P.; Chakraborty, A.K.; Choudhury, C.; Bais, M.V.; Mishra, P.; Misra, A. Clinical Data Investigation Identifies MARK3 as an Oncogenic Driver in Castration-Resistant Prostate Cancer. Biochem. Biophys. Rep. 2025, 42, 102003. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, J.-M.; Mahammedi, H.; Gravis, G.; Roubaud, G.; Beuzeboc, P.; Largillier, R.; Borchiellini, D.; Linassier, C.; Ebran, N.; Pace-Loscos, T.; et al. Abigene, a Prospective, Multicentric Study of Abiraterone Acetate Pharmacogenetics in Metastatic Castration-Resistant Prostate Cancer. Pharmaceutics 2023, 15, 651. [Google Scholar] [CrossRef] [PubMed]
- Golla, R.; Jaiswal, S.; Jayan, A.; Cheemanapalli, S. Transcriptomic Analysis of Human Castration-Resistant Prostate Cancer: Insights into Novel Therapeutic Strategies. Comput. Biol. Chem. 2025, 118, 108459. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Jiang, Z.; Ma, T.; Li, J.; Chen, J.; Ye, P.; Feng, L. Integrating Transcriptomics and Network Analysis-Based Multiplexed Drug Repurposing to Screen Drug Candidates for M2 Macrophage-Associated Castration-Resistant Prostate Cancer Bone Metastases. Front. Immunol. 2022, 13, 989972. [Google Scholar] [CrossRef]
- Zhang, W.; Maeser, D.; Lee, A.; Huang, Y.; Gruener, R.F.; Abdelbar, I.G.; Jena, S.; Patel, A.G.; Huang, R.S. Integration of Pan-Cancer Cell Line and Single-Cell Transcriptomic Profiles Enables Inference of Therapeutic Vulnerabilities in Heterogeneous Tumors. Cancer Res. 2024, 84, 2021–2033. [Google Scholar] [CrossRef]
- Lee, D.; Lee, S.; Kim, Y.; Park, S.; Bae, S.-M.; Cho, E.; Park, E.-J.; Park, H.; Kim, S.-Y.; So, I.; et al. Cyclosporin A Inhibits Prostate Cancer Growth through Suppression of E2F8 Transcription Factor in a MELK-dependent Manner. Oncol. Rep. 2023, 50, 218. [Google Scholar] [CrossRef]
- Yan, Y.; Mao, X.; Zhang, Q.; Ye, Y.; Dai, Y.; Bao, M.; Zeng, Y.; Huang, R.; Mo, Z. Molecular Mechanisms, Immune Cell Infiltration, and Potential Drugs for Prostate Cancer. Cancer Biomark. 2021, 31, 87–96. [Google Scholar] [CrossRef]
- Hongo, H.; Kosaka, T.; Suzuki, Y.; Mikami, S.; Fukada, J.; Oya, M. Topoisomerase II Alpha Inhibition Can Overcome Taxane-Resistant Prostate Cancer through DNA Repair Pathways. Sci. Rep. 2021, 11, 22284. [Google Scholar] [CrossRef]
- Hongo, H.; Kosaka, T.; Suzuki, Y.; Oya, M. Discovery of a New Candidate Drug to Overcome Cabazitaxel-Resistant Gene Signature in Castration-Resistant Prostate Cancer by in Silico Screening. Prostate Cancer Prostatic Dis. 2023, 26, 59–66. [Google Scholar] [CrossRef]
- Hongo, H.; Kosaka, T.; Takayama, K.-I.; Baba, Y.; Yasumizu, Y.; Ueda, K.; Suzuki, Y.; Inoue, S.; Beltran, H.; Oya, M. G-Protein Signaling of Oxytocin Receptor as a Potential Target for Cabazitaxel-Resistant Prostate Cancer. PNAS Nexus 2023, 3, pgae002. [Google Scholar] [CrossRef]
- Huang, R.-H.; Hong, Y.-K.; Du, H.; Ke, W.-Q.; Lin, B.-B.; Li, Y.-L. A Machine Learning Framework Develops a DNA Replication Stress Model for Predicting Clinical Outcomes and Therapeutic Vulnerability in Primary Prostate Cancer. J. Transl. Med. 2023, 21, 20. [Google Scholar] [CrossRef]
- Puhr, M.; Eigentler, A.; Handle, F.; Hackl, H.; Ploner, C.; Heidegger, I.; Schaefer, G.; Brandt, M.P.; Hoefer, J.; Van Der Pluijm, G.; et al. Targeting the Glucocorticoid Receptor Signature Gene Mono Amine Oxidase-A Enhances the Efficacy of Chemo- and Anti-Androgen Therapy in Advanced Prostate Cancer. Oncogene 2021, 40, 3087–3100. [Google Scholar] [CrossRef]
- Huang, L.; Xie, Y.; Jiang, S.; Liu, K.; Ming, Z.; Shan, H. Elucidating the Role of Pyrimidine Metabolism in Prostate Cancer and Its Therapeutic Implications. Sci. Rep. 2025, 15, 2003. [Google Scholar] [CrossRef]
- Tian, S.; Liao, X.; Cao, W.; Wu, X.; Chen, Z.; Lu, J.; Wang, Q.; Zhang, J.; Chen, L.; Zhang, W. GSFM: A Genome-Scale Functional Module Transformation to Represent Drug Efficacy for in Silico Drug Discovery. Acta Pharm. Sin. B 2025, 15, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Granata, I.; Barboro, P. Identification of Molecular Markers Associated with Prostate Cancer Subtypes: An Integrative Bioinformatics Approach. Biomolecules 2024, 14, 87. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Ge, Q.; Zhao, W.; Yu, C.; Bai, H.; Wu, X.; Tao, J.; Xu, W.; Qiu, Y.; Chen, L.; et al. Integrative Multi-Omics Analysis and Machine Learning Refine Global Histone Modification Features in Prostate Cancer. Front. Mol. Biosci. 2025, 12, 1557843. [Google Scholar] [CrossRef] [PubMed]
- Bacolod, M.D.; Barany, F. A Unified Transcriptional, Pharmacogenomic, and Gene Dependency Approach to Decipher the Biology, Diagnostic Markers, and Therapeutic Targets Associated with Prostate Cancer Metastasis. Cancers 2021, 13, 5158. [Google Scholar] [CrossRef]
- Rydzewski, N.R.; Peterson, E.; Lang, J.M.; Yu, M.; Laura Chang, S.; Sjöström, M.; Bakhtiar, H.; Song, G.; Helzer, K.T.; Bootsma, M.L.; et al. Predicting Cancer Drug TARGETS—TreAtment Response Generalized Elastic-neT Signatures. NPJ Genom. Med. 2021, 6, 76. [Google Scholar] [CrossRef]
- Vasciaveo, A.; Arriaga, J.M.; De Almeida, F.N.; Zou, M.; Douglass, E.F.; Picech, F.; Shibata, M.; Rodriguez-Calero, A.; De Brot, S.; Mitrofanova, A.; et al. OncoLoop: A Network-Based Precision Cancer Medicine Framework. Cancer Discov. 2023, 13, 386–409. [Google Scholar] [CrossRef]
- Yadav, S.S.; Stockert, J.A.; Hackert, V.; Yadav, K.K.; Tewari, A.K. Intratumor Heterogeneity in Prostate Cancer. Urol. Oncol. Semin. Orig. Investig. 2018, 36, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Wang, L.; Khaki, A.R.; Jimenez, R.E.; Mauer, E.; Stoppler, M.C.; Chao, C.Y.; Kohli, M. Homologous Recombination Repair (HRR) Mutation Concordance between Liquid Biopsy (LB) and Tumor Tissue by NGS in a Real-World Prostate Cancer (PC) Database. J. Clin. Oncol. 2023, 41, 260. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Taylor, A.; Haffner, M.C.; Abida, W.; Bryce, A.H.; Karsh, L.I.; Tagawa, S.T.; Twardowski, P.; Serritella, A.V.; Lang, J.M. Germline and Somatic Testing for Homologous Repair Deficiency in Patients with Prostate Cancer (Part 1 of 2). Prostate Cancer Prostatic Dis. 2024, 27, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cyrta, J.; Prandi, D.; Arora, A.; Hovelson, D.H.; Sboner, A.; Rodriguez, A.; Fedrizzi, T.; Beltran, H.; Robinson, D.R.; Gopalan, A.; et al. Comparative Genomics of Primary Prostate Cancer and Paired Metastases: Insights from 12 Molecular Case Studies. J. Pathol. 2022, 257, 274–284. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Wang, Y.; Zhu, F.; Shi, H.; Zhang, J.; Wang, Z.; Qu, M.; Zhang, H.; Wang, T.; et al. Intratumor Heterogeneity and Clonal Evolution Revealed in Castration-Resistant Prostate Cancer by Longitudinal Genomic Analysis. Transl. Oncol. 2022, 16, 101311. [Google Scholar] [CrossRef]
- Fan, Y.; Liu, Z.; Chen, Y.; He, Z. Homologous Recombination Repair Gene Mutations in Prostate Cancer: Prevalence and Clinical Value. Adv. Ther. 2024, 41, 2196–2216. [Google Scholar] [CrossRef]
- Vescovo, M.; Raspollini, M.R.; Nibid, L.; Castiglione, F.; Nardi, E.; De Biase, D.; Massari, F.; Giunchi, F.; Pepe, F.; Troncone, G.; et al. Storage Time and DNA Quality Determine BRCA1/2 Sequencing Success in Prostate Cancer: A Multicentre Analysis with Therapeutic Implications. Cancers 2025, 17, 1705. [Google Scholar] [CrossRef]
- Pandey, S.; Yadav, P. Liquid Biopsy in Cancer Management: Integrating Diagnostics and Clinical Applications. Pract. Lab. Med. 2025, 43, e00446. [Google Scholar] [CrossRef]
- Ionescu, F.; Zhang, J.; Wang, L. Clinical Applications of Liquid Biopsy in Prostate Cancer: From Screening to Predictive Biomarker. Cancers 2022, 14, 1728. [Google Scholar] [CrossRef]
- Morrison, G.J.; Goldkorn, A. Development and Application of Liquid Biopsies in Metastatic Prostate Cancer. Curr. Oncol. Rep. 2018, 20, 35. [Google Scholar] [CrossRef]
- Brinkmann, C.; Baum, R.P.; Stargardt, T. Cost-Utility Analysis of 177Lu-PSMA-617 Radioligand Therapy in Second-Line and Third-Line Treatment for Metastatic Castration-Resistant Prostate Cancer (mCRPC) in Germany. Eur. J. Nucl. Med. Mol. Imaging, 2025; in press. [Google Scholar] [CrossRef]
- Teppala, S.; Scuffham, P.A.; Tuffaha, H. The Cost-Effectiveness of Germline BRCA Testing-Guided Olaparib Treatment in Metastatic Castration Resistant Prostate Cancer. Int. J. Technol. Assess. Health Care 2024, 40, e14. [Google Scholar] [CrossRef]
- Xu, C.; Cai, J.; Zhuang, J.; Zheng, B.; Chen, L.; Sun, H.; Zheng, G.; Wei, X.; Liu, M. Cost-Effectiveness of Olaparib, a PARP Inhibitor, for Patients with Metastatic Castration-Resistant Prostate Cancer in China and United States. Ann. Transl. Med. 2022, 10, 830. [Google Scholar] [CrossRef]
- Das, S.; Ganguly, S.C.; Bera, S.; Kundu, M. Advance in Prostate Cancer Biomarker Discovery: Bridging Detection, Prognosis and Therapeutics. Discov. Oncol. 2025, 16, 954. [Google Scholar] [CrossRef]
- Lin, Y.; Zhao, X.; Miao, Z.; Ling, Z.; Wei, X.; Pu, J.; Hou, J.; Shen, B. Data-Driven Translational Prostate Cancer Research: From Biomarker Discovery to Clinical Decision. J. Transl. Med. 2020, 18, 119. [Google Scholar] [CrossRef]
- Ferber, D.; El Nahhas, O.S.M.; Wölflein, G.; Wiest, I.C.; Clusmann, J.; Leßmann, M.-E.; Foersch, S.; Lammert, J.; Tschochohei, M.; Jäger, D.; et al. Development and Validation of an Autonomous Artificial Intelligence Agent for Clinical Decision-Making in Oncology. Nat. Cancer 2025, 1–13. [Google Scholar] [CrossRef]
- Ankolekar, A.; Boie, S.; Abdollahyan, M.; Gadaleta, E.; Hasheminasab, S.A.; Yang, G.; Beauville, C.; Dikaios, N.; Kastis, G.A.; Bussmann, M.; et al. Advancing Breast, Lung and Prostate Cancer Research with Federated Learning. A Systematic Review. NPJ Digit. Med. 2025, 8, 314. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, L.; Bao, X.; Han, Z.; Wang, Z.; Nie, S.; Gu, Y.; Gong, J. Short-Term Peri- and Intra-Tumoral CT Radiomics to Predict Immunotherapy Response in Advanced Non-Small Cell Lung Cancer. Transl. Lung Cancer Res. 2025, 14, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Garje, R.; Riaz, I.B.; Naqvi, S.A.A.; Rumble, R.B.; Taplin, M.-E.; Kungel, T.M.; Herchenhorn, D.; Zhang, T.; Beckermann, K.E.; Vapiwala, N.; et al. Systemic Therapy in Patients With Metastatic Castration-Resistant Prostate Cancer: ASCO Guideline Update. J. Clin. Oncol. 2025, 43, 2311–2334. [Google Scholar] [CrossRef] [PubMed]

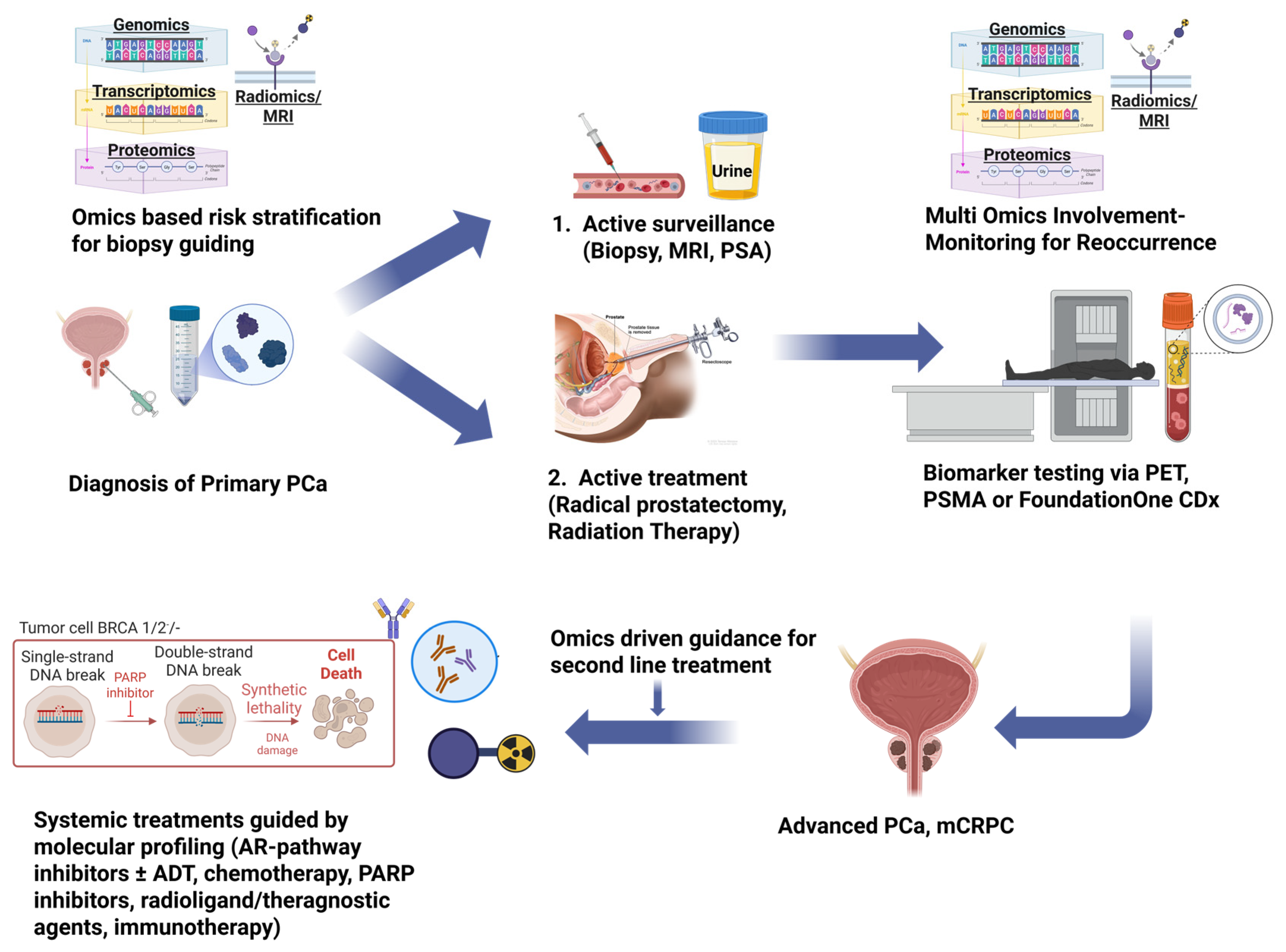
| Combination Therapy (Dose) | Genomic Requirement (Companion Diagnostic) | Pivotal Trial (s) | Key Treated Population (n) | Key Efficacy Outcome | p-Value (Primary Endpoint †) | Notable Points |
|---|---|---|---|---|---|---|
| Olaparib (300 mg) vs. abiraterone/enzalutamide (monotherapy) [44,48] | BRCA1/2 or ATM mutations by FoundationOne CDx, BRACAnalysis CDx or qualified liquid CDx | PROfound (phase III) | Cohort A, n = 245 (387 total) | rPFS 7.4 vs. 3.6 mo, HR 0.34; OS 19.1 vs. 14.7 mo, HR 0.69 | <0.001 | 66% crossover; 33% screen-failure; grade ≥ 3 anaemia 23% |
| Rucaparib (600 mg) (monotherapy) [54] | Deleterious germline/somatic BRCA1/2 (central or validated local NGS) | TRITON2 (phase II, single-arm) → TRITON3 (phase III) | TRITON2 BRCA n = 115 (62 RECIST-evaluable); TRITON3 BRCA n = 302 | ORR 44% (TRITON2); rPFS 11.2 vs. 6.4 mo, HR 0.50 (TRITON3) | TRITON2—(single-arm study; no comparator, therefore no p value) TRITON3 < 0.0001 (rPFS); TRITON3 | Accelerated approval awaiting OS verification; 47% tissue-plasma discordance; grade ≥ 3 anaemia ≈ 25% |
| Olaparib (300 mg) + Abiraterone (1000 mg)/Prednisone (5 mg) (combination therapy) [59] | None (all-comers); preplanned BRCA1/2 subgroup | PROpel (phase III) | ITT n = 796; BRCA subset n = 85 | ITT rPFS 24.8 vs. 16.6 mo, HR 0.66; BRCA HR 0.24; interim OS HR 0.30 | <0.0001 (rPFS ITT) | Label restricted to BRCA; grade ≥ 3 anaemia 15–16%; 14% discontinuation; first PARP–NHAA doublet approval |
| Niraparib 200 mg and Abiraterone acetate (1000 mg) + prednisone/prednisolone (10 mg) (combination therapy) [62] | HRR-positive by tissue/plasma NGS; label restricted to BRCA1/2 | MAGNITUDE (phase III) | ITT n = 423; BRCA subgroup n = 225 | rPFS (BRCA) 16.6 vs. 10.9 mo, HR 0.53; HRR-pos HR 0.73 | 0.0014 (rPFS in BRCA) | OS neutral; grade ≥ 3 anaemia 29.7%; 15% discontinuation; volumetric PSMA metrics complementary |
| Talazoparib (0.5 mg) + Enzalutamide (160 mg) (combination therapy) [63] | HRR-deficiency mutation confirmed via FoundationOne CDx | TALAPRO-2 (phase III) | ITT n = 805; HRR mutation subset n = 399 | ITT rPFS NR vs. 21.9 mo, HR 0.63; HRR subset HR 0.45 | <0.0001 (rPFS ITT) | FDA approved on 20 June 2023, for HRR-mutated mCRPC.
|
| Combination/Intervention (Dose) | Genomic/Molecular Requirement (Companion Assay) | Trial (Phase) | Key Treated Population (n) | Key Efficacy Outcome | p-Value (Primary End-Point) | Notable Points |
|---|---|---|---|---|---|---|
| Ipatasertib (400 mg) with abiraterone vs. placebo with abiraterone [84] | PTEN-loss by IHC (FoundationOne® genomic confirmatory subset) | IPATential150 (III) | PTEN-loss subset ≈ 521 of 1101 | rPFS 18.5 vs. 16.5 mo (HR 0.77) | 0.034 | OS HR 0.94; NS; grade ≥ 3 AEs 70% vs. 39% |
| Intermittent docetaxel (withheld when plasma mGSTP1 clears) vs. continuous docetaxel (75 mg m−2 q3w) [85] | Detectable mGSTP1 ctDNA at baseline and clearance after 2 cycles (mSTRAT assay) | GUIDE (II) | Target 28 (was 120); 6 randomised to date | Ongoing—primary rPFS | NS | First epigenetic ctDNA-adaptive chemotherapy; slow accrual, trial now closed to recruitment |
| Evolocumab (420 mg sc q4w) + SOC therapy [88] | PCPro lipidomic score > −1.1903 (5-analyte ceramide/TRG/CH total) | Evolocumab-PCPro (II, single-arm) | PCPro-positive pts (target ≈ 40) | Primary: PCPro re-classification at wk 12 (ongoing) | NS | Precision metabolomics strategy; endpoints include PSA50 and broad lipidomic shifts |
| Nivolumab and ipilimumab (two schedules) [89] | Immunogenic signature (ImS+): MMR-d, DDR-d or high TILs ≥ 20% | NEPTUNES (II) | C1 35, C2 36 | Composite response rate 40% vs. 25% (overall 32%) | (≥40% predefined as meaningful) | Higher grade 3–4 AEs in IPI-intense arm; responders enriched for MMR-d & BRCA/ATM loss |
| AZD4635 75 mg qd ± durvalumab 1500 mg q4w [90] | Baseline 14-gene AdenoSig (exploratory, not selection) | NCT02740985 (Ia/b) | 108 total (mono 65; combo 43) | ORR 5% mono, 16% combo; PSA50 22% combo; AdenoSig-high rPFS 21 vs. 8.7 wks (HR ≈ 0.46) | NS | First-in-human adenosinergic blockade; benefit confined to AdenoSig-high tumors |
| Early docetaxel 75 mg m−2 q3w + ADT vs. ADT alone [91] | PAM50 luminal-basal subtype & Decipher GC (RNA) | CHAARTED correlative (III) | Analytic set n = 160 | Luminal B OS benefit HR 0.45 (p = 0.007); GC-high greatest 3-yr OS gain (+25%) | 0.007 (luminal B) | Transcriptomics refines who benefits from upfront docetaxel in mHSPC |
| Outcome-adaptive ARPI (abiraterone/enzalutamide) vs. taxane (docetaxel/cabazitaxel) [92] | Real-time 78-gene ctDNA panel → 5 molecular signatures (AR–/TP53-WT, TP53-alt, TMPRSS2-ERG, HRD, unselected) | ProBio platform (adaptive RCT) | First 218 randomisations (193 pts) | ARPI vs. taxane: TTNLCB† 11.1 vs. 6.9 mo (STR 1.60); OS 38.7 vs. 21.7 mo | Bayesian adaptive (no fixed α) | Greatest ARPI benefit in AR–/TP53-WT & TMPRSS2-ERG; proof-of-concept for same-day ctDNA-guided randomisation |
| Biomarker (Analytical Platform/Biospecimen) | Registration/Approval or Investigational Status (as of 2025) | Matched Therapeutic Scheme |
|---|---|---|
| BRCA1/2 pathogenic mutation (NGS/tissue or plasma) [51,56,63] | Approved—FDA-cleared companion tests (FoundationOne CDx, BRACAnalysis) | PARP inhibitors: olaparib with/without abiraterone, rucaparib niraparib combined with abiraterone, talazoparib combined with enzalutamide |
| High PSMA expression on ^68Ga-PSMA-11 PET/CT test (molecular imaging) [71] | Approved-Phase III (VISION), imaging-based test as a companion to therapy | Radioligand therapy lutetium-177 vipivotide tetraxetan (Pluvicto™) |
| PTEN-loss (IHC or NGS assay/blood plasma samples) [84] | Phase III (IPATential150) | Ipatasertib 400 mg daily plus abiraterone |
| PCPro ceramide lipidomic score (high) (Ceramide biomarker panels/Blood plasma sample) [88] | Phase II (PCPro) | Evolocumab (PCSK9 inhibitor) added to standard therapy |
| Immunogenic signature assay (ImS+): MMR-d, DDR-d or high TILs ≥ 20%, (Blood specimens; plasma or serum) [89] | Phase II (NEPTUNES; ongoing) | Nivolumab (PD-1 inhibitor) in combination with ipilimumab (CTLA4 inhibitor) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fatima, Y.; Jobre, K.N.; Gomez-Gomez, E.; Małkiewicz, B.; Vlahou, A.; Mokou, M.; Mischak, H.; Frantzi, M.; Jankowski, V. Omics-Mediated Treatment for Advanced Prostate Cancer: Moving Towards Precision Oncology. Int. J. Mol. Sci. 2025, 26, 7475. https://doi.org/10.3390/ijms26157475
Fatima Y, Jobre KN, Gomez-Gomez E, Małkiewicz B, Vlahou A, Mokou M, Mischak H, Frantzi M, Jankowski V. Omics-Mediated Treatment for Advanced Prostate Cancer: Moving Towards Precision Oncology. International Journal of Molecular Sciences. 2025; 26(15):7475. https://doi.org/10.3390/ijms26157475
Chicago/Turabian StyleFatima, Yasra, Kirubel Nigusu Jobre, Enrique Gomez-Gomez, Bartosz Małkiewicz, Antonia Vlahou, Marika Mokou, Harald Mischak, Maria Frantzi, and Vera Jankowski. 2025. "Omics-Mediated Treatment for Advanced Prostate Cancer: Moving Towards Precision Oncology" International Journal of Molecular Sciences 26, no. 15: 7475. https://doi.org/10.3390/ijms26157475
APA StyleFatima, Y., Jobre, K. N., Gomez-Gomez, E., Małkiewicz, B., Vlahou, A., Mokou, M., Mischak, H., Frantzi, M., & Jankowski, V. (2025). Omics-Mediated Treatment for Advanced Prostate Cancer: Moving Towards Precision Oncology. International Journal of Molecular Sciences, 26(15), 7475. https://doi.org/10.3390/ijms26157475